
Abstract
Emerging insights into cellular senescence highlight the relevance of senescence in musculoskeletal disorders, which represent the leading global cause of disability. Cellular senescence was initially described by Hayflick et al. in 1961 as an irreversible nondividing state in in vitro cell culture studies. We now know that cellular senescence can occur in vivo in response to various stressors as a heterogeneous and tissue-specific cell state with a secretome phenotype acquired after the initial growth arrest. In the past two decades, compelling evidence from preclinical models and human data show an accumulation of senescent cells in many components of the musculoskeletal system. Cellular senescence is therefore a defining feature of age-related musculoskeletal disorders, and targeted elimination of these cells has emerged recently as a promising therapeutic approach to ameliorate tissue damage and promote repair and regeneration of the skeleton and skeletal muscles. In this review, we summarize evidence of the role of senescent cells in the maintenance of bone homeostasis during childhood and their contribution to the pathogenesis of chronic musculoskeletal disorders, including osteoporosis, osteoarthritis, and sarcopenia. We highlight the diversity of the senescent cells in the microenvironment of bone, joint, and skeletal muscle tissue, as well as the mechanisms by which these senescent cells are involved in musculoskeletal diseases. In addition, we discuss how identifying and targeting senescent cells might positively affect pathologic progression and musculoskeletal system regeneration.
Similar content being viewed by others

Introduction
The musculoskeletal system (MSK), including bone, cartilaginous tissues, skeletal muscle, tendon, ligament, intervertebral disc, and others, is the structural framework for the body and enables movement. Bones are held in position by ligaments. Skeletal muscles power the movement of bones. Muscles and bones are connected through tendons. These tissues together form the articulating joints. In addition to providing physical support, MSK tissues have endocrine functions and communicate with other tissues to affect organism homeostasis and overall physiological health. The skeleton is a highly dynamic structure that changes in shape and composition throughout life. Skeletal muscle fiber also has high remodeling plasticity on demand. Not surprisingly, bone, cartilage, and skeletal muscle cell homeostasis is tightly controlled, as is the maintenance of tissue structure and mass. Aging, characterized by a gradual functional decline, is the greatest risk factor for many chronic musculoskeletal conditions, such as osteoporosis, osteoarthritis, and sarcopenia. As common musculoskeletal complications, these three disorders share genetic, endocrine, and mechanical risk factors, and are also closely connected both mechanically and metabolically. These age-associated musculoskeletal conditions leads to MSK structural degeneration, mechanical pain, decreased mobility, and limited function.1,2 Particularly, among patients aged 65 years or older, chronic skeletal diseases, such as osteoporosis and associated bone fracture and osteoarthritis, are the most prevalent conditions leading to frailty and a decline in mobility. An estimated 10 million Americans have osteoporosis, and another 43 million have low bone density. Osteoporosis will be responsible for more than $25 billion in annual health care spending by 2025 in the United States.3 Osteoarthritis is the most prevalent chronic joint disease affecting the knees, hands, hips, and spine, and is the fourth most common cause of hospitalization among adults in the United States, resulting in enormous health care costs.4 Sarcopenia is present in an estimated 5%–13% of individuals older than 60 and 50% of persons older than 80,5 and can cause severe adverse clinical outcomes.6 Despite substantial social demand, no medications for osteoarthritis or sarcopenia disease treatment have been approved by the US Food and Drug Administration. Related to their structural function, MSK tissues are susceptible to trauma and injury, and aging also adversely affects the tissue regenerative capacity, resulting in delayed tissue repair, disability, and pain. In the past few years, the MSK research community has been considerably interested in cellular senescence, which represents a series of diverse, dynamic, and heterogeneous cellular states with the senescence‐associated secretory phenotype (SASP). In this review, we provide a comprehensive summary of cellular senescence known to be involved in MSK homeostasis, disease, and regeneration. As senescent cells (SnCs) appear to have dual roles in physiologic tissue homeostasis/repair as well as pathologic responses, this review highlights the functional heterogeneity and the molecular pathways responsible for senescence induction in distinct physiological settings.
Pathways triggering cellular senescence and the SASP
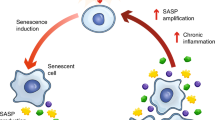
Cellular senescence, initially described by Hayflick et al. in 1961 as limited replicative potential of normal cultured human fibroblasts,7,8 was thought to be an in vitro phenomenon. Starting at the beginning of the twenty-first century, work by multiple groups showed that these SnCs are also abundant in vivo. Based on the consensus reached by the International Cell Senescence Association, cellular senescence is now viewed as a cell state triggered by stressful insults and certain physiologic processes, characterized by a prolonged—and generally irreversible—cell cycle arrest with secretory features, macromolecular damage, and altered metabolism (Fig. 1).9,10,11,12,13,14 The SnCs can affect their local tissue environments through the SASP. Importantly, removal of SnCs in adult mice led to major improvements in health span and lifespan. The recent generation of senescence reporter/ablation mouse models and the development of senotherapies have advanced our knowledge of the phenotypic features and pathogenesis of the SnCs, as well as their contributions to physiologic and pathologic processes.

