
Abstract
Disc degeneration primarily contributes to chronic low back and neck pain. Consequently, there is an urgent need to understand the spectrum of disc degeneration phenotypes such as fibrosis, ectopic calcification, herniation, or mixed phenotypes. Amongst these phenotypes, disc calcification is the least studied. Ectopic calcification, by definition, is the pathological mineralization of soft tissues, widely studied in the context of conditions that afflict vasculature, skin, and cartilage. Clinically, disc calcification is associated with poor surgical outcomes and back pain refractory to conservative treatment. It is frequently seen as a consequence of disc aging and progressive degeneration but exhibits unique molecular and morphological characteristics: hypertrophic chondrocyte-like cell differentiation; TNAP, ENPP1, and ANK upregulation; cell death; altered Pi and PPi homeostasis; and local inflammation. Recent studies in mouse models have provided a better understanding of the mechanisms underlying this phenotype. It is essential to recognize that the presentation and nature of mineralization differ between AF, NP, and EP compartments. Moreover, the combination of anatomic location, genetics, and environmental stressors, such as aging or trauma, govern the predisposition to calcification. Lastly, the systemic regulation of calcium and Pi metabolism is less important than the local activity of PPi modulated by the ANK-ENPP1 axis, along with disc cell death and differentiation status. While there is limited understanding of this phenotype, understanding the molecular pathways governing local intervertebral disc calcification may lead to developing disease-modifying drugs and better clinical management of degeneration-related pathologies.
Similar content being viewed by others

Back pain and Intervertebral disc degeneration
Chronic low back pain (cLBP) and neck pain are the top causes of years lived with disability and health care costs worldwide.1,2 Despite the critical need for clinical approaches to facilitate the social and medical impact, a complete understanding of the etiology and pathophysiology of this painful condition remains elusive.2,3 Among several identified factors, disc degeneration is considered a primary contributing factor to cLBP.3 Disc degeneration, also described as discarthrosis, has the potential to lead to spine instability, stenosis, spondylolisthesis, and deformity, all described as causes of radiculopathy, chronic back and neck pain, paresthesia, or even neurological deficits.4,5,6 In the US alone, over $100 billion is estimated to be spent annually on direct and indirect costs of disc degeneration and low back/neck pain.2 Despite the high socioeconomic burden of disc degeneration, there are no disease-modifiable therapies, and surgery is still the only suboptimal solution for partial symptomatic relief when conservative treatment fails.7
The intervertebral disc and adjacent vertebrae comprise a functional motion segment, allowing flexibility, range of motion, and biomechanical properties of the spinal column.8 Notably, the interaction between the disc compartments is essential to promote a hydraulic mechanism for handling the loading and mechanical stress intrinsic to spine movement.9 The nucleus pulposus (NP) is the central tissue derived from the embryonic notochord and is rich in hydrophilic aggrecan molecules that give the disc its hydration.10 The annulus fibrosus (AF) surrounds the NP and restricts the NP swelling with its circumferential and concentric lamellae made of collagen fibers.11 Finally, the endplate (EP) comprising a cartilaginous and bony region borders the NP and AF cranially and caudally, allowing the infusion of nutrients and oxygen to cells in these compartments [10].
Disc degeneration sub-phenotypes
The disc degenerative process is multifactorial and influenced by genetics, obesity, type II diabetes, and lifestyle choices, such as activity status, smoking and nicotine use, and abnormal loading.12,13,14,15 Moreover, aging is also considered one of the leading risk factors in the progression of disc degeneration and has become more relevant with the increase in life expectancy.16 There is ample evidence implicating an essential role of genetics in the predisposition to disc degeneration.17 In a twin study, Battié and colleagues showed that genetic background was the top determinant of disc degeneration, surpassing aging and mechanical loading.18 Additionally, several single-nucleotide polymorphisms related to matrix anabolism, catabolism, inflammation, and cell signaling have exhibited a strong correlation to disc degeneration.19,20,21 Finally, abnormal spine mechanics in pathologies afflicting the vertebral column, such as scoliosis, kyphosis, and spino-pelvic malalignment, also promote disc degeneration.22,23,24
Irrespective of the etiology, disc degeneration is a heterogeneous pathology and comprises a broad spectrum of phenotypes such as fibrosis, ectopic calcification, herniation, or mixed phenotypes.5,25,26 Unlike degenerative pathologies of other related skeletal tissues, the distinction among these phenotypes is often not precise, and there is still some controversy about different stages of degeneration vs. different degenerative phenotypes. Recent animal and human studies suggest that each degenerative phenotype presents a unique molecular and structural signature.27 Loss of disc viscoelastic properties characterizes fibrotic phenotypes due to a shift in biosynthesis from proteoglycan rich-matrix towards a stiffer extracellular matrix (ECM) rich in collagens, along with hypertrophic chondrocyte-like differentiation of resident cells and a decrease in disc height.28,29,30 Disc herniation, on the other hand, results from the protrusion of NP content through AF or endplate, with possible neurological symptoms and local inflammation with or without pronounced immune cell ingress.31 Disc calcification is possibly the least understood among these phenotypes, usually characterized by calcium phosphate mineral deposition in one or all disc compartments with pronounced cell death.32
Intervertebral disc calcification
Disc calcification is highly prevalent in older individuals and is associated with disc aging25 and higher grades of degeneration.33 Intriguingly, despite a higher propensity of decline at levels C5/6, T6/7, and L4/5, the distribution of disc calcification throughout the spine exhibits variability among studies.25,27,34,35 Whether this process is a response to altered biomechanics or a consequence of a degenerative cascade is still unknown. Additionally, there is an association between disc calcification with systemic diseases such as ochronosis, hemochromatosis, chondrocalcinosis, ankylosing spondylitis, pseudogout, hyperparathyroidism, as well as spine deformities.25,36,37 However, the true prevalence of disc calcifications may be underdiagnosed because of the low sensitivity of routine spine imaging techniques such as X-rays and MRI scans.36,38 CT scan is the ideal modality for diagnosing and quantifying this phenotype, but no studies have yet characterized the incidence of calcifications seen on CT at the population level.38
Histological studies of cadaveric human tissues and animal models suggest disc calcification may occur in all three disc compartments, associated with increased calcium levels, inorganic phosphate, and elevated collagen (COL) 10 and tissue non-specific alkaline phosphatase (TNAP) activity.27,32 Interestingly, disc degeneration with a higher prevalence of disc calcification is also linked to increased expression of osterix, RUNX2, bone morphogenic protein (BMP) 2, and osteocalcin (OCN), all known markers of osteogenic differentiation.27,39,40 A recent study showed that deficiency of progranulin (PGRN), a pleiotropic growth factor, increases tartrate-resistant acid phosphatase (TRAP), Cathepsin, COL10, matrix metallopeptidase (MMP) 13, and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) 5 levels, leading to dystrophic calcification, disc narrowing, and degeneration in mice.41 Disc cells respond to local tissue ossification, but whether they are the initiator of calcification needs to be better established. Interestingly, during pathologies that afflict spinal stability, such as spondylolisthesis, scoliosis, or degenerative progression of the disc, osteophyte formation is usually seen as a physiological response to attain local fusion and, possibly, to promote motion segment stabilization.42 Similar to disc degeneration, calcification correlates with the incidence of scoliosis, with prominent endplate involvement, suggesting vertebra-disc co-remodeling to achieve fusion and stability.32,43
Unlike intervertebral discs, in large joints, such as the knee, calcium phosphate, and calcium pyrophosphate dihydrate are the most common types of calcium crystals.44 Knee meniscus calcification, for example, is found in nearly 20% of the elderly, even without a history of joint disorder. However, 80% of meniscal calcification is associated with cartilage lesions in age-controlled patients.45,46 In osteoarthritic menisci, calcification shows a different extracellular matrix composition and higher content of calcium phosphate in comparison to primary chondrocalcinosis, characterized by calcium pyrophosphate dehydrate deposits.47 Although the trigger for joint calcification is unknown, there are some well-established hallmarks of the disease: hypertrophic chondrocytes, presenting the high expression of COL10;39,48 TNAP, ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), and progressive ankylosis gene (ANK) upregulation;49,50 and local inflammation.51 Despite the prevalence of cartilage calcification and its close link to OA and cartilage trauma, the pathophysiology in the knee, similar to the intervertebral disc, is still poorly understood, which hinders the development of disease-modifying drugs.
Ectopic mineralization-–molecular and cellular pathways
Mineralization, the ordered deposition of hydroxyapatite, is a tightly controlled physiological process restricted to specific locations of the body.52 The pathological deposition of hydroxyapatite outside the boundaries of the rigid skeleton is known as ectopic mineralization, a condition that affects millions of people worldwide. Ectopic mineralization is linked to clinical pathologies such as trauma, aging, cancer, diabetes, and autoimmune diseases, all significant causes of morbidity and mortality.16,52 Although ectopic mineralization can occur in any tissue, it is often associated with blood vessels, tumors, ligamentous/cartilaginous tissues, and joints.49,53,54 Our understanding of how the systems that prevent ectopic mineralization are regulated under physiological conditions still needs to be completed, resulting in an overall lack of success in developing effective treatments for this pathology.
