
Abstract
Senescent cells activate genetic programmes that irreversibly inhibit cellular proliferation, but also endow these cells with distinctive metabolic and signalling phenotypes. Although senescence has historically been considered a protective mechanism against tumourigenesis, the activities of senescent cells are increasingly being associated with age-related diseases, including cancer. An important feature of senescent cells is the secretion of a vast array of pro-inflammatory cytokines, chemokines, and growth factors collectively known as the senescence-associated secretory phenotype (SASP). Recent research has shown that SASP paracrine signalling can mediate several pro-tumourigenic effects, such as enhancing malignant phenotypes and promoting tumour initiation. In this review, we summarise the paracrine activities of senescent cells and their role in tumourigenesis through direct effects on growth and proliferation of tumour cells, tumour angiogenesis, invasion and metastasis, cellular reprogramming and emergence of tumour-initiating cells, and tumour interactions with the local immune environment. The evidence described here suggests cellular senescence acts as a double-edged sword in cancer pathogenesis, which demands further attention in order to support the use of senolytic or SASP-modulating compounds for cancer treatment.
Similar content being viewed by others


Introduction
The field of senescence has greatly expanded since the sensencent cell state was first observed in normal human fibroblasts, by Hayflick and Moorhead, over half a century ago.1 Initially referring to the finite proliferative capacity of cells in vitro, senescence is now defined as a cellular state of stable and long-term loss of proliferative capacity, but with the retention of normal metabolic activity and viability. It is characterised by specific changes in morphology (e.g. enlarged and flat cells), metabolism (e.g. increased glycolysis over mitochondrial oxidative phosphorylation), and cell physiology (e.g. resistance to apoptosis).2,3,4,5
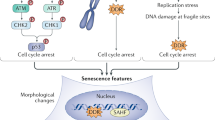
Senescence serves as a response to stress, and several inducing stimuli have now been identified including chemotherapeutic, radiation and oxidative stress, amongst others (Fig. 1). Activation of the senescence programme leads to cellular and molecular changes such as proliferation arrest, chromatin remodelling, elevated expression of cell cycle inhibitors (such as p16INK4A or p21CIP1), activation of a DNA damage response, enlargement of the lysosomal compartment, and activation of a senescence-associated secretory phenotype (SASP).5,6 The SASP mediates the paracrine activities of senescent cells through the secretion of a myriad of factors including cytokines and chemokines (e.g. IL1α, IL1β, IL6, IL8, CXCL1, CXCL2), growth factors (e.g. amphiregulin, EGF, BMPs, FGFs, VEGF, WNTs), extracellular matrix (ECM) components (e.g. fibronectin), and proteases (e.g. MMPs, plasminogen activators), as well as exosome-like small extracellular vesicles.7,3,8,9,10,11 The composition and intensity of the SASP response can be affected by several factors including the senescence-inducing mechanism, cell type, and the amount of time passed since senescence initiation, indicating that there is no singular SASP.12,13,14,15,16,17

Overview of senescence inducers, changes in cell physiology, and activation of the senescence-associated secretory phenotype (SASP). The senescence programme can be activated by different stress stimuli (shown in blue) such as: cytotoxic chemotherapeutic drugs, replicative stress (which occurs due to deficiencies in the DNA replication machinery or maintenance of cell cycle checkpoints), ionising radiation, oncogenic signalling, and oxidative stress. The main cellular and molecular effects are shown in red and include an expansion of the lysosomal compartment, metabolic and mitochondrial alterations, accumulation of DNA damage and rearrangement of the chromatin landscape, resistance to apoptosis, and an irreversible arrest of the cell cycle. Most senescent cells also activate a senescence-associated secretory phenotype (SASP), which is composed of growth factors, cytokines, chemokines, and metalloproteinases. Examples of common SASP factors are shown. These secreted factors can signal in an autocrine fashion to reinforce the senescence phenotype, or paracrinally with multiple effects on neighbouring cells. EGF epithelial growth factor, FGFs fibroblast growth factors, BMPs bone morphogenetic proteins, IL1 interleukin 1, IL6 interleukin 6, IL8 interleukin 8, CCL2 C–C motif chemokine ligand 2, MMP2 matrix metallopeptidase 2, MMP3 matrix metallopeptidase 3