Overview of senescence. Several factors can induce senescence in different tissues, such as tissue injury, telomere shortening, and oncogenic signaling that all lead to DNA damage, the DNA damage response (DDR), and consequent cell cycle arrest by activation of p16/pRB signaling and/or p53/p21 signaling. Nucleolar stress and ribosome biogenesis defects can also induce RPS14 accumulation in the nucleus and activates Rb by inhibiting the CDK4/cyclin D1 complex, leading to cell cycle arrest. Senescent cells also exhibit increased senescence-associated β-galactosidase (SA-β-gal) production, reactive oxygen species (ROS) accumulation, and anti-apoptotic factors such as BCL-XL and BCL-2. Senescent cells exhibit several phenotypic changes such as a resistance to apoptosis, oxidative stress and damage, metabolic changes, morphological changes, cell cycle arrest, and extracellular vesicle secretion containing SASP factors such as IL-1, IL-6, TNF, MMP13, and various growth factors. This SASP can either feedback in an autocrine manner to the senescent cell or in a paracrine manner influence and promote senescence and inflammation in the surrounding cells and tissues
SnC cycle arrest and related senescence biomarkers
Irreversible cell cycle arrest is a defining characteristic of cellular senescence despite its many facets. Earlier studies of the mechanisms of the replicative senescence that causes stable cell cycle arrest found that replicative senescence in human cells is provoked by telomere erosion after extensive serial passaging.15,16,17 Cells can also undergo senescence independent of telomere shortening, and senescence can be induced when cells are exposed to genotoxic agents, such as reactive oxygen species, deoxyribonucleic acid (DNA)-damaging agents, hypoxia, mitochondria dysfunction, certain activated oncogenes, and epigenetic alteration.18,19,20,21 Two main tumor suppressor-mediated signaling pathways, p53/p21CIP1 and p16INK4a/pRB, are responsible for the growth arrest of SnCs.22,23,24,25 The p53/p21CIP1 pathway is activated downstream of the DNA damage response from DNA double-strand breaks or uncapped telomeres. p16INK4a/pRB signaling initiates and maintains permanent cell cycle arrest. The INK4A/ARF locus is normally epigenetically silenced by polycomb repressive complexes. Disrupting the activities of polycomb repressive complexes derepresses p16INK4a and induces senescence.26,27,28 p16INK4A activation inhibits CDK4 and CDK6 activity, which leads to RB hypophosphorylation, blockade of S-phase entry, and cell cycle arrest.29 Ribosome biogenesis defect is another feature of SnCs and also an important contributor to cell cycle arrest. The involvement of ribosome biogenesis perturbation in cellular senescence was first reported in a study showing that excessive rRNA transcription in oncogene-induced senescence or inhibition of rRNA processing in replicative senescence generates nucleolar stress, which drives cell cycle arrest through p53.30 The mechanisms of how ribosome biogenesis defect drives cellular senescence have been further revealed recently. Specifically, accumulation of the ribosomal 40S subunit protein RPS14 is a key contributor to senescence in response to multiple stimuli, and the capacity of RPS14 to trigger cellular senescence is CDK4/Rb-dependent and p53-independent.31 RPL22/eL22, another ribosomal protein, is sufficient to induce a cellular senescent phenotype by inhibiting CDK4-Cyclin D1 to decrease RB phosphorylation.32 Moreover, inhibition of the reaction catalyzed by LSG1, a GTPase involved in the biogenesis of the 60S ribosomal subunit, leads to a robust induction of cellular senescence that is associated with perturbation of endoplasmic reticulum homeostasis.33 We recently found that reduced/loss of expression of a ribonuclease angiogenin (ANG) in osteoclasts in long bone metaphysis leads to cellular senescence of neighboring vascular cells by inducing ribosomal biogenesis deficit.34 This finding provides more evidence on the role of ribosome biogenesis damage as a trigger of cellular senescence.