The most common mineral pathologically deposited in soft tissues is hydroxyapatite, composed of calcium phosphate crystals Ca10(PO4)6(OH)2. It can, however, also consist of calcium oxalates and octacalcium phosphate, as for instance, often seen in kidney stones.55 Two main processes responsible for ectopic calcification are dystrophic and heterotopic ossification, which diverge histologically and in composition.56,57 Dystrophic calcification is characterized by amorphous, unorganized calcium phosphate crystals interspersed with necrotic debris, not associated with ECM such as collagens, shown by a high ratio of phosphate to protein content.27,56,57 In opposition, heterotopic ossification is characterized by mineralized mature bone, with minerals associated with local ECM, presenting low ratio phosphate to protein, and elevated local activity of TNAP and TRAP associated with osteoblast and osteoclast-like cells.56,57
The soft tissues are usually protected against uncontrolled mineralization by two complementary systems: proteins and low molecular weight compounds (<1 000 Da). The first group includes proteins like fetuin-A, osteopontin, and Matrix Gla Protein (MGP).58 In contrast, the best-known example of the second group is inorganic pyrophosphate (PPi), which consists of two phosphate groups linked by a hydrolyzable ester bond.50,53,59,60,61 PPi is thought to inhibit mineralization by direct interaction with calcium phosphate ion clusters or nanocrystals, thereby preventing their further growth and keeping them in solution for removal by fetuin-A-dependent mechanisms.62 Fetuin-A knockout mice highlight the importance of the interplay between PPi and proteinaceous mineralization inhibitors and do not show an obvious phenotype unless plasma PPi concentrations are substantially decreased.63 In contrast, Mgp−/− mice show extensive vascular and cartilage calcifications without reduced PPi levels, but the phenotypes are rescued when plasma levels of inorganic phosphate (Pi) are reduced.64 Interestingly, in addition to the direct interaction with calcium phosphate clusters, a recent study suggests that PPi may partly exert its biological effects by signaling through G-protein coupled receptor (GPCR) in osteoblasts and osteoclasts.65 It is also important to note that PPi is not the only endogenous small molecule in the anti-mineralization network protecting soft connective tissues against pathological deposition of calcium phosphates. Other molecules involved in the modulation of ectopic mineralization include magnesium and citrate.63,66,67,68
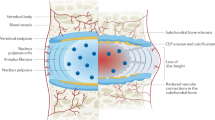
All plasma PPi is derived from adenosine triphosphate (ATP) and other nucleoside triphosphates released into circulation and converted into adenosine monophosphate (AMP) and PPi by ENPP1.69 The two most essential proteins mediating the cellular efflux of ATP underlying PPi formation in plasma are ATP binding cassette subfamily C member 6 (ABCC6) and ANK. ABCC6-dependent ATP release from hepatocytes is responsible for approximately 60%–70% of plasma PPi, with the more ubiquitous membrane protein ANK contributing another 20% by sustaining the cellular efflux of ATP from peripheral tissues.70,71,72,73 Importantly, this multi-pass transmembrane protein ANK provides much of the extracellular ATP needed to form PPi in poorly perfused tissues in addition to mediating the release of citrate and succinate, with the former directly inhibiting mineralization while the latter interacting with specific receptors on the plasma membrane.73 In bones and teeth, PPi is hydrolyzed into Pi by phosphatases, of which TNAP is the most prominent.71 An additional layer of regulation is afforded by the ectonucleotidase CD73, which cleaves AMP into Pi and adenosine.74 The latter inhibits TNAP and lowers PPi degradation. An overview of the factors currently known to participate in extracellular PPi homeostasis is shown in Fig. 1.

Schematic showing intradiscal calcification pathways. AF and EP present a heterotopic ectopic calcification nature – modulated by PPi metabolism - vs. NP, which shows a dystrophic nature, blinded to PPi metabolism changes. Bold proteins and lines – promote calcification. Dotted lines – inhibit calcification. Cross – no role in intradiscal calcification. Question mark – pathway still not explored in the disc. Proteins involved in extracellular PPi homeostasis. All PPi detected in the circulation originate from ENPP1-mediated conversion of extracellular ATP into AMP and PPi. The two main proteins involved in cellular ATP release for ENPP1-mediated PPi formation in plasma are ABCC6, primarily expressed in the liver, and ANK, ubiquitous. PPi is hydrolyzed to inorganic phosphate (Pi) in the periphery by the ecto-enzyme Tissue Non-specific Alkaline Phosphatase (TNAP). CD73 converts AMP into Pi and adenosine. The latter factor inhibits TNAP activity and, subsequently, PPi degradation. Although ENPP1 is only shown in the liver, this ecto-enzyme is ubiquitously expressed and released into blood plasma
ENPP1 plays a central role in PPi generation from ATP. A complete loss of ENPP1 activity results in Generalized Arterial Calcification of Infancy (GACI), a rare condition that, in many cases, leads to death shortly after birth due to massive calcification of the major arteries.49 GACI patients have virtually no PPi in their plasma, which explains their severe and acute form of vascular calcification.75 On the other hand, Pseudoxanthoma elasticum (PXE) is a milder ectopic mineralization disorder caused by mutations in ABCC6, with clinical manifestations noted in various tissues.70 Considering the vital contribution of ANK in PPi homeostasis in tissues that are not well perfused, it is not surprising that loss-of-function mutations in ANK predominantly result in calcification of joint space, articular cartilage, and the disc, eventually resulting in a severe form of ankylosis.76,77 Likewise, inactivating mutations in CD73 result in late-onset ectopic mineralization of periarticular ligaments and the arteries of the lower extremities.78,79 Finally, the absence of functional TNAP results in hypophosphatasia due to increased local levels of PPi and, consequently, hypomineralization of bones.80
In addition to these molecular regulators, several cellular processes participate in pathological tissue mineralization. Inflammation, usually seen in degenerative processes, can trans-differentiate soft tissue cell types such as smooth muscle cells, tenocytes, endothelial cells, and chondrocytes into cells exhibiting osteoblast-like characteristics.49,81,82,83 Extracellular vesicles (EV) secreted by these cells are associated with pathological calcification. In fact, during homeostasis, EV usually contains MGP and fetuin-A, which inhibit local calcification. However, cellular stress causes a shift of EV enriched with calcification makers and calcium phosphate crystals, which promote local calcification by inducing osteogenic and proinflammatory genetic programs and TNAP activity.84,85,86
Similarly, endoplasmic stress response (ER) and autophagy prevent ectopic calcification by promoting cell viability, controlling the secretion of healthy ECM, and facilitating proper protein turnover and intracellular calcium regulation.87,88 For example, vascular calcification shows the involvement of activated PERK-eIF2-ATF4-CHOP complex in different mouse models, which further promotes osteogenic differentiation and mineralization through BMP and RUNX2 pathways.87,89,90 Similarly, autophagy, a key recycling mechanism active during physiological stress, plays a protective role in the presence of high concentrations of Pi by suppressing mammalian target of rapamycin (mTOR) signaling, counteracting reactive oxygen species (ROS), and protecting against apoptosis, ameliorating ectopic calcification.91,92 Finally, cellular fate modulates dystrophic calcification processes. Apoptosis significantly contributes to ectopic calcification by promoting the generation of local calcium phosphate deposits and diminishing the local total capacity of Pi uptake.93,94 On the other hand, cellular differentiation is common in ectopic calcification, characterized by upregulation of BMP2, TNAP, and RUNX2.33,84,95,96
Disc calcification: insight from animal models
The most common animal models used in intervertebral disc biology are bovine, dogs, rabbits, rats, and murine (Tables 1–2). Despite the apparent differences, namely the persistence of notochordal-NP cells, quadrupedal, additional vertebrae, and some variations in the extracellular matrix composition, their contribution to understanding molecular and cellular mechanisms of disc degeneration is well accepted.97 In fact, due to relatively low cost, availability of various molecular genetics tools, and biological similarities with the human skeleton, mouse models have successfully helped to study and understand several common and rare human conditions and diseases that afflict the skeleton.60,98,99,100 Concerning intervertebral disc, for example, the mouse models have been instrumental in our understanding of the embryonic development and nature of cells within each of the disc compartments.101,102,103,104 Studies have shown that, despite differences in gait and posture, the distribution of loads on the vertebrae is similar between quadrupeds and bipeds.105 Indeed, the orientation of vertebral trabeculae is identical between humans and mice, suggesting a similar loading axis.105 While these models need careful interpretation of the findings when drawing parallels to human pathology, they have become valuable tools for understanding various disc degeneration phenotypes, including dystrophic calcification, and even exploring potential therapeutical approaches.27,106 Recent advances in histological methods, scoring systems, and the increased availability of genetically modified mice have further contributed to new insights into pathological intervertebral disc calcification (Table 1).107,108,109
Mouse models of disc calcification
LG/J and SM/J inbred strains
The link between genetic background and age-dependent progression of disc degeneration in mice has recently been explored in a study utilizing three different inbred mouse strains, C57BL/6, LG/J, and SM/J, which showed distinct degenerative phenotypes, namely, fibrosis, disc calcification, and apoptosis with cell fate change, respectively.27 The idea that genetic makeup is an essential contributor to degenerative sub-phenotypes is also supported by twin cohort studies, which showed the strongest correlation between familiar aggregation and disc degeneration, followed by aging and mechanical loading.17,18
We recently studied LG/J and SM/J, super and poor cartilage healing mouse strains with similar ancestry, to investigate age-dependent disc degeneration due to their opposing regenerative phenotypes. C57BL6, one of the most widely used mouse strains in research, was also used for comparison.110,111,112 While SM/J mice showed early disc degeneration, starting at eight weeks, LG/J and C57BL6 showed a healthier phenotype at skeletal maturity.27,110 However, around 18 months of age, unlike C57BL6 and SM/J strains, LG/J mice exhibited intradiscal calcifications in the caudal spine, which became increasingly prevalent at 23 months, with 80% of studied discs evidencing calcification. Interestingly, despite better cartilage healing potential, LG/J is also shown to have a higher predisposition of synovial and meniscus calcifications following knee trauma,113 which further supports a hypothesis that abnormal systemic response to stress, such as aging and trauma, on LG/J genetic background promotes ectopic calcification.