SASP effects can be beneficial or deleterious for normal physiology depending on its composition, intensity, and the local tissue microenvironment. Furthermore, the SASP is involved in valuable physiological processes such as promoting tissue repair,18,19,20 fine-tuning the development of embryonic structures,21,22,23 and stimulating immune surveillance.24,25 However, the deleterious consequences that result from ineffective clearance of senescent cells and their over-accumulation in tissues can promote age-related diseases and cancer.2,26,27,28,29,30 Supporting this notion, the burden of senescent cells in tissues increases significantly with age in mice, primates, and humans,27 and they can be found in both benign and malignant tumours.31,32,33,34,35 Importantly, genetic or chemical ablation of senescent cells in mouse models delays the onset of age-related disorders, including cancer, leading to increased life-spans and promoting tissue rejuvenation in late life.36,37,38
Senescence was traditionally considered an innate anti-cancer mechanism as it can serve to eliminate damaged cells,3,5 whereby activation of the senescence programme in cells harbouring oncogenic mutations serves as a tumour suppressor mechanism, preventing the expansion of these mutated cells and progression into malignancies.3,5 However, the role of senescence in tumourigenesis has been revised in recent years. There is mounting evidence that dysregulation or inappropriate activation of senescence contributes to tumour progression and malignancy.5,7,17 This review will discuss the paracrine effects of senescent cells on different aspects of tumour cell behaviour including: (i) direct effects on growth and proliferation of tumour cells; (ii) tumour angiogenesis, invasion, and metastasis; (iii) cellular reprogramming and emergence of tumour-initiating cells; and (iv) tumour interactions with the local immune environment (Fig. 2). These subdivisions of the senescence-associated activities are mainly conceptual; senescent cells exert compounded effects and it is not easy to distinguish between some of these activities through current experimental approaches, especially in an in vivo context. For in-depth discussion of the functions of cellular senescence in physiological processes, such as embryological development and tissue repair, as well as in ageing, we refer the reader to other reviews in the field.2,3

Summary of the paracrine effects of the SASP in promoting tumourigenesis
Growth and proliferation of tumour cells
Cells present in the tumour microenvironment, such as fibroblasts, can become senescent and promote the growth and proliferation of tumour cells.7,39,40 This has been demonstrated both in vitro and in vivo. Co-culture of senescent fibroblasts, induced by various stimuli (e.g. radiation, DNA damage, replicative exhaustion), can promote the growth and proliferation of benign, pre-malignant, and malignant cells from a range of tumour types.9,12,26,41,42,43,44 For example, in co-culture assays, radiation-induced senescent fibroblasts sustained the growth of mammary epithelial cells that had dysregulated cell cycle and cell death pathways.44 These in vitro observations have been further substantiated in vivo, where co-injection of senescent fibroblasts has been shown to increase tumourigenicity in xenograft models, including primary breast cancer tissues.26,45,46,47
The contributions of specific SASP components have been demonstrated using genetic knockdown, siRNAs, and other molecular inhibitors.12,26,41,45,46,48,49 The use of siRNA and blocking antibodies against amphiregulin (AREG) reduced the growth of benign prostate epithelial cells induced by conditioned media from senescent fibroblasts.48 Furthermore, a critical role for SASP in the promotion of obesity-associated liver cancer has been demonstrated using elegant genetic approaches,50 where deletion of IL1β (Il1b) was sufficient to reduce the expression of IL6 and CXCL1 in the liver, as well as the number and size of liver tumours.
Finally, there is evidence showing that the expression of growth factors alone, including some that fall under the SASP umbrella, can induce tumours independently in a paracrine/non-cell autonomous manner.51 For example, expression of fibroblast growth factor 10 (FGF10) by urogenital mesenchymal cells results in the induction of multifocal prostatic adenocarcinoma in epithelial cells.52 Similarly, expression of fibroblast growth factor 19 (FGF19) by skeletal muscle cells has been shown to induce hepatocellular carcinomas, which acquire somatic mutations in β-catenin (Ctnnb1).53 These experiments collectively demonstrate that the SASP can promote cancer cell growth, challenging the view that senescence is primordially a beneficial process involved in preventing cancer progression.
