
Abstract
Background
Obesity and related factors have been implicated as possible aetiological factors for the development of glioma in epidemiological observation studies. We used genetic markers in a Mendelian randomisation framework to examine whether obesity-related traits influence glioma risk. This methodology reduces bias from confounding and is not affected by reverse causation.
Methods
Genetic instruments were identified for 10 key obesity-related risk factors, and their association with glioma risk was evaluated using data from a genome-wide association study of 12,488 glioma patients and 18,169 controls. The estimated odds ratio of glioma associated with each of the genetically defined obesity-related traits was used to infer evidence for a causal relationship.
Results
No convincing association with glioma risk was seen for genetic instruments for body mass index, waist-to-hip ratio, lipids, type-2 diabetes, hyperglycaemia or insulin resistance. Similarly, we found no evidence to support a relationship between obesity-related traits with subtypes of glioma–glioblastoma (GBM) or non-GBM tumours.
Conclusions
This study provides no evidence to implicate obesity-related factors as causes of glioma.
Similar content being viewed by others


Introduction
Glioma is the most common primary intracranial tumour, accounting for around 80% of all malignant brain tumours.1 Thus far, few established risk factors for the development of glioma have been robustly identified.2
Obesity-related factors are increasingly being recognised as risk determinants for the development many of common cancers, such as those of the breast and colorectum.3 Evidence from epidemiological observational studies, for obesity-related traits being a risk factor for the development of glioma have, however been inconsistent, with only a subset of studies reporting a significant association.4,5,6,7,8,9 Furthermore, in contrast to most cancers, some studies have reported diabetes to be protective against glioma.10,11,12,13 Obesity-related exposures are however inherently interrelated,14, 15 and in traditional epidemiological studies it can be problematic to isolate specific risk factors that may exert a causal influence on disease from those that are merely associated with an underlying causal factor (i.e. confounded). In addition, findings can be affected by reverse causation.
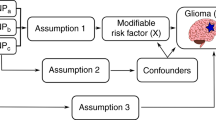
Mendelian randomisation (MR) is an analytical approach to the traditional epidemiological study whereby genetic markers are used as proxies or instrumental variables (IVs) of environmental and lifestyle-related risk factors.16 Such genetic markers cannot be influenced by reverse causation and can act as unconfounded markers of exposures provided the variants are not associated with the disease through an alternative mechanism.16 Under these circumstances, the association between a genetic variant (or set of variants) and outcome of interest implies a causal relationship between the risk factor and outcome. MR has therefore been compared to a natural randomised controlled trial, circumventing some of the limitations of epidemiological observational studies.17 However, as IVs used in MR often explain a small proportion of the exposure phenotypic variance, large sample sizes are required to have sufficient power.18
To gain insight into the aetiology of glioma, we have examined the role of obesity-related risk factors in glioma using an MR-based framework. Specifically, we identified genetic variants associated with 10 key obesity-related risk factors from external genetic association studies. We implemented two-sample MR19 to estimate associations between these genetic variants with glioma risk using genome-wide association study (GWAS) data from the Glioma International Case-Control Consortium study (GICC).20
Materials and methods
Two-sample MR was undertaken using GWAS data. Ethical approval was not sought for this specific project because all data came from the summary statistics of published GWAS, and no individual-level data were used.