While we observed that LG/J mice possessed widespread calcifications in the NP and AF, no calcification of spinal ligaments was noted, as is seen during ankylosing spondylitis.60 Additionally, elevated levels of free calcium, not associated with mineralization nodules or collagen, and increased focal apoptosis suggested the dystrophic nature of this mineralization. Notably, the calcification was restricted to the caudal spine, with a higher incidence seen at Ca6-7, Ca7-8, and Ca8-9, suggesting that crosstalk between anatomic location, local mechanical environment, and genetic background is essential for the predisposition of disc calcification.34 While LG/J mice showed normal systemic levels of free Ca2+ and phosphorous, TNAP levels were elevated with aging in both LG/J and SM/J mice.53 Surprisingly, unlike LG/J mice, SM/J mice did not show any signs of disc calcification despite very low levels of fetuin-A, a known calcification inhibitor, and high levels of TNAP, a promoter of calcification.61 These findings suggested that systemic changes in regulators of mineral metabolism alone have little effect on intradiscal calcification.
Mechanistic, transcriptomic, and protein analyses of these three strains suggested a contribution of cell death to elevated free local calcium and phosphate to AF tissue mineralization.114 Additionally, resident cells showed an altered differentiation program characterized by increased expression of endochondral genes Bmp6, Tgfb2, Runx2, Fgf2, and Postn with a decrease in terminal osteoblastic differentiation markers Sp7, Bglap, and Alpl.115 An increase in macrophage markers CD14, CD68, and CD163 was also noted, which strongly links macrophage activation with dystrophic calcification in LG/J mice.116 Before leaving this topic, it is essential to note that similar to enriched pathways in old LG/J mice, a subset of human NP samples showed an enrichment of genes in pathways concerning response to stress, wound healing, cell death, endochondral bone, inflammation, cell division, phosphorous metabolism, and extracellular matrix organization.
Mouse models with altered PPi metabolism
ANK is a biological inhibitor of calcified mineralization. Mutations of Ank gene in mice promote abnormal mineral deposition in joints and eventually fusion.76,117 In humans, the loss-of and gain-of-function mutations in Ank are associated with craniometaphyseal dysplasia, ankylosis, and familial chondrocalcinosis, respectively, which are characterized by abnormal facial and long bone growth, skeletal fusions affecting spinal and small articular joints, and hearing ossicles, and in case of chondrocalcinosis, chronic arthropathy resulting from calcium pyrophosphate dihydrate (CPPD) crystal deposition.118
Ank/Ank mice, which carry a nonsense mutation in exon 11 of the murine ortholog Ank (p.E440X), resulting in the C-terminal truncation of the ANK protein by 53 aa and thus a complete loss of ANK function, show severe skeletal phenotypes and disc calcification76,77 The discs show altered extracellular matrix composition with decreased abundance of COLI and COMP but increased COL10.77 Together with upregulated COLX, there are no changes in MMP13 levels, which indicates that AF cells’ differentiation into a true hypertrophic chondrocyte-like phenotype is unlikely. Noteworthy, the mineralization process observed in the intermediate AF and outer AF is different regarding mechanism, composition, and local cell survival. The intermediate AF was acellular, with high radio-opacity and peripheral cells undergoing apoptosis, suggesting a dystrophic calcium deposition. The outer region presented similar X-ray density to bone and densely residing cells, indicative of a more organized mineralized process. Additionally, the outer cell showed TNAP and TRAP activity, with OCN staining without evidence of monocytes, macrophages, and osteoclasts, suggesting the acquisition of an osteoblastic-like phenotype by resident cells of the AF in ank mice.77 These results indicate that different tissues and zones may present a divergent cellular and possibly signaling response to local PPi and calcium deposition, ultimately determining the local mineralization process and nature.65
Noteworthy, transcriptomic analysis highlighted the consequences of Ank loss in affecting ectopic calcification-linked pathways, such as autophagy, with downregulating genes related to protein homeostasis and heat shock protein suggesting proteostasis.92 Additionally, we observed Basic Helix-Loop-Helix ARNT Like 1 (BMAL1)/ Clock circadian regulator (CLOCK) dysregulation in the NP and AF in ank mice, involving several key regulators of the circadian clock – Cry2, Nr1d1, and Per3. Like ank mice, Bmal1−/− mice show disc calcification in the AF compartment and cortical and trabecular bone loss.77,119,120 These results suggest that different tissues may show a divergent cellular response to changes in local PPi levels and calcium deposition, ultimately determining the local mineralization status. Importantly, in addition to controlling local PPi and citrate levels ANK may play a central role in disc calcification by modulating BMAL1/CLOCK pathways, cell fate, autophagy, local inflammation, and matrix remodeling which, are known promoters of ectopic calcification.
Another key regulator of local PPi levels is ENPP1, an enzyme that hydrolyzes ATP to AMP and PPi.121 Asj-2J mice with a spontaneous mutation of ENPP1 have been used as a model of generalized arterial calcification of infancy (GACI). These mice show ectopic calcification in several tissues, including ears, muzzle skin, trachea, aorta, and vertebrae.121 Ectopic calcification in asj-2J mice was only noted in the CEP and AF compartments, without associated osteoblastic and osteoclastic activity, as early as 4–11 weeks of age.121 Together with the phenotype seen in ank mice, the results indicate that PPi levels in AF and CEP compartments are critical to prevent ectopic calcification in the disc.77,121 Considering the dystopic nature of the calcification processes and the presence of these phenotypes only in collagen-rich tissues, we speculate that the higher content of proteoglycans and lower levels of collagens characteristic of the NP compartment may be protective for calcium phosphate deposition, even under conditions of low PPi levels.77,121,122
Similar to ANK, ABCC6 inhibits ectopic mineralization by regulating circulating levels of extracellular ATP, which is converted to AMP and PPi by ENPP1.70 The loss-of-function mutations of ABCC6 cause pseudoxanthoma elasticum (PXE), an autosomal recessive metabolic disorder characterized by ectopic mineralization in elastin-rich tissues.123 Despite ectopic calcification being seen in Abcc6−/− mice in elastin- and collagen-rich connective tissues such as eye, skin, and arteries along with reduced vertebral bone quality, there was no evidence of calcification in the disc.124 It is plausible that the avascular environment of the disc is less sensitive to systemic changes in calcium and PPi levels, a conclusion also supported by the phenotypic assessment of ank/ank, LG/J, and SM/J mice. Furthermore, the unique ECM composition, containing cartilage oligomeric matrix protein (COMP) and proteoglycan aggrecan, known inhibitors of calcification,54,125 may be sufficient to prevent disc mineralization in Abcc6−/− mice.
Slc29a1 −/− mice
Null mutation of the Slc29a1 locus, which encodes equilibrative nucleoside transporter1 (ENT1) – a transmembrane transporter of hydrophilic nucleosides, such as adenosine – associates with ectopic mineralization of the spine in humans.126 Similarly, ENT1−/− mice develop ectopic mineralization of the fibrous connective tissues of the spine, including the AF and adjacent ligaments.60 Consequently, these mice have been successfully used as a model of Diffuse Idiopathic Skeletal Hyperostosis (DISH), a noninflammatory spondyloarthropathy characterized by ectopic calcification of spinal tissues.60,127 Noteworthy, ENT1−/− mice follow the spatial, temporal, and anatomical ectopic mineralization pattern seen in humans with DISH.60 It is important to note that calcification is first observed in ENT1−/− mice between 6 and 8 weeks of age in the paraspinal connective tissues of the cervical vertebrae, developing gradually and caudally until becoming symptomatic by 8 months.