Tumour angiogenesis, invasion, and metastasis
Senescent cells can contribute to the acquisition of invasive and metastatic properties of cancer cells, as well as the induction of tumour-associated angiogenesis.7,39 Tumour invasion and metastasis frequently involve an epithelial to mesenchymal shift in cellular phenotype (epithelial–mesenchymal transition, EMT). During EMT, epithelial cells attain key aspects enabling tumour invasion, including loss of cellular polarity and cell-to-cell adhesion, and gain of both migratory and invasive properties. Importantly, it is known that conditioned media from senescent cells can induce EMT in cell lines derived from many tumour types, including non-aggressive breast cancer, mesothelioma, and melanoma, as evidenced by decreased expression of epithelial markers (e.g. E-cadherin, cytokeratins) and increased expression of mesenchymal markers (e.g. vimentin).12,54,55 In addition, individual SASP components can contribute to induce EMT phenotypes; IL6, for example, has been shown to have cell-adhesion disrupting actions, which is an important component of invasion.56 Senescent cells and the SASP can also guide and promote cancer cell migration/invasion in models of thyroid and skin cancers.57,58 Furthermore, ablation of senescent cells after chemotherapy can prevent or delay cancer relapse and spread to distal tissues.59 Tumour invasion and metastasis also involve disruption of the basement membrane and remodelling of the ECM by matrix metalloproteinases (MMPs), which are often expressed as SASP factors.7 Indeed, the invasive properties of several epithelial cell types are enhanced by MMPs secreted by senescent cells, such as MMP2 and MMP3.41,43,44
A large number of proangiogenic factors are also known to be secreted by senescent cells, whereas angiostatic molecules have not been found to be secreted.27,60 In particular, IL6 has been reported to promote tumour-supportive angiogenesis in a Ras-driven tumour model.61 Similarly, co-injection of senescent fibroblasts or peritoneal mesothelial cells with cancer cells in xenograft models results in significantly greater tumour angiogenesis.62,63 These data suggest that the paracrine activities of senescent cells are involved in the acquisition of malignant and metastatic phenotypes by signalling to transformed cells or their microenvironment.
Cellular reprogramming of cells and emergence of tumour-initiating cells in culture
Tumour cells may exhibit loss of differentiation and may also attain stem cell characteristics; both features of cancer progression. In benign tumours and well-differentiated cancers, the histology of a tumour typically recapitulates the histology of the tissue of origin. In contrast, undifferentiated cancers have abnormal histology and typically exhibit more aggressive behaviour, as less differentiated cells are usually more proliferative.
Interestingly, the SASP is able to inhibit differentiation both in vitro and in vivo, while in some cases leads to acquisition of stem cell characteristics.41,44,54,55,64,65,66 Exposure of keratinocytes to the culture medium from senescent cells promotes expression of tumour stem cell markers, such as CD44, and leads to a greater regenerative capacity in vivo. 65 Similarly, co-culturing undifferentiated myeloma cells in conditioned media from senescent myeloma cells promotes the emergence, maintenance, and migration of cancer stem-like cells.64 Higher in vivo expression of stem cell markers has also been observed in the liver in close association with GFP-labelled RAS-induced senescent cells.65 In addition, induction of senescence and SASP in mesothelioma cells led to the emergence of a subpopulation of highly clonogenic cells with enhanced ability to form tumours when xenografted in mice.55 Furthermore, cellular reprogramming, which is the process by which adult differentiated cells can be induced to become functionally equivalent to embryonic stem cells, can be induced in vivo by senescent cells through SASP activation, and this can be stimulated in different models of tissue damage. While senescence is a barrier to reprogramming in vitro, the paracrine activities of senescent cells can promote the expression of stem cell markers and proliferation of neighbouring cells in vivo, 66,67,68 and IL6 is a key player in driving this process.
The molecular mechanisms underpinning the paracrine induction of cancer stem cell features have been variably addressed. For instance, non-tumourigenic melanoma cells exposed to IL6 or chemokine ligand-2 (CCL2) develop tumourigenic potential in vivo in a STAT3-dependent manner.54 In vitro, co-culture experiments showed that SASP induced the expression of critical reprogramming factors NANOG, SOX2, and OCT4.54 Indeed, it has further been shown that increased IL6 expression, through induction of senescence either genetically or from tissue damage, can create a tissue context that increases reprogramming efficiency in vivo. 66 In this sense, a crucial role for the mechanistic target of rapamycin (mTOR) complex has recently been unveiled, whereby it can either counteract or facilitate reprogramming by cell-intrinsic and cell-extrinsic mechanisms, respectively.69 Together, these data suggest that senescent cells, through their SASP, can induce undifferentiated cellular states; depending on the context, this can be beneficial (e.g. tissue regeneration) or harmful (e.g. promotion of tumour-initiating cells).