Genetic instruments for obesity and related risk factors
Genetic instruments were identified as a panel of single-nucleotide polymorphisms (SNPs) identified from recent meta-analyses or largest studies published to date. Specifically: (i) SNPs for body mass index (BMI) and waist-to-hip ratio (WHR) were identified from the Genetic Investigation of ANthropometric Traits (GIANT) consortium;21, 22 (ii) SNPs for circulating high-density and low-density lipoprotein cholesterol (HDL and LDL), total cholesterol and triglycerides, were identified from the Global Lipids Genetic Consortium (GLGC);23 (iii) SNPs for factors related to hyperglycaemia and hyperinsulinemia—fasting glucose, fasting insulin and 2-h post-challenge glucose, were obtained from the Meta-Analysis of Glucose and Insulin related traits Consortium (MAGIC)24 and (iv) SNPs for type-2 diabetes were identified from.25 For each SNP, we recovered the chromosome position, the effect estimate expressed in standard deviations (SD) of the trait per-allele along with the corresponding standard error (Supplementary Table 1). We restricted our analysis to SNPs associated at genome-wide significance (i.e. P ≤ 5.0 × 10−8) in individuals with European ancestry. To avoid co-linearity between SNPs for each trait, we excluded SNPs that were correlated (i.e. r2 ≥ 0.01) within each trait, and only considered the SNPs with the strongest effect on the trait for inclusion in genetic risk scores (Supplementary Table 2). For type-2 diabetes, linkage disequilibrium (LD) scores with rs140730081 were calculated via a proxy SNP rs2259835 (r2 = 0.48). After imposing these criteria, we obtained 7 SNPs for 2-h post-challenge glucose, 75 for BMI, 33 for fasting glucose, 13 for fasting insulin, 54 for HDL cholesterol, 26 for LDL cholesterol, 38 for type-2 diabetes, 39 for total cholesterol, 25 for triglycerides and 33 for WHR.
Glioma association results
To evaluate the association of each genetic instrument with glioma risk, we made use of data from the most recent meta-analysis of GWAS in glioma, comprising >10 million genetic variants (after imputation) in 12,488 glioma patients and 18,169 controls from eight independent GWAS data sets of individuals of European descent (Supplementary Table 3).20 Comprehensive details of the genotyping and quality control of the seven GWAS have been previously reported.20 To limit the effects of cryptic population stratification, association test statistics for six of the glioma GWAS were generated using principal components as previously detailed.20 Gliomas are heterogeneous and different tumour subtypes, defined in part by malignancy grade (e.g. pilocytic astrocytoma World Health Organization (WHO) grade I, diffuse ‘low-grade’ glioma WHO grade II, anaplastic glioma WHO grade III and GBM WHO grade IV) can be distinguished.26 For the sake of diagnostic brevity, we considered gliomas as being either GBM or non-GBM tumours.
Statistical analysis
The odds ratios (OR) of glioma per unit of SD increment for each obesity-related trait, were estimated using generalised summary data-based Mendelian randomisation (GSMR).27 This approach performs a multi-SNP MR analysis, which is more powerful than other existing summary data-based MR methodologies.28 Separation of signals of causality from horizontal pleiotropy (a single locus influencing affecting multiple phenotypes, also referred to as type-II pleiotropy) is a recognised issue in MR analyses and we therefore used a HEIDI-outlier test27 to detect and eliminate genetic instruments that have apparent pleiotropic effects on both the obesity-related trait and glioma. A P value threshold of 0.01 for the HEIDI-outlier test was utilised as recommended by Zhu et al. The HEIDI-outlier test may also in theory detect additional violations of the assumptions of MR such as the exclusion restriction assumption. Given that glioma is a binary outcome and type-2 diabetes a binary exposure, the resulting causal effect estimate in this scenario represents the odds for glioma risk per unit increase in the log OR for type-2 diabetes.
For each statistical test, we considered a global significance level of P < 0.05 as being satisfactory to derive conclusions. To assess the robustness of our conclusions, we imposed a Bonferroni-corrected significance threshold of 0.0017 (i.e. 0.05/30, to correct for testing 10 traits over three outcomes). We considered a P value > 0.05 as non-significant (i.e. no association), a P value ≤ 0.05 as evidence for a potential causal association, and a P value ≤ 0.0017 as significant evidence for an association. Additionally, we defined the Bayesian false null probability (BFNP) using the Bayesian false discovery probability (BFDP) as per Wakefield29 by BFNP = 1 − BFDP. Then to assess whether null results found could be considered reliable, we calculated the minimum prior probability of the alternative hypothesis for which the BFNP was >10%. The power of an MR investigation depends greatly on the proportion of variance in the risk factor that is explained by the respective IV. We estimated study power a priori using the methodology of Burgess.30 Statistical analyses were undertaken using R software (Version 3.1.2).