Interestingly, this process occurs without inflammation and, as expected, with significantly elevated plasma adenosine levels. Of note, expression of Mgp, Enpp1, Ank, and Spp1113,128 were significantly downregulated in the discs of ENT1 null mice. This suggests a crosstalk between ENT1, ENPP1, and ANK in AF cells, further supporting their importance in inhibiting disc mineralization. The authors of this study noted that the expression of Alpl, responsible for the hydrolysis PPi, was decreased in the ENT1−/− mice and interpreted this finding as a compensatory down-regulation in response to ectopic mineralization within the disc. However, we opine that this observation could reflect increased plasma adenosine levels, as adenosine is known to inhibit Alpl expression. Surprisingly, however, in contrast to mRNA levels, micro mass cultures of AF cells from ENT1−/− mice under conditions that promote mineralization showed elevated TNAP activity, an observation further supported by higher TNAP activity staining in discs in situ, suggesting a possible local source of mineralization and higher tissue-specific potential to mineralize.127
Sparc −/− mice
Secreted protein, acidic and rich in cysteine (SPARC), also known as osteonectin, is a glycoprotein secreted by osteoblasts during mineralization that binds to calcium and promotes crystal formation.129 Interestingly, SPARC levels decrease during disc degeneration and aging.130 To follow-up on this, Gruber and coworkers first described the disc phenotype of Sparc mutants, which showed accelerated disc degeneration with increased lumbar herniations and endplate calcification and sclerosis.130 Additionally, Sparc null mice develop chronic back pain, demonstrating similar behavioral radiculopathy symptoms observed in humans.131 While disc herniation is directly linked to radiculopathy, endplate changes including calcification and sclerosis also promote chronic back pain.35 Notably, spinal hypersensitivity seen during aging or lumbar spine instability is promoted by osteoclast-dependent sensory innervation in porous areas of the sclerotic endplates.132 Accordingly, inhibiting osteoclastic activity or formation by deletion of Netrin1 in osteoclasts or of Rankl in the osteocytes, respectively, prevents sensory innervation and hypersensitivity.131 Likewise, increasing osteogenesis in sclerotic, porous endplates of mice with lumbar spine instability by resveratrol treatment leads to decreased hyperalgesia.133 SPARC is also known to induce the expression of metalloproteases and collagen, which may affect local tissue homeostasis.134 Sparc knockdown decreased the collagen I expression by AF cells, which may explain the susceptibility to disc herniation in Sparc null mice.129 Significantly, changes in the disc ECM are associated with altered mechanical loading, leading to endplate calcification.35 However, possible dysfunction of endplate cells caused by Sparc modulation may also be involved in this process, as shown in aortic ectopic calcification, by disrupting BMPR-II/p-p38 signaling.135 Overall, these studies underscore the uniqueness of each disc tissue regarding the mineralization process.
Bmal1 −/− mice
The intervertebral disc function depends on the circadian clock to maintain tissue homeostasis.119,136 Mutations in Bmal1, a core clock gene, accelerate disc degeneration and modulate the local inflammatory state.119 Bmal1−/− mice show ectopic calcification restricted to AF, suggesting divergent functions of this pathway across disc tissues. Notably, disruption of BMAL1 also promotes heterotopic calcification of tendons and ligaments with aging via activation of TGF/BMP signaling.137 The functional and structural similarities between AF and tendons/ligaments suggest the critical role of BMAL1 in preventing calcification of ligamentous and fibrocartilaginous tissues. Consequently, during aging, dysregulation and lower levels of BMAL1 may explain the increase in the prevalence of disc calcification by activating TGF/BMP signaling, as noted in ligamentous tissues.138 Supporting this crucial role of Bmal1 contribution to disc calcification, ank mutant mice with dysregulated BMAL1/CLOCK signaling showed similar dystrophic mineralization of the AF. Further studies are needed to explore the possible downstream effectors and molecular targets of BMAL1 signaling to prevent this mineralization process.77,119
Non-murine models of disc calcification
There are clear advantages of murine models in studying the molecular regulation of biological processes through genetic and injury manipulation.97 Nevertheless, intervertebral disc calcification has been well documented in several non-murine models, including ovine,139 chondrodystrophic dogs,140 and sand rats (Psammomys obesus) (Table 2).141 The sand rats show a prominent EP calcification as one of the main features during the age-dependent progression of disc degeneration.141 Melrose et al. described the presence of disc calcification in all three compartments on the ovine discs during aging, with different distribution along the spine, presenting maximal incidence at the lower lumbar level and no deposits in the lumbosacral and lower thoracic disc.139 These results further support the contribution of the interplay between the mechanical factors and anatomical location to disc calcification in line with observations in humans and aged LG/J mice.27,32 Moreover, the mineral deposits in aged ovine discs were composed of hydroxyapatite, showing larger crystallite size than cortical bone, associated with no COL10, osteopontin, and osteonectin, suggesting a dystrophic process, similar to the aged LG/J mice. Furthermore, proteoglycans, known to inhibit calcification, were lower in the calcified disc, possibly justifying the increased susceptibility of ectopic calcification during disc aging.27,139
Genome-wide studies of chondrodystrophic dogs have identified a significant association between disc degeneration, namely with calcification and a polymorphism causing overexpression of FGF4 associated with insertion of FGF4 retrogene on domestic dog (Canis familiaris, CFA) chromosome 12 and 18 - CFA12 and CFA18.140 However, other models have not explored the molecular basis of this phenomenon. While these models fail to study the causative relation between molecular mechanisms and disc calcification processes, they underscore the evolutionary nature of this pathophysiological process, suggesting that aging-associated discal calcification is conserved across vertebrates.
Challenges posed by disc calcification in clinical medicine
From a clinical perspective, few studies have systematically explored the consequences of this disc degeneration sub-phenotype on disease progression, treatment options, and prognosis. Prior studies have described the prevalence of calcified intervertebral disc in the general adult population as approximately 5% on chest radiographs and 6% on abdominal radiographs.142 While the prevalence is even higher in elderly patients, symptomatic disc calcifications have classically been considered rare, especially in adults. However, disc calcification has now become associated with various disease processes contributing to pain, stiffness, and altered spine biomechanics, ultimately leading to significant patient morbidity.
One of the main challenges in clinical medicine is distinguishing the extent of symptoms caused by a calcified disc versus concurrent pathologies affecting the spine. Broadly, it appears that disc calcification can be grouped into 3 potential pathophysiologic processes: inflammatory, mechanical, or degenerative.35 Some of the inflammatory or systemic processes that have been described include ochronosis, hemochromatosis, and chondrocalcinosis. Ochronosis results from an enzyme deficiency leading to excess homogentisic acid deposited in joints and the disc space. It demonstrates some of the most impressive radiographic features for patients later in life, including AF and NP calcification. Hemochromatosis and chondrocalcinosis also have disc calcification, but their appearance is less pronounced on radiography, and much of what we know about their presence in the disc space comes from cadaveric studies.37
The two other critical processes associated with calcification, mechanical and degenerative, are manifest in many spinal disease states and have significant overlap. Scoliosis is one of the most common diseases in which we see disc calcification. Scoliosis is a three-dimensional curvature of the spine, resulting in abnormal mechanical loading and deformity if allowed to progress. Hristova et al. discuss how the calcification seen in scoliotic patients occurs more often in the cells of the endplates and how the extent of calcification can differ between convex and concave parts of a curve due to differences in proteoglycan content. Calcium deposits and collagen 10 in intervertebral disc are also present in scoliotic and disc degeneration patients but are absent in control discs. Zehra et al. postulate that these disc architecture and calcification patterns similarities suggest that even adolescent idiopathic scoliosis may be a premature degenerative process.35 While abnormal loading mechanics are characteristic of scoliotic discs, there remains no clinical consensus on whether the disc calcification in these patients contributed to or was a result of their pathology. Regardless, disc calcification has also been shown to predict spine flexibility and segment instability and should be further studied for its potential role in deformity.143,144
Prior case reports have associated disc calcification with discogenic back pain, one of the most challenging diagnoses to manage, even without calcification. Nogueira-Barbosa and Azizaddini et al. present acute thoracolumbar and cervical pain cases, respectively, in patients who had a calcified disc in the location of their pain without any neurologic deficits.142,145 Both cases involved middle-aged patients with unremarkable symptom onset and were managed nonoperatively with pain medication and physical therapy. Interestingly, in both cases, the calcification reached complete resolution after follow-up, with the cervical study documenting radiographic disappearance up to 6 months after symptom onset.
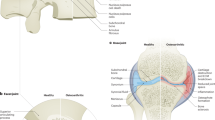
Another challenging aspect of treating disc calcification is when it is present with herniation (Fig. 2). There have been several reports on this phenomenon, and it is also one of the few disc calcification presentations with a surgical solution. Calcified herniated discs are still challenging to treat as they are associated with myelopathy and intradural extension.146 Even after surgical resection, they can still be related to poor outcomes and complications postoperatively. Depending on their size and location, thoracic calcified disc herniations require unique preoperative planning. Some approaches are performed through the thoracic cavity to achieve adequate resection and decompression of the neural elements.147,148

Clinical presentation of disc calcification - X-ray and MRI images. 72-year-old male who presented with a calcified thoracic disc herniation for pre-surgical workup. a Preoperative lateral view x-ray demonstrating intervertebral disc calcification (arrow) in the thoracic spine. b CT sagittal reconstruction of thoracic spine demonstrating disc calcification (white arrow) and herniation of calcified material extending posteriorly (black arrow). c Sagittal T2-weighted MRI demonstrating disc herniation into the spinal canal (white arrow); calcified material within the disc space is not clearly visualized
Radiographic and advanced imaging studies have demonstrated that no single imaging study can adequately characterize all disc calcifications (Table 3). Most clinicians will begin with a conventional radiograph, but as discussed, the prevalence of calcifications seen on X-rays in patients with symptoms of pain, stiffness, and spinal deformity can be low. Computed tomography (CT) scans have also been described in numerous studies as a method to confirm disc calcifications.142,145 One issue with utilizing CT scans is the higher radiation exposure, which is an important consideration when evaluating pediatric patients. The other concern is that while it may provide detail on the nature and location of calcification, it gives little piece on the spinal cord’s or nerve roots' state. Technetium bone scan was used in a case report that demonstrated increased uptake at the level of a thoracic disc calcification; however, it proved to be non-specific, with the entire vertebral body adjacent to the disc also showing increased uptake.142 Magnetic Resonance Imaging (MRI) can indirectly assess the content of the disc through water content; however, there are no clear criteria for distinguishing calcifications. Zehra et al. also discuss that high signal intensity on T1-weighted MRI associated with disc calcification was challenged and may be associated more with fatty infiltration.36,149,150 A more recent MRI technique has sparked interest, called ultra-short time-to-echo (UTE) MRI. Specifically, the “UTE Disc Sign,” or UDS, was established by comparing UTE MRI studies to radiographs of patients with IVD calcifications. The study authors further proposed that UTE MRI may be able to identify subtle and unique disc changes that may not be revealed on plain radiographs or conventional MRI.