Modulation of local immune response and immune evasion by senescent cells
The relationship between senescence, tumourigenesis, and the immune system is complex and remains incompletely understood. Cells undergoing damage-induced senescence are often cleared by the immune system, as several SASP factors are cytokines and chemokines that can modulate the local immune environment.2,3,5,70,71 In this regard, the SASP has been shown to promote inflammation.7,72
Immune surveillance refers to the removal of pathogens, as well as pre-malignant and malignant cells, by the immune system. In some cases, it has been shown that senescent cells are involved in these processes. For example, senescent cells promote their own clearance through the secretion of CCL2, which attracts and activates NK-T cells.73,74 Using a mouse model of liver carcinoma, p53-deficient RAS-driven tumours induced to senesce through re-establishment of p53 function exhibited innate immune cells migrating into the vicinity of the senescent tumour area, leading to complete tumour regression.24 Such senescence-induced activation of the local immune system has also been shown to activate the clearance of pre-malignant hepatocytes.75
In contrast, senescent cells can also promote tumour evasion of immune surveillance.76,77 During ageing of the skin, senescent stromal cells and their SASP (particularly IL6) drive an increase in the number of suppressive myeloid cells in mice and humans. Furthermore, it was shown that this leads to the inhibition of anti-tumour T-cell responses and enhanced tumour growth.77 Further research is required to clarify the factors that control the pro- and anti-tumour surveillance activities of senescent cells.
Conclusion
There is increasing evidence indicating that, in addition to their cell- and non-cell autonomous tumour-suppressive activities, the paracrine signals derived from senescent cells have detrimental roles in aging-related pathogenesis and cancer. Since senescent cells are generally abundant in benign tumours and also present at low numbers in several malignancies,31,32,33,34,57 their paracrine activities could contribute to tumour progression and cancer metastasis. Moreover, it is possible that these activities may also be involved in the initial steps of oncogenic transformation of normal cells and tumour initiation, as recently suggested in a mouse model of a human brain tumour.78 Promising translational opportunities have emerged in the use of molecules that selectively target and eliminate senescent cells (termed senolytics), or those that modulate the SASP and its negative effects (Table 1).79 In this regard, the elimination of senescent cells or targeting the SASP represents a potential strategy for stopping or slowing tumour progression, as many activities of senescent cells promote tumour growth and malignant progression. It may be expected that the same paracrine activities capable of enhancing the cancerous phenotype of cells harbouring oncogenic mutations in vitro and in vivo, could also contribute to the initial epigenetic and genetic alterations that fuel the appearance of tumour-initiating cells in normal, non-transformed cells.78 If so, early ablation of senescent cells in pre-malignant lesions using senolytic compounds or neutralisation of the SASP may provide a plausible approach to prevent cancer.
References
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Munoz-Espin, D. & Serrano, M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Wiley, C. D. & Campisi, J. From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab. 23, 1013–1021 (2016).
Rodier, F. & Campisi, J. Four faces of cellular senescence. J. Cell Biol. 192, 547–556 (2011).
van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
Coppe, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev. Pathol. 5, 99–118 (2010).
Kirkland, J. L. & Tchkonia, T. Cellular senescence: a translational perspective. EBioMedicine 21, 21–28 (2017).
Takasugi, M. et al. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 8, 15729 (2017).
Lehmann, B. D. et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 68, 7864–7871 (2008).
Hernandez-Segura, A. et al. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–60.e4 (2017).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Maciel-Baron, L. A. et al. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age 38, 26 (2016).
Coppe, J. P. et al. Tumor suppressor and agingbiomarkerp16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 286, 36396–36403 (2011).
Kipling, D. et al. A transcriptomic analysis of the EK1.Br strain of human fibroblastoid keratocytes: the effects of growth, quiescence and senescence. Exp. Eye Res. 88, 277–285 (2009).
Hoare, M. et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 18, 979–992 (2016).
Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J. & Kirkland, J. L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 123, 966–972 (2013).
Jun, J. I. & Lau, L. F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 12, 676–685 (2010).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell. 31, 722–733 (2014).