Results
In our data sets, there were missing data for one fasting insulin SNP (rs1530559), four type-2 diabetes SNPs (rs2972156, rs34706136, rs11257658, rs144613775) and one total cholesterol SNP (rs7570971). These SNPs were excluded from our analysis. Performing HEIDI-outlier analysis on the instruments for each trait identified two SNPs as violating the assumptions of MR with respect to horizontal pleiotropy, rs11603023 for total cholesterol and rs5756931 for triglyceride, which were further excluded. Both SNPs are in LD with the lead SNP in glioma risk loci.
Subsequently, Table 1 details the number of SNPs used as an IV for each of the obesity-related traits, the mean and SD of the risk factor in the original discovery study, and the proportion of variance explained for each factor by the corresponding genetic instruments. Effect estimates for each SNP used as genetic instruments for each risk factor and disease risk are detailed in Supplementary Table 1. For BMI and LDL, the SNPs rs12016871 and rs9411489 have since merged with the SNPs rs9581854 and rs635634, respectively, and it is from these subsequent SNPs the associations with glioma were derived. Figure 1 shows the statistical power of genetic instruments for different levels of predicted ORs for each obesity-related trait.

Study power against OR for each obesity-related trait and all glioma (P = 0.05, two-sided). A line indicating a power of 80% is shown. BMI body mass index, HDL high-density lipoprotein, LDL low-density lipoprotein, OR odds ratio
Figure 2 shows a plot of the association of each IV with exposure against the association with glioma, together with the resulting GSMR estimate of the log OR. For each of the obesity-related traits under investigation, an approximately null estimate for effect was obtained, with the strongest association being shown by fasting insulin. Setting a threshold of P ≤ 0.05, no statistically significant associations were shown for 2-h post-challenge glucose (ORSD = 1.25, 95% confidence interval (CI) = 0.93–1.67), BMI (ORSD = 0.91, 95% CI = 0.77–1.07), fasting glucose (ORSD = 1.00, 95% CI = 0.78–1.3), fasting insulin (ORSD = 1.32, 95% CI = 0.71–2.46), HDL cholesterol (ORSD = 1.01, 95% CI = 0.98–1.05), LDL cholesterol (ORSD = 1.00, 95% CI = 0.95–1.05), type-2 diabetes (ORSD = 1.04, 95% CI = 0.97–1.11), total cholesterol (ORSD = 0.98, 95% CI = 0.88–1.09), triglycerides (ORSD = 1.01, 95% CI = 0.97–1.06) and WHR (ORSD = 1.11, 95% CI = 0.84–1.46).

SNP-specific effects for risk of all glioma. For each figure, the effect size of the respective measure for: a 2-h post-challenge glucose, b BMI, c fasting glucose, d fasting insulin, e HDL cholesterol, f LDL cholesterol, g type-2 diabetes, h total cholesterol, i triglycerides and j WHR is plotted against the effect for all glioma. Error bars represent one SD. The GSMR estimate is plotted as a dashed line for reference. BMI body mass index, GSMR generalised summary data-based Mendelian randomisation, HDL high-density lipoprotein, LDL low-density lipoprotein, SD standard deviation, WHR waist–hip ratio
We explored the possibility that a relationship between an obesity-related trait and glioma might be subtype-specific, considering GBM and non-GBM separately. Figures 3 and 4 show corresponding plots of the association of each IV with exposure against the association with GBM and non-GBM glioma. The strongest association was provided by the relationship between increased triglyceride level and risk of non-GBM glioma (ORSD = 1.07, 95% CI = 1.00–1.13, P = 0.044), albeit non-significant after adjustment for multiple testing (Table 2). Table 3 presents the minimum prior probabilities of an association required for each trait to have a BFNP ≥ 0.1. Where possible, the maximum likely OR has been taken from the largest value reported in observational studies.7, 12, 31 In the event that this was not possible, an upper bound of 2 was chosen. If the ‘true’ maximum likely OR were lower, then the smallest required prior probability would in fact be lower. There is no current precedent for what value should be taken for the prior probability of an association, indeed attempting to sample published papers would produce an over estimation due to winners curse, but it is noted that a value of 10% would ensure all the results reported would have significance.