In the context of back pain, disc calcification may be responsible for changes in local mechanics, inflammatory reactions, and accelerating disc degeneration.35,151 Furthermore, disc calcification can also predict spine flexibility and segment instability. Knowledge of these associations and advancements in imaging techniques will help determine appropriate indications and allow providers to select the best treatment, be it nonoperative or a variety of surgical approaches (Table 3).35,144
Disc calcification therapeutical solutions
Several approaches have been explored to regenerate and prevent the progression of disc degeneration with minimal success.152 A critical aspect of this challenge is the environment of the disc space itself, which has avascular, acidic, and hypoxic properties.153 The cartilaginous endplate, which facilitates the diffusion of nutrients, begins to undergo progressive calcification throughout degeneration. This results in worsening nutrient diffusion. Cellular metabolism also shifts towards more anaerobic pathways, further decreasing the pH of the disc. These factors compromise the function of existing NP cells but also act as a barrier to the proliferation of regenerative cells.154
Regarding disc calcification, there are no specific therapeutic modalities available yet. While mesenchymal cell transplantation may represent a promising approach to treat degeneration in other joints,155 chondrogenic differentiation of cells often leads to the hypertrophic rather than articular phenotype, promoting local mineralization.156,157 Recently, Abcc6−/− mice treated with potassium citrate showed a dose-dependent decrease in TRAP levels and improvement in vertebral bone health parameters. This approach may be used to treat disc mineralization by promoting direct chelation of Ca2+ by citrate and the dissolution of intradiscal calcium deposits by inducing metabolic acidosis, as well as ameliorating local mineralization signaling and promoting cell survival.124,158
A case report of a 40-year-old man with chronic low back pain due to ochronosis presented a symptomatic decrease in calcium deposition after treatment with anakinra, an IL-1 antagonist drug.159 This approach has already been successfully used for treating refractory calcium pyrophosphate deposition-induced (CPPD) arthritis.160 In treating dystrophic vascular calcification, a few compounds have shown promising results in delaying or decreasing ectopic calcification, such as bisphosphonates;161 Vitamin D162 and Vitamin K163 and chelating drugs such as EDTA164 and citrate.158 These agents may hold some promise in treating disc degeneration accompanied by prominent calcification phenotype.
Recombinant human growth and differentiation factor-5 (rhGDF-5) has also been explored in phase 2 clinical trials as an intradiscal therapy for degenerative disc disease, showing promising results (NCT00813813 and NCT01124006). Experiments in rabbit models of disc degeneration showed that intradiscal injections of rhGDF-5 can increase disc height and hydration. Guo et al. have proposed that it can also inhibit calcification of the endplates and annulus fibrosus, thereby further promoting nutrient supply to the disc.165 However, clinical results have yet to demonstrate the treatment’s effect on disc calcification.
Conclusions
Disc calcification is a poorly understood degenerative phenotype and poses a significant challenge in the clinical management of patients. Since the personalized approach to treating spine disease is gaining traction, and with an increase in the aging population, the urgency to better understand the different degenerative sub-phenotypes to optimize the therapeutic approach has never been more pressing. The need for well-accepted animal models poses challenges in understanding the etiology and fundamental mechanisms driving disc calcification. However, based on the recent findings from various animal models, disc calcification appears to be a complex pathophysiology with different presentations according to the disc compartment (Tables 1–2).
These studies indicate that AF and EP are more susceptible to ectopic calcification in scenarios where local PPi homeostasis is compromised.49,77,127,130 Interestingly, this process in the AF is usually followed by an increase in local TNAP and TRAP activity and resident cell differentiation into osteoblastic-like phenotype.77,127 Altogether, these data show that the calcification of the AF and EP may share similarities with mineralization mechanisms noted in other collagen-rich tissues such as ligaments and skin.49,52,53,56,77 On the other hand, systemic modulation of free calcium and PPi do not seem to play an essential role in driving disc calcification.27,124 Finally, similar to human patients, NP calcium deposits are only noted in aging models, including mice and sheep. This phenotype is generally associated with decreased proteoglycan levels, increased cell death, and increased levels of COL10.27,32,139 While genetic background seems to play an essential role in the susceptibility to disc calcification, we found an increased prevalence of calcification in lumbar and caudal levels in both ovine and mouse models, respectively.18,27,139 In human studies, approximately ~6% of degenerated human discs present discal calcification, with a higher prevalence in aged discs and the AF compartment.25 Like animals, an increased prevalence in a specific region, namely the lower thoracic, is noted, implying that the interaction of biomechanical factors with aging and genetic predisposition promotes disc mineralization.25
In summary, anatomic location, genetic predisposition, and environmental stress, such as aging or trauma, strongly influence intervertebral disc calcification phenotype. Efforts must be made to understand the unique regulation of the intervertebral disc as an organ system to improve the outcome of new treatments in the future.
References
Mokdad, A. H. et al. The state of US health, 1990-2016. JAMA 319, 1444–1472 (2018).
Katz, J. N. Lumbar disc disorders and low-back pain: socioeconomic factors and consequences. J. Bone Jt. Surg. Ser. A 88, 21–24 (2006).
Cheung, K. M. C. et al. Prevalence and pattern of lumbar magnetic resonance imaging changes in a population study of one thousand forty-three individuals. Spine (Philos. Pa 1976) 34, 934–940 (2009).
Chou, R. et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann. Intern. Med. 147, 478–491 (2007).
Roberts, S., Evans, H., Trivedi, J. & Menage, J. Histology and pathology of the human intervertebral disc. J. Bone Jt. Surg. Ser. A 88, 10–14 (2006).
Airaksinen, O. et al. Chapter 4: European guidelines for the management of chronic nonspecific low back pain. Eur. Spine J. 15, S192–300 (2006).
Dowdell, J. et al. Intervertebral disk degeneration and repair. Clin. Neurosurg. 80, S46–S54 (2017).
Dvořák, J., Vajda, E. G., Grob, D. & Panjabi, M. M. Normal motion of the lumbar spine as related to age and gender. Eur. Spine J. 4, 18–23 (1995).
Adams, M. A. & Hutton, W. C. The effect of posture on the role of the apophysial joints in resisting intervertebral compressive forces. J. Bone Jt. Surg. Ser. B 62, 358–362 (1980).
Huang, Y.-C., Urban, J. P. G. & Luk, K. D. K. Intervertebral disc regeneration: do nutrients lead the way? Nat. Rev. Rheumatol. 10, 1–6 (2014).
Tsantrizos, A., Ito, K., Aebi, M. & Steffen, T. Internal strains in healthy and degenerated lumbar intervertebral discs. Spine (Philos. Pa 1976) 30, 2129–2137 (2005).
Uematsu, Y., Matuzaki, H. & Iwahashi, M. Effects of nicotine on the intervertebral disc: an experimental study in rabbits. J. Orthop. Sci. 6, 77–82 (2001).
Vo, N. et al. Differential effects of nicotine and tobacco smoke condensate on human annulus fibrosus cell metabolism. J. Orthop. Res. 29, 1585–1591 (2011).
Singh, D., Park, W., Hwang, D. & Levy, M. S. Severe obesity effect on low back biomechanical stress of manual load lifting. Work 51, 337–348 (2015).
Vadalà, G. et al. Early intervertebral disc degeneration changes in asymptomatic weightlifters assessed by T1ρ’ -magnetic resonance imaging. Spine (Philos. Pa 1976) 39, 1881–1886 (2014).
UN. World Population Prospects 2019 (Department of Economic and Social Affairs, 2019).
Munir, S. et al. Endplate defect is heritable, associated with low back pain and triggers intervertebral disc degeneration: a longitudinal study from Twinsuk. Spine (Philos. Pa 1976) 43, 1496–1501 (2018).
Battié, M. C. et al. The twin spine study: contributions to a changing view of disc degeneration. Spine J. 9, 47–59 (2009).
Toktaş, Z. O. et al. Association of collagen I, IX and vitamin D receptor gene polymorphisms with radiological severity of intervertebral disc degeneration in Southern European Ancestor. Eur. Spine J. 24, 2432–2441 (2015).
Takahashi, M. et al. The association of degeneration of the intervertebral disc with 5a/6a polymorphism in the promoter of the human matrix metalloproteinase-3 gene. J. Bone Jt. Surg. Ser. B 83, 491–495 (2001).