Krizhanovsky, V. et al. Senescence of activated stellate cells limits liver fibrosis. Cell 134, 657–667 (2008).
Storer, M. et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130 (2013).
Munoz-Espin, D. et al. Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118 (2013).
Davaapil, H., Brockes, J. P. & Yun, M. H. Conserved and novel functions of programmed cellular senescence during vertebrate development. Development 144, 106–114 (2017).
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Lujambio, A. To clear, or not to clear (senescent cells)? That is the question. Bioessays 38(Suppl. 1), S56–S64 (2016).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. Y. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA 98, 12072–12077 (2001).
Campisi, J. Aging, cellular senescence, and cancer. Annu Rev. Physiol. 75, 685–705 (2013).
Lecot, P., Alimirah, F., Desprez, P. Y., Campisi, J. & Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 114, 1180–1184 (2016).
Kuilman, T. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
Perez-Mancera, P. A., Young, A. R. & Narita, M. Inside and out: the activities of senescence in cancer. Nat. Rev. Cancer 14, 547–558 (2014).
Collado, M. et al. Tumour biology: senescence in premalignant tumours. Nature 436, 642 (2005).
Haugstetter, A. M. et al. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 103, 505–509 (2010).
Vizioli, M. G. et al. Evidence of oncogene-induced senescence in thyroid carcinogenesis. Endocr. Relat. Cancer 18, 743–757 (2011).
Reimann, M. et al. Tumor stroma-derived TGF-beta limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell. 17, 262–272 (2010).
Roberson, R. S., Kussick, S. J., Vallieres, E., Chen, S. Y. & Wu, D. Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 65, 2795–2803 (2005).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Baker, D. J. et al. Naturallyoccurringp16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Alspach, E., Fu, Y. & Stewart, S. A. Senescence and the pro-tumorigenic stroma. Crit. Rev. Oncog. 18, 549–558 (2013).
Liu, S. et al. Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci. Rep. 5, 17895 (2015).
Parrinello, S., Coppe, J. P., Krtolica, A. & Campisi, J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J. Cell Sci. 118(Pt 3), 485–496 (2005).
Dilley, T. K., Bowden, G. T. & Chen, Q. M. Novel mechanisms of sublethal oxidant toxicity: induction of premature senescence in human fibroblasts confers tumor promoter activity. Exp. Cell Res. 290, 38–48 (2003).
Liu, D. & Hornsby, P. J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 67, 3117–3126 (2007).
Tsai, K. K., Chuang, E. Y., Little, J. B. & Yuan, Z. M. Cellular mechanisms for low-dose ionizing radiation-induced perturbation of the breast tissue microenvironment. Cancer Res. 65, 6734–6744 (2005).
Capell, B. C. et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 30, 321–336 (2016).
Laberge, R. M. et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 (2015).
Jeon, H. Y. et al. Irradiation induces glioblastoma cell senescence and senescence-associated secretory phenotype. Tumour Biol. 37, 5857–5867 (2016).
Bavik, C. et al. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 66, 794–802 (2006).
Herranz, N. et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 17, 1205–1217 (2015).
Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013).
Martinez-Barbera, J. P. & Andoniadou, C. L. Concise review: paracrine role of stem cells in pituitary tumors: a focus on adamantinomatous craniopharyngioma. Stem Cells 34, 268–276 (2016).
Memarzadeh, S. et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell. 12, 572–585 (2007).
Nicholes, K. et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am. J. Pathol. 160, 2295–2307 (2002).
Ohanna, M. et al. Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget 4, 2212–2224 (2013).
Canino, C. et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 31, 3148–3163 (2012).
Tamm, I., Kikuchi, T., Cardinale, I. & Krueger, J. G. Cell-adhesion-disrupting action of interleukin 6 in human ductal breast carcinoma cells. Proc. Natl Acad. Sci. USA 91, 3329–3333 (1994).
Kim, Y. H. et al. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat. Commun. 8, 15208 (2017).
Farsam, V. et al. Senescent fibroblast-derived Chemerin promotes squamous cell carcinoma migration. Oncotarget 7, 83554–83569 (2016).
Demaria, M. et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176 (2017).
Oubaha, M. et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 8, 362ra144 (2016).
Ancrile, B., Lim, K. H. & Counter, C. M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719 (2007).