SNP-specific effects for risk of GBM glioma. For each figure, the effect size of the respective measure for a 2-h post-challenge glucose, b BMI, c fasting glucose, d fasting insulin, e HDL cholesterol, f LDL cholesterol, g type-2 diabetes, h total cholesterol, i triglycerides and j WHR is plotted against the effect for GBM glioma. Error bars represent one SD. The GSMR estimate is plotted as a dashed line for reference. BMI body mass index, GBM glioblastoma mulitforme, GSMR generalised summary data-based Mendelian randomisation, HDL high-density lipoprotein, LDL low-density lipoprotein, SD standard deviation, WHR waist–hip ratio

SNP-specific effects for risk of non-GBM glioma. For each figure, the effect size of the respective measure for a 2-h post-challenge glucose, b BMI, c fasting glucose, d fasting insulin, e HDL cholesterol, f LDL cholesterol, g type-2 diabetes, h total cholesterol, i triglycerides and j WHR, is plotted against the effect for non-GBM glioma. Error bars represent one SD. The GSMR estimate is plotted as a dashed line for reference. BMI body mass index, GBM glioblastoma mulitforme, GSMR generalised summary data-based Mendelian randomisation, HDL high-density lipoprotein, LDL low-density lipoprotein, SD standard deviation, WHR waist–hip ratio
Discussion
There is an abundance of studies that have implicated obesity and related traits (notably diabetes), as risk factors for all of the major common cancers, including breast, colorectal, oesophageal, pancreatic, ovarian and renal.3 Furthermore, there is increasing evidence that obesity is likely to also be a risk factor for many of the less common tumours, such as those of the haematopoietic system.3, 32 The mechanistic basis of how obesity and diabetes affects an increased cancer risk is poorly understood. The long-term metabolic consequences of obesity and its related traits are complex and several mechanisms have been suggested, including increased insulin and insulin-like growth factor signalling, chronic inflammation and signalling via adipokines.33 Such mechanisms would be compatible with obesity and related traits having a generic effect on cancer risk.
Evidence for obesity influencing risk of glioma from previous observational studies has been mixed.4, 6, 9 Intriguingly, in contrast to other cancers, an inverse relationship between both diabetes and increased HbA1c with risk of glioma has been reported in some but not all studies.4,5,6,7, 9 Furthermore, in so far as it has been studied, anti-diabetic treatment has been reported to not influence glioma risk.12 In terms of the wider spectrum of the metabolic syndrome, a study has linked elevated levels of triglyceride to risk of developing glioma.9
Our findings do not support a causal role for higher BMI and related metabolic risk factors, including diagnosis of type-2 diabetes and blood lipid levels, in influencing glioma risk. An important strength of our analysis is that by utilising the random allocation of genetic variants, we were able to overcome potential confounding, for example, from other interrelated traits.14, 15 Furthermore, reverse causation and selection bias may have biased estimates from previously published observational studies. By exploiting data from large genetic consortia for multiple obesity-related traits and glioma risk has enabled us to more precisely test study hypotheses than if we had been reliant on individual-level data from a small study. The only obesity-related trait with a first-stage F-statistic <10 was WHR (F = 6.75) and therefore weak instrument bias for other traits is unlikely.34 In addition, given that a poor outcome from glioma is almost universal, it is unlikely that survival bias will have influenced study findings materially. Finally, we have employed a Bayesian approach to interpret the significance of the null results while comparing our findings to published observational epidemiological studies. There is currently no precedent within the MR community as to what value is an accurate representation of the prior probability of association. If the true value is ~20%, then the null findings for 2 h post-challenge glucose, BMI, fasting glucose, fasting insulin and WHR all have a >10% chance of being false.