Huang, X. et al. Interleukin 6 (IL-6) and IL-10 promoter region polymorphisms are associated with risk of lumbar disc herniation in a Northern Chinese han population. Genet. Test. Mol. Biomark. 21, 17–23 (2017).
Guerin, H. L. & Elliott, D. M. Quantifying the contributions of structure to annulus fibrosus mechanical function using a nonlinear, anisotropic, hyperelstic model. J. Orthop. Res. 25, 508–516 (2007).
O’Connell, G. D., Jacobs, N. T., Sen, S., Vresilovic, E. J. & Elliott, D. M. Axial creep loading and unloaded recovery of the human intervertebral disc and the effect of degeneration. J. Mech. Behav. Biomed. Mater. 4, 933–942 (2011).
Roberts, S. Disc morphology in health and disease. Biochem. Soc. Trans. 30, 864–869 (2002).
Chanchairujira, K. et al. Intervertebral disk calcification of the spine in an elderly population: radiographic prevalence, location, and distribution and correlation with spinal degeneration. Radiology 230, 499–503 (2007).
Boos, N. et al. Classification of age-related changes in lumbar intervertebral discs: 2002 Volvo Award in basic science. Spine (Philos. Pa 1976) 27, 2631–2644 (2002).
Novais, E. J. et al. Comparison of inbred mouse strains shows diverse phenotypic outcomes of intervertebral disc aging. Aging Cell 19, e13148 (2020).
Yee, A. et al. Fibrotic-like changes in degenerate human intervertebral discs revealed by quantitative proteomic analysis. Osteoarthr. Cartil. 24, 503–13 (2016).
Roberts, S., Bains, M. A., Kwan, A., Menage, J. & Eisenstein, S. M. Type X collagen in the human invertebral disc: an indication of repair or remodelling? Histochem. J. 30, 89–95 (1998).
Vo, N. et al. Accelerated aging of intervertebral discs in a mouse model of progeria. J. Orthop. Res. 28, 1600–1607 (2010).
Bachmeier, B. E. et al. Matrix metalloproteinase expression levels suggest distinct enzyme roles during lumbar disc herniation and degeneration. Eur. Spine J. 18, 1573–1586 (2009).
Hristova, G. I. et al. Calcification in human intervertebral disc degeneration and scoliosis. J. Orthop. Res. 29, 1888–95 (2011).
Shao, J. et al. Differences in calcification and osteogenic potential of herniated discs according to the severity of degeneration based on Pfirrmann grade: a cross-sectional study. BMC Musculoskelet. Disord. 17, 191 (2016).
Teraguchi, M. et al. Prevalence and distribution of intervertebral disc degeneration over the entire spine in a population-based cohort: the Wakayama Spine Study. Osteoarthr. Cartil. 22, 104–110 (2014).
Zehra, U. et al. Mechanisms and clinical implications of intervertebral disc calcification. Nat. Rev. Rheumatol. 18, 352–362 (2022).
Bangert, B. A. et al. Hyperintense disks on T1-weighted MR images: correlation with calcification. Radiology 195, 437–443 (1995).
Weinberger, A. & Myers, A. R. Intervertebral disc calcification in adults: a review. Semin. Arthritis Rheum. 8, 69–75 (1978).
Stigen, Ø., Ciasca, T. & Kolbjørnsen, Ø. Calcification of extruded intervertebral discs in dachshunds: a radiographic, computed tomographic and histopathological study of 25 cases. Acta Vet. Scand. 61, 13 (2019).
Park, J. et al. Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol. Open 4, 608–621 (2015).
Jin, L. et al. Annulus fibrosus cell characteristics are a potential source of intervertebral disc pathogenesis. PLoS One 9, e96519 (2014).
Zhao, Y. P. et al. Progranulin knockout accelerates intervertebral disc degeneration in aging mice. Sci. Rep. 5, 9102 (2015).
Stokes, I. A. F. & Iatridis, J. C. Mechanical conditions that accelerate intervertebral disc degeneration: Overload versus immobilization. Spine 29, 2724–2732 (2004).
Roberts, S., Menage, J. & Eisenstein, S. M. The cartilage end‐plate and intervertebral disc in scoliosis: calcification and other sequelae. J. Orthop. Res. 11, 747–757 (1993).
Sun, Y. & Mauerhan, D. R. Meniscal calcification, pathogenesis and implications. Curr. Opin. Rheumatol. 24, 152–157 (2012).
Mitrovic, D. R. et al. The prevalence of chondrocalcinosis in the human knee joint. An autopsy survey. J. Rheumatol. 15, 633–641 (1988).
Mitrovic, D. et al. Anatomic incidence of meniscochondrocalcinosis of the knee. Rev. Rhum. Mal. Osteoartic. 49, 495–499 (1982).
Meyer, F. et al. Chondrocytes from osteoarthritic and chondrocalcinosis cartilage represent different phenotypes. Front. Cell Dev. Biol. 26, e622287 (2021).
Le Graverand, M. P. H. et al. Formation and phenotype of cell clusters in osteoarthritic meniscus. Arthritis Rheum. 44, 1808–1818 (2001).
Zhang, J. et al. Ectopic mineralization of cartilage and collagen-rich tendons and ligaments in Enpp1asj-2J mice. Oncotarget 7, 12000–12009 (2016).
Li, Q. et al. Inhibition of tissue-nonspecific alkaline phosphatase attenuates ectopic mineralization in the Abcc6−/− mouse model of PXE but not in the Enpp1 mutant mouse models of GACI. J. Investig. Dermatol. 139, 360–368 (2019).
Jiang, S., Zhang, C., Lu, Y. & Yuan, F. The molecular mechanism research of cartilage calcification induced by osteoarthritis. Bioengineered 13, 13082–13088 (2022).
Kempf, H., Komarova, S. & Murshed, M. Editorial: ectopic mineralization of tissues: mechanisms, risk factors, diseases, and prevention. Front. Cell Dev. Biol. 9, 759702 (2021).
Ziegler, S. G. et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci. Transl. Med. 9, eaal1669 (2017).
Du, Y. et al. Cartilage oligomeric matrix protein inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2. Circ. Res. 108, 917–928 (2011).
Rosenthal, A. K. Basic calcium phosphate crystal-associated musculoskeletal syndromes: an update. Curr. Opin. Rheumatol. 30, 168–172 (2018).
Fournier, D. E., Kiser, P. K., Beach, R. J., Dixon, S. J. & Séguin, C. A. Dystrophic calcification and heterotopic ossification in fibrocartilaginous tissues of the spine in diffuse idiopathic skeletal hyperostosis (DISH). Bone Res. 8, 16 (2020).
Moore, S. N. et al. Validation of a radiography-based quantification designed to longitudinally monitor soft tissue calcification in skeletal muscle. PLoS One 11, e0159624 (2016).
Teebi, A. S. et al. Keutel syndrome: further characterization and review. Am. J. Med. Genet. 78, 182–187 (1998).
Oyoung, J. et al. Matrix gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 133, 18406–18412 (2011).
Warraich, S. et al. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J. Bone Miner. Res. 28, 1135–49 (2013).
Schäfer, C. et al. The serum protein α2-Heremans-Schmid glycoprotein/ fetuin-A is a systemically acting inhibitor of ectopic calcification. J. Clin. Investig. 112, 357–66 (2003).
Johnson, K. & Terkeltaub, R. Inorganic pyrophosphate (PPI) in pathologic calcification of articular cartilage. Front. Biosci. 1, 988–997 (2005).
Babler, A. et al. Microvasculopathy and soft tissue calcification in mice are governed by fetuin-A, magnesium and pyrophosphate. PLoS One 15, e0228938 (2020).
Murshed, M., Harmey, D., Millán, J. L., McKee, M. D. & Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 19, 1093–1104 (2005).
Bourne, L. E. et al. Evidence that pyrophosphate acts as an extracellular signalling molecule to exert direct functional effects in primary cultures of osteoblasts and osteoclasts. Bone 176, 116868 (2023).
ter Braake, A. D., Shanahan, C. M. & de Baaij, J. H. F. Magnesium counteracts vascular calcification. Arterioscler Thromb. Vasc. Biol. 37, 1431–1445 (2017).
Rimer, J. D., Sakhaee, K. & Maalouf, N. M. Citrate therapy for calcium phosphate stones. Curr. Opin. Nephrol. Hypertens. 28, 130–139 (2019).
Szeri, F. et al. The membrane protein ANKH is crucial for bone mechanical performance by mediating cellular export of citrate and ATP. PLoS Genet. 16, e1008884 (2020).
Rutsch, F. et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 34, 379–381 (2003).
Jansen, R. S. et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation - brief report. Arterioscler Thromb. Vasc. Biol. 34, 1985–1989 (2014).
Harmey, D. et al. Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank: an integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 164, 1199–1209 (2004).
Nürnberg, P. et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat. Genet. 28, 37–41 (2001).
Szeri, F. et al. The mineralization regulator ANKH mediates cellular efflux of ATP, not pyrophosphate. J. Bone Miner. Res. 37, 1024–1031 (2022).