Coppe, J. P., Kauser, K., Campisi, J. & Beausejour, C. M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 281, 29568–29574 (2006).
Mikula-Pietrasik, J. et al. Senescent peritoneal mesothelium induces a pro-angiogenic phenotype in ovarian cancer cells in vitro and in a mouse xenograft model in vivo. Clin. Exp. Metastasis 33, 15–27 (2016).
Cahu, J., Bustany, S. & Sola, B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 3, e446 (2012).
Ritschka, B. et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 31, 172–183 (2017).
Mosteiro, L. et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 354, (2016).
Chiche, A. et al. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell. Stem Cell. 20, 407–14.e4 (2017).
Mosteiro, L., Pantoja, C., de Martino, A. & Serrano, M. Senescence promotes in vivo reprogramming through p16(INK)(4a) and IL-6. Aging Cell (2017). PubMed PMID: 29280266. Epub 2017/12/28. eng. https://doi.org/10.1111/acel.12711.
Aarts, M. et al. Coupling shRNA screens with single-cell RNA-seq identifies a dual role for mTOR in reprogramming-induced senescence. Genes Dev. 31, 2085–2098 (2017).
Vicente, R., Mausset-Bonnefont, A. L., Jorgensen, C., Louis-Plence, P. & Brondello, J. M. Cellular senescence impact on immune cell fate and function. Aging Cell 15, 400–16 (2016).
Velarde, M. C., Demaria, M. & Campisi, J. Senescent cells and their secretory phenotype as targets for cancer therapy. Interdiscip. Top. Gerontol. 38, 17–27 (2013).
Ruhland, M. K., Coussens, L. M. & Stewart, S. A. Senescence and cancer: an evolving inflammatory paradox. Biochim. Biophys. Acta 1865, 14–22 (2016).
Iannello, A., Thompson, T. W., Ardolino, M., Lowe, S. W. & Raulet, D. H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 210, 2057–2069 (2013).
Sagiv, A. & Krizhanovsky, V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology 14, 617–628 (2013).
Kang, T. W. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479, 547–551 (2011).
Toso, A. et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 9, 75–89 (2014).
Ruhland, M. K. et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 7, 11762 (2016).
Gonzalez-Meljem, J. M. et al. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat. Commun. 8, 1819 (2017).
Childs, B. G. et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 16, 718–735 (2017).
Yosef, R. et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 7, 11190 (2016).
Moiseeva, O. et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 12, 489–498 (2013).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Orjalo, A. V., Bhaumik, D., Gengler, B. K., Scott, G. K. & Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl Acad. Sci. USA 106, 17031–17036 (2009).
Fuhrmann-Stroissnigg, H. et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 8, 422 (2017).
Acknowledgements
The authors would like to thank Dr. Cynthia L. Andoniadou and Scott Haston for their useful comments and critical appraisal during the preparation of the manuscript. This work was supported by the Medical Research Council (MRC) (Grants MR/M000125/1), Great Ormond Street Hospital for Children Charity/Children with Cancer UK (GOSHCC/CWCUK) (Grant W1055) and by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children National Health Service Foundation Trust and University College London. J.M.G.-M. is supported by a CONACYT-UCL Postgraduate Fellowship and a CONACYT-REDESD travel fellowship. J.R.A. is supported by a Cancer Research UK Clinical Research Training Fellowship. J.P.M.-B. is a Great Ormond Street Hospital for Children’s Charity Principal Investigator.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Note: This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gonzalez-Meljem, J.M., Apps, J.R., Fraser, H.C. et al. Paracrine roles of cellular senescence in promoting tumourigenesis. Br J Cancer 118, 1283–1288 (2018). https://doi.org/10.1038/s41416-018-0066-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-018-0066-1
This article is cited by
-
Human cytomegalovirus infection triggers a paracrine senescence loop in renal epithelial cells
Communications Biology (2024)
-
Aging microenvironment and antitumor immunity for geriatric oncology: the landscape and future implications
Journal of Hematology & Oncology (2023)
-
The roles of bone remodeling in normal hematopoiesis and age-related hematological malignancies
Bone Research (2023)
-
Role of senescent tumor cells in building a cytokine shield in the tumor microenvironment: mathematical modeling
Journal of Mathematical Biology (2023)
-
Cellular senescence in cancer: clinical detection and prognostic implications
Journal of Experimental & Clinical Cancer Research (2022)