There are however potential limitations in our analysis that warrant further discussion. Firstly, the use of summary test statistics in two-sample MR analyses requires consideration of sample overlap, the winner’s curse and genotype uncertainty.35, 36 Sample overlap between the association studies of the exposure traits and outcome trait has the potential of inflating the type I error rate. The number of controls shared between the glioma GWAS and the anthropometric and lipid GWAS are, however <2% of the respective exposure sample size. Although we are unable to calculate an exact number of glioma cases sampled in the exposure GWAS, given the lifetime risk of glioma is only 0.24%, very few numbers of glioma cases will have been analysed in the exposure trait studies. Hence, such sample overlap is unlikely to contribute to type I error rate inflation.36 As the instrumental variables were discovered in the data used in this two-sample MR analysis, weak instrument bias will be accentuated due to winner’s curse, thus attenuating the causal effect estimate towards the null.36 Uncertainty with respect to genotyping or disease associations may diminish causal effect estimates.36 However IVs used in this analysis are robust and only SNPs passing stringent quality control thresholds were used in the analysis. Secondly, MR is limited in the extent to which it can explore different life course models, such as when an exposure has a temporal relationship to the outcome risk.35 Finally, our study does have limitations related to power. However, based on the relatively sizable fraction of variance explained by the genetic instruments for the majority of the obesity-related factors (Table 1), typically there was sufficient statistical power (>80%) to detect even modest odds ratios of 1.43, and close to complete statistical power (99%) to detect relative risks of 1.72 (Fig. 1).
In conclusion, our findings shed light on an issue for which the evidence to date has been mixed. Specifically, they provide evidence against obesity and related traits as significant risk factors for the development of glioma.
Availability of data and material
Genotype data from the GICC GWAS are available from the database of Genotypes and Phenotypes (dbGaP) under accession phs001319.v1.p1. In addition, genotypes from the GliomaScan GWAS can be accessed through dbGaP accession phs000652.v1.p1.
References
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. NeuroOncology 15, ii1–56 (2013).
Ostrom, Q. T. et al. The epidemiology of glioma in adults: a “state of the science” review. NeuroOncology 16, 896–913 (2014).
Kyrgiou, M. et al. Adiposity and cancer at major anatomical sites: umbrella review of the literature. BMJ 356, j477 (2017).
Kaplan, S., Novikov, I. & Modan, B. Nutritional factors in the etiology of brain tumors: potential role of nitrosamines, fat, and cholesterol. Am. J. Epidemiol. 146, 832–841 (1997).
Niedermaier, T. et al. Body mass index, physical activity, and risk of adult meningioma and glioma: a meta-analysis. Neurology 85, 1342–1350 (2015).
Sergentanis, T. N. et al. Obesity and risk for brain/CNS tumors, gliomas and meningiomas: a meta-analysis. PLoS ONE 10, e0136974 (2015).
Wiedmann, M. et al. Body mass index and the risk of meningioma, glioma and schwannoma in a large prospective cohort study (The HUNT Study). Br. J. Cancer 109, 289–294 (2013).
Dai, Z.-F., Huang, Q.-L. & Liu, H.-P. Different body mass index grade on the risk of developing glioma: a meta-analysis. Chin. Neurosurg. J. 1, 7 (2015).
Edlinger, M. et al. Blood pressure and other metabolic syndrome factors and risk of brain tumour in the large population-based Me-Can cohort study. J. Hypertens. 30, 290–296 (2012).
Kitahara, C. M. et al. Personal history of diabetes, genetic susceptibility to diabetes, and risk of brain glioma: a pooled analysis of observational studies. Cancer Epidemiol. Biomark. Prev. 23, 47–54 (2014).
Schwartzbaum, J. et al. Associations between prediagnostic blood glucose levels, diabetes, and glioma. Sci. Rep. 7, 1436 (2017).
Seliger, C. et al. Diabetes, use of anti-diabetic drugs, and the risk of glioma. NeuroOncology 18, 340–349 (2016).
Zhao, L., Zheng, Z. & Huang, P. Diabetes mellitus and the risk of glioma: a meta-analysis. Oncotarget 7, 4483–4489 (2016).
Brown, C. D. et al. Body mass index and the prevalence of hypertension and dyslipidemia. Obes. Res. 8, 605–619 (2000).
GBD 2015 Obesity Collaborators et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 377, 13–27 (2017).
Davey Smith, G. & Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–R98 (2014).
Nitsch, D. et al. Limits to causal inference based on Mendelian randomization: a comparison with randomized controlled trials. Am. J. Epidemiol. 163, 397–403 (2006).