Moorhead, W. J. et al. Dysregulation of FOXO1 (Forkhead Box O1 Protein) drives calcification in arterial calcification due to deficiency of CD73 and is present in peripheral artery disease. Arterioscler Thromb. Vasc. Biol. 40, 1680–1694 (2020).
Ferreira, C. R. et al. Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet. Med. 23, 396–407 (2021).
Ho, A. M., Johnson, M. D. & Kingsley, D. M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 289, 265–70 (2000).
Ohnishi, T. et al. Loss of function mutation in Ank causes aberrant mineralization and acquisition of osteoblast-like-phenotype by the cells of the intervertebral disc. Cell Death Dis. 14, 447 (2023).
Cudrici, C. D. et al. Multifocal calcific periarthritis with distinctive clinical and radiological features in patients with CD73 deficiency. Rheumatology 61, 163–173 (2022).
Ichikawa, N. et al. Arterial calcification due to deficiency of CD73 (ACDC) as one of rheumatic diseases associated with periarticular calcification. J. Clin. Rheumatol. 21, 216–220 (2015).
Millán, J. L. & Whyte, M. P. Alkaline phosphatase and hypophosphatasia. Calcified Tissue Int. 98, 398–416 (2016).
Lee, S. J., Lee, I. K. & Jeon, J. H. Vascular calcification—new insights into its mechanism. Int. J. Mol. Sci. 21, 2685 (2020).
Henze, L. A. et al. Impact of C-reactive protein on osteo-/chondrogenic transdifferentiation and calcification of vascular smooth muscle cells. Aging 11, 5445–5462 (2019).
Tschaffon, M. E. A. et al. A novel in vitro assay to study chondrocyte-to-osteoblast transdifferentiation. Endocrine 75, 266–275 (2022).
Nishimura, R. et al. Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification. J. Biol. Chem. 287, 33179–33190 (2012).
Kapustin, A. N. et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ. Res. 109, e1–e12 (2011).
Yang, W. et al. Extracellular vesicles in vascular calcification. Clin. Chim. Acta 499, 118–122 (2019).
Furmanik, M. et al. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78 (glucose-regulated protein, 78 kDa)-loaded extracellular vesicles. Arterioscler Thromb. Vasc. Biol. 41, 898–914 (2021).
Grootaert, M. O. J. et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 11, 2014–2032 (2015).
Duan, X., Zhou, Y., Teng, X., Tang, C. & Qi, Y. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem. Biophys. Res. Commun. 387, 694–699 (2009).
Miyazaki-Anzai, S. et al. Endoplasmic reticulum stress effector CCAAT/enhancer-binding protein homologous protein (CHOP) regulates chronic kidney disease-induced vascular calcification. J. Am. Heart Assoc. 24, e000949 (2014).
Hsu, Y. J. et al. Hyperphosphatemia induces protective autophagy in endothelial cells through the inhibition of Akt/mTOR signaling. J. Vasc. Surg. 62, 210–221 (2015).
Frauscher, B. et al. Autophagy protects from uremic vascular media calcification. Front. Immunol. 9, 1866 (2018).
Kim, H. et al. α-Lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of Gas6/Axl/Akt survival pathway. J. Cell Mol. Med. 16, 273–86 (2012).
Proudfoot, D. et al. Apoptosis regulates human vascular calcification in vitro. Circ. Res. 87, 1055–62 (2000).
Li, X., Yang, H. Y. & Giachelli, C. M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 199, 271–277 (2008).
Lin, M. E. et al. Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc. Res. 112, 606–616 (2016).
Lotz, J. C. Animal models of intervertebral disc degeneration: lessons learned. Spine 29, 2742–2750 (2004).
Glasson, S. S., Blanchet, T. J. & Morris, E. A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 15, 1061–1069 (2007).
Kim, H. N. et al. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 18, e12923 (2019).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Choi, K. S., Cohn, M. J. & Harfe, B. D. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev. Dyn. 237, 3953–8 (2008).
Chan, W. C. W., Au, T. Y. K., Tam, V., Cheah, K. S. E. & Chan, D. Coming together is a beginning: the making of an intervertebral disc. Birth Defects Res. C. Embryo Today 102, 83–100 (2014).
McCann, M. R., Tamplin, O. J., Rossant, J. & Seǵuin, C. A. Tracing notochord-derived cells using a Noto-cre mouse: implications for intervertebral disc development. DMM Dis. Models Mech. 5, 73–82 (2012).
Aszódi, A., Chan, D., Hunziker, E., Bateman, J. F. & Fässler, R. Collagen II is essential for the removal of the notochord and the formation of intervertebral discs. J. Cell Biol. 143, 1399–1412 (1998).
Smit, T. H. The use of a quadruped as an in vivo model for the study of the spine - biomechanical considerations. Eur. Spine J. 11, 137–44 (2002).
Novais, E. J. et al. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 12, 5213 (2021).
Tessier, S., Tran, V. A., Ottone, O. K. & Novais, E. J. TonEBP-deficiency accelerates intervertebral disc degeneration underscored by matrix remodeling, cytoskeletal rearrangements, and changes in proinflammatory gene expression. Matrix Biol. 87, 94–111 (2019).
Novais, E. J., Diekman, B. O., Shapiro, I. M. & Risbud, M. V. p16 Ink4a deletion in cells of the intervertebral disc affects their matrix homeostasis and senescence associated secretory phenotype without altering onset of senescence. Matrix Biol. 82, 54–70 (2019).
Gorth, D. J., Shapiro, I. M. & Risbud, M. V. A new understanding of the role of IL-1 in age-related intervertebral disc degeneration in a murine model. J. Bone Miner. Res. 34, 1531–1542 (2019).
Choi, H. et al. A novel mouse model of intervertebral disc degeneration shows altered cell fate and matrix homeostasis. Matrix Biol. 70, 102–122 (2018).
Rai, M. F. et al. Heritability of articular cartilage regeneration and its association with ear-wound healing. Arthritis Rheum. 64, 2300–2310 (2013).
Hrbek, T., de Brito, R. A., Wang, B., Pletscher, L. S. & Cheverud, J. M. Genetic characterization of a new set of recombinant inbred lines (LGXSM) formed from the intercross of SM/J and LG/J inbred mouse strains. Mamm. Genome 17, 417–429 (2006).
Rai, M. F., Schmidt, E. J., Hashimoto, S., Cheverud, J. M. & Sandell, L. J. Genetic loci that regulate ectopic calcification in response to knee trauma in LG/J by SM/J advanced intercross mice. J. Orthop. Res. 33, 1412–23 (2015).
Priante, G. et al. Cell death in ectopic calcification of the kidney. Cell Death Dis. 10, 466 (2019).
Patel, J. J. et al. Differing calcification processes in cultured vascular smooth muscle cells and osteoblasts. Exp. Cell Res. 380, 100–113 (2019).
Chen, Q. et al. HMGB1 induces secretion of matrix vesicles by macrophages to enhance ectopic mineralization. PLoS One 11, e0156686 (2016).
Hakim, F. T. et al. Hereditary joint disorder in progressive ankylosis (ank/ank) mice I. association of calcium hydroxyapatite deposition with inflammatory arthropathy. Arthritis Rheum. 27, 1411–1420 (1984).
Morava, E. et al. Autosomal recessive mental retardation, deafness, ankylosis, and mild hypophosphatemia associated with a novel ANKH mutation in a consanguineous family. J. Clin. Endocrinol. Metab. 96, E189–98 (2011).
Dudek, M. et al. The intervertebral disc contains intrinsic circadian clocks that are regulated by age and cytokines and linked to degeneration. Ann. Rheum. Dis. 76, 576–584 (2017).
Samsa, W. E., Vasanji, A., Midura, R. J. & Kondratov, R. V. Deficiency of circadian clock protein BMAL1 in mice results in a low bone mass phenotype. Bone 84, 194–203 (2016).
Siu, S. Y. et al. Variable patterns of ectopic mineralization in Enpp1 asj-2J mice, a model for generalized arterial calcification of infancy. Oncotarget 7, 83837–83842 (2016).
Ohnishi, T., Novais, E. J. & Risbud, M. V. Alterations in ECM signature underscore multiple sub-phenotypes of intervertebral disc degeneration. Matrix Biol. 6–7, 100036 (2020).
Borst, P., Váradi, A. & van de Wetering, K. PXE, a mysterious inborn error clarified. Trends Biochem. Sci. 44, 125–140 (2019).
Boneski, P. K. et al. Abcc6 null mice—a model for mineralization disorder PXE shows vertebral osteopenia without enhanced intervertebral disc calcification with aging. Front. Cell Dev. Biol. 3, 823249 (2022).
Eanes, E. D., Hailer, A. W., Midura, R. J. & Hascall, V. C. Proteoglycan inhibition of calcium phosphate precipitation in liposomal suspensions. Glycobiology 2, 571–8 (1992).
Daniels, G. et al. Lack of the nucleoside transporter ENT1 results in the Augustine-null blood type and ectopic mineralization. Blood 125, 3651–3654 (2015).
Ii, H. et al. Disruption of biomineralization pathways in spinal tissues of a mouse model of diffuse idiopathic skeletal hyperostosis. Bone 90, 37–49 (2016).
Terkeltaub, R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signal. 2, 371–377 (2006).