Freeman, G., Cowling, B. J. & Schooling, C. M. Power and sample size calculations for Mendelian randomization studies using one genetic instrument. Int. J. Epidemiol. 42, 1157–1163 (2013).
Pierce, B. L. & Burgess, S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 178, 1177–1184 (2013).
Melin, B. S. et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 49, 789–794 (2017).
Locke, A. E. et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015).
Shungin, D. et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015).
Willer, C. J. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
Scott, R. A. et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 44, 991–1005 (2012).
Gaulton, K. J. et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type-2 diabetes susceptibility loci. Nat. Genet. 47, 1415–1425 (2015).
Louis, D. N. et al. The 2016 World Health Organization Classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016).
Zhu Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9, 224 (2018).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Wakefield, J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet. 81, 208–227 (2007).
Burgess, S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int. J. Epidemiol. 43, 922–929 (2014).
Derr, R. L. et al. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 27, 1082–1086 (2009).
Yang, T. O. et al. Body size in early life and risk of lymphoid malignancies and histological subtypes in adulthood. Int J. Cancer 139, 42–49 (2016).
Font-Burgada, J., Sun, B. & Karin, M. Obesity and cancer: the oil that feeds the flame. Cell Metab. 23, 48–62 (2016).
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Lawlor, D. A. Commentary: two-sample Mendelian randomization: opportunities and challenges. Int. J. Epidemiol. 45, 908–915 (2016).
Burgess, S., Davies, N. M. & Thompson, S. G. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 40, 597–608 (2016).
Acknowledgements
L.D.-H. was supported by a Wellcome Trust Summer Student bursary. A.S. is supported by a Cancer Research UK clinical fellowship and The Royal Marsden Hospital Haematology Research Fund. In the UK, funding was provided by Cancer Research UK (C1298/A8362 supported by the Bobby Moore Fund). The GICC was supported by grants from the National Institutes of Health, Bethesda, Maryland (R01CA139020, R01CA52689, P50097257, P30CA125123). The UK Interphone Study was supported by the European Commission Fifth Framework Program ‘Quality of Life and Management of Living Resources’ and the UK Mobile Telecommunications and Health Programme. The Mobile Manufacturers Forum and the GSM Association provided funding for the study through the scientifically independent International Union against Cancer (UICC).
Author information
Authors and Affiliations
Contributions
R.S.H., A.S. and A.J.C. managed the project. L.D.-H., A.S., P.J.L., A.J.C. and R.S.H. drafted the manuscript. L.D.-H. performed statistical analyses. B.K., K.L., A.J.S. and R.H.S. provided UK data. M. Simon, P.H., M.M.N. and K.-H.J. provided German data. Q.T.O., J.E.E.-P., G.N.A., E.B.C., D.I., J.S., J.S.B.-S., S.H.O., J.L.B., R.K.L., C.J., R.B.J., B.S.M., M.R.W., M.L.B. and R.S.H. provided GICC data. S.C. and P.R. provided National Cancer Institute (NCI) data. M. Sanson provided French data. All authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Ethics approval and consent to participate
Two-sample MR was undertaken using GWAS data. Ethical approval was not sought for this specific project because all data came from the summary statistics of published GWAS, and no individual-level data were used.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Disney-Hogg, L., Sud, A., Law, P.J. et al. Influence of obesity-related risk factors in the aetiology of glioma. Br J Cancer 118, 1020–1027 (2018). https://doi.org/10.1038/s41416-018-0009-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-018-0009-x
This article is cited by
-
Causal relationship between type 2 diabetes and glioblastoma: bidirectional Mendelian randomization analysis
Scientific Reports (2024)
-
Modifiable risk factors for glioblastoma: a systematic review and meta-analysis
Neurosurgical Review (2023)
-
Systematic review of Mendelian randomization studies on risk of cancer
BMC Medicine (2022)
-
Searching for causal relationships of glioma: a phenome-wide Mendelian randomisation study
British Journal of Cancer (2021)
-
Testing for causality between systematically identified risk factors and glioma: a Mendelian randomization study
BMC Cancer (2020)