Zhu, Y., Sen, Gu,Y., Jiang, C. & Chen, L. Osteonectin regulates the extracellular matrix mineralization of osteoblasts through P38 signaling pathway. J. Cell Physiol. 235, 2220–2231 (2020).
Gruber, H. E. et al. Targeted deletion of the SPARC gene accelerates disc degeneration in the aging mouse. J. Histochem. Cytochem. 53, 1131–1138 (2005).
Millecamps, I., Tajerian, M., Sage, E. H. & Stone, L. S. Behavioral signs of chronic back pain in the SPARC-null mouse. Spine 36, 95–102 (2011).
Ni, S. et al. Sensory innervation in porous endplates by Netrin-1 from osteoclasts mediates PGE2-induced spinal hypersensitivity in mice. Nat. Commun. 10, 5643 (2019).
Lv, X. et al. Resveratrol‐enhanced SIRT1‐mediated osteogenesis in porous endplates attenuates low back pain and anxiety behaviors. FASEB J. 35, e21414 (2021).
Barker, T. H. et al. SPARC regulates extracellular matrix organization through its modulation of integrin-linked kinase activity. J. Biol. Chem. 280, 36483–36493 (2005).
Wang, Y. et al. SPARC-related modular calcium binding 1 regulates aortic valve calcification by disrupting BMPR-II/p-p38 signalling. Cardiovasc. Res. 118, 913–928 (2022).
Suyama, K., Silagi, E. S., Choi, H., Sakabe, K. & Mochida, J. Circadian factors BMAL1 and RORα control HIF-1α transcriptional activity in nucleus pulposus cells: implications in maintenance of intervertebral disc health. Oncotarget 7, 23056–23071 (2016).
Liang, Q. et al. Disruption of the mouse Bmal1 locus promotes heterotopic ossification with aging via TGF-beta/BMP signaling. J. Bone Min. Metab. 40, 40–55 (2022).
Yang, P. et al. The role of bone morphogenetic protein signaling in vascular calcification. Bone 141, 115542 (2020).
Melrose, J. et al. Calcification in the ovine intervertebral disc: a model of hydroxyapatite deposition disease. Eur. Spine J. 18, 479–489 (2009).
Brown, E. A. et al. FGF4 retrogene on CFA12 is responsible for chondrodystrophy and intervertebral disc disease in dogs. Proc. Natl. Acad. Sci. USA 114, 11476–11481 (2017).
Gruber, H. E., Johnson, T., Norton, H. J. & Hanley, E. N. The sand rat model for disc degeneration: radiologic characterization of age-related changes: cross-sectional and prospective analyses. Spine 27, 230–234 (2002).
Nogueira-Barbosa, M. H., da Silva Herrero, C. F. P., Pasqualini, W. & Defino, H. L. A. Calcific discitis in an adult patient with intravertebral migration and spontaneous remission. Skeletal. Radiol. 42, 1161–1164 (2013).
Zehra, U. et al. Spinopelvic alignment predicts disc calcification, displacement, and Modic changes: evidence of an evolutionary etiology for clinically-relevant spinal phenotypes. JOR Spine 3, e1083 (2020).
Yao, G. et al. Characterization and predictive value of segmental curve flexibility in adolescent idiopathic scoliosis patients. Spine (Philos. Pa 1976) 42, 1622–1628 (2017).
Azizaddini, S., Arefanian, S., Redjal, N., Walcott, B. P. & Mollahoseini, R. Adult acute calcific discitis confined to the nucleus pulposus in the cervical spine: case report. J. Neurosurg. Spine 19, 170–173 (2013).
Court, C., Mansour, E. & Bouthors, C. Thoracic disc herniation: surgical treatment. Orthop. Traumatol. Surg. Res. 104, S31–S40 (2018).
Roelz, R. et al. Giant central thoracic disc herniations: surgical outcome in 17 consecutive patients treated by mini-thoracotomy. Eur. Spine J. 25, 1443–1451 (2016).
Börm, W. et al. Surgical treatment of thoracic disc herniations via tailored posterior approaches. Eur. Spine J. 20, 1684–1690 (2011).
Malghem, J. et al. High signal intensity of intervertebral calcified disks on T1-weighted MR images resulting from fat content. Skeletal. Radiol. 34, 80–6 (2005).
Tyrrell, P. N., Davies, A. M., Evans, N. & Jubb, R. W. Signal changes in the intervertebral discs on MRI of the thoracolumbar spine in ankylosing spondylitis. Clin. Radiol. 50, 377–83 (1995).
Zehra, U. et al. The association of lumbar intervertebral disc calcification on plain radiographs with the UTE Disc Sign on MRI. Eur. Spine J. 27, 1049–1057 (2018).
Orozco, L. et al. Intervertebral disc repair by autologous mesenchymal bone marrow cells: a pilot study. Transplantation 92, 822–828 (2011).
Vadalà, G., Ambrosio, L., Russo, F., Papalia, R. & Denaro, V. Interaction between mesenchymal stem cells and intervertebral disc microenvironment: from cell therapy to tissue engineering. Stem Cells Int. 2019, 2376172 (2019).
Canseco, J. A., Kanhere, A. P., Schroeder, G. D., Vaccaro, A. R. & Kepler, C. K. Intradiscal therapies for lumbar degenerative disk disease. J. Am. Acad. Orthop. Surg. 30, e1084–e1094 (2022).
Muthu, S. et al. Failure of cartilage regeneration: emerging hypotheses and related therapeutic strategies. Nat. Rev. Rheumatol. 19, 403–416 (2023).
van Gool, S. A. et al. Fetal mesenchymal stromal cells differentiating towards chondrocytes acquire a gene expression profile resembling human growth plate cartilage. PLoS One 7, e44561 (2012).
Vadalà, G. et al. Mesenchymal stem cells injection in degenerated intervertebral disc: cell leakage may induce osteophyte formation. J. Tissue Eng. Regen. Med. 6, 348–355 (2012).
Ou, Y. et al. Citrate attenuates vascular calcification in chronic renal failure rats. APMIS 125, 452–458 (2017).
Boleto, G., Allanore, Y. & Wipff, J. Ochronosis of the spine mimicking ankylosing spondylitis successfully treated with anakinra. Jt. Bone Spine 87, 368–369 (2020).
Ottaviani, S. et al. Efficacy of anakinra in calcium pyrophosphate crystal-induced arthritis: a report of 16 cases and review of the literature. Jt. Bone Spine 80, 178–182 (2013).
Persy, V., De Broe, M. & Ketteler, M. Bisphosphonates prevent experimental vascular calcification: treat the bone to cure the vessels? Kidney Int. 70, 1537–1538 (2006).
Lau, W. L. et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 82, 1261–1270 (2012).
Spronk, H. M. H. et al. Tissue-specific utilization of menaquinone-4 results in the prevention of arterial calcification in warfarin-treated rats. J. Vasc. Res. 40, 531–537 (2003).
Maniscalco, B. S. & Taylor, K. A. Calcification in coronary artery disease can be reversed by EDTA-tetracycline long-term chemotherapy. Pathophysiology 11, 95–101 (2004).
Guo, S. et al. The mechanisms and functions of GDF-5 in intervertebral disc degeneration. Orthop. Surg. 13, 734–741 (2021).
Zhang, Y. et al. Early onset of disc degeneration in SM/J mice is associated with changes in ion transport systems and fibrotic events. Matrix Biol. 70, 123–139 (2018).
Lappalainen, A. K., Vaittinen, E., Junnila, J. & Laitinen-Vapaavuori, O. Intervertebral disc disease in Dachshunds radiographically screened for intervertebral disc calcifications. Acta Vet. Scand. 56, 89 (2014).
Mogensen, M. S. et al. Genome-wide association study in dachshund: identification of a major locus affecting intervertebral disc calcification. J. Heredity 102, S81–S86 (2011).
Chang, E. Y., Du, J. & Chung, C. B. UTE imaging in the musculoskeletal system. J. Magn. Reson Imaging 41, 870–83 (2015).
Gates, G. F. SPECT bone scanning of the spine. Semin. Nucl. Med. 28, 78–94 (1998).
Acknowledgements
The authors acknowledge support by R01AR055655, R01AR074813, and R01 AG073349 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and the National Institute on Aging (NIA). K.v.d.W. is supported by PXE International.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
M.V.R., E.N., R.N., J.C., K.v.d.W., C.K., and A.H. have no conflicts to disclose. A.V. has received royalty payments from Medtronic, Stryker Spine, Globus, Aesculap, Thieme, Jaypee, Elsevier, and Taylor Francis/Hodder and Stoughton. He has stock/stock option ownership interests in Replication Medica, Globus, Paradigm Spine, Stout Medical, Progressive Spinal Technologies, Advanced Spinal Intellectual Properties, Spine Medica, Computational Biodynamics, Spinology, In Vivo, Flagship Surgical, Cytonics, Bonovo Orthopaedics, Electrocore, Gamma Spine, Location Based Intelligence, FlowPharma, R.S.I., Rothman Institute and Related Properties, Innovative Surgical Design, and Avaz Surgical.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Novais, E.J., Narayanan, R., Canseco, J.A. et al. A new perspective on intervertebral disc calcification—from bench to bedside. Bone Res 12, 3 (2024). https://doi.org/10.1038/s41413-023-00307-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-023-00307-3