Despite different pathways identified in mediating SnC cycle arrest, no consensus exists regarding the biomarkers of the SnCs.35 Generally, none of the commonly used senescence-associated markers are specific or universal for all cell types, making identification of SnCs challenging. For example, senescence-associated beta-galactosidase was the first documented biomarker of senescence;36 however, it can also be detected in non-senescent macrophages,37 osteoclasts,38 and some other cell types, especially those in stressed states.39 p16INK4a and p21WAF1/Cip1 are also commonly used markers of cellular senescence, but they are not expressed in all SnCs,35 and their expressions depend on the specific type of senescence program.40 γ-H2AX and 53BP1, DNA damage response proteins, are also often used as SnC markers, but they also appear in transient DNA-damaged non-SnCs.41 Quantification of senescence-associated distension of satellites (SADS) (i.e., unraveled centromeres)42,43,44 and telomere-associated foci (i.e., sites of DNA damage within telomeres)45 have also been used to evaluate cellular senescence. Ribosomal protein RPL29 accumulates in nucleoli in response to cellular senescence stimuli, and therefore can serve as a new biomarker for cellular senescence.31 Reduced or loss of expression of cell-proliferating proteins Ki6746 and nestin,47,48,49 and loss of Lamin B150 and HMGB151 in the nucleus are also important features of SnCs. However, Ki67 and nestin loss are also observed in quiescent or terminally differentiated cells. A recent study reported that nuclear nestin deficiency drives tumor senescence via Lamin A/C-dependent nuclear deformation, providing a mechanism by which nestin loss may be involved in cellular senescence.52 Because none of the genes described above appear to be expressed uniquely by SnCs, the levels of multiple transcripts should be measured at the same time and in the same sample to avoid false positives. Importantly, it has been shown, by comparing whole-transcriptome datasets from different types of SnCs, that there is a “core” senescence signature—a set of genes that are commonly differentially expressed in the SnCs.53,54 A newly developed two-phase algorithmic assessment for quantification of various senescence-associated parameters in the same specimen55 will be helpful to overcome the limitation of measuring individual SnC markers.
SASP
In addition to stable cell cycle arrest, the SASP or senescence-messaging secretome is another key feature of SnCs that distinguishes the cells from other cell cycle-arrested cells. SnCs secrete hundreds of factors that include proinflammatory cytokines, chemokines, growth factors, and proteases.9,56,57,58,59 A recent study reported that oxylipins, a class of biologically active lipids that arise from the oxygenation of polyunsaturated fatty acids, are a component of the SASP.60 Not only do SnCs with SASP induce autocrine reinforcement, but they also communicate with neighboring cells in a paracrine manner to propagate the stress response in the tissue microenvironment.56,61,62 Of note, the paracrine effects of the SASP can be beneficial or detrimental depending on the tissue context.63 On the one hand, the SASP prevents cancer progression by inducing paracrine senescence in neighboring cells,61,62 promoting embryogenesis, tissue repair, and regeneration,58,64,65 and activating the immune system, which facilitates clearance of damaged cells.66,67 On the other hand, the SASP promotes tumorigenic processes such as angiogenesis and invasion56,68 and induces chronic inflammation that drives aged-related abnormalities in various tissues.69,70 The SASP factors produced by SnCs are highly cell specific and vary substantially even in the same type of cells, depending on the duration of senescence and origin of the pro-senescence stimulus.53 Through mass spectrometry, a core set of SASP proteins that are commonly produced by different types of SnCs were identified,54 which is helpful for the prediction of senescence-associated functions. It is important to note that despite the existence of certain core proteins, the expression and secretion of most SASP factors remain variable and context dependent. SASP induction and cell proliferative arrest appear to be regulated through separate signaling pathways because SASP is not induced by the expression of master cell cycle arrest regulatory genes p16Ink4a or p21Waf1/Cip1.57,71 Reportedly, transcriptional activation, chromatin remodeling, damaged DNA sensing, and extracellular vesicles, such as exosomes, may cause the activation of the SASP gene and promote the development of SASP. This aspect of SnCs has been summarized in several recent review articles.72,73,74,75,76 Although multiple pathways have been reported for SASP initiation and regulation of SnCs, the precise mechanisms regulating SASP induction are far from understood. Further defining the senescent secretome and its heterogeneity in function in various biological contexts and better understanding the temporospatial regulation of the SASP will help identify more biomarkers of SnCs, enabling the development of specific senescence modulatory therapies.
Cellular senescence in bone homeostasis, disease, and repair
Childhood bone homeostasis and diseases
Human bones change throughout the lifespan. Bone growth is characterized by a sharp increase during early puberty and deceleration and eventual cessation during late puberty. As growth in length accelerates, bone mass accrual also increases markedly during childhood and adolescence but gradually slows and eventually ceases during late puberty and young adulthood77,78 (Fig. 2a). At the cellular level, the growth plate at the ends of long bones and the adjacent primary spongiosa undergo substantial changes during late puberty to adapt to the much slower bone growth/accrual during this period. Vascular endothelial cells that form invaded blood vessels and mesenchymal stem/progenitor cells that replenish bone-forming osteoblasts are highly proliferative during the rapid bone-growth period, but these cells likely stop proliferating or are replaced by other cell types when bone growth stops. We uncovered a bone-growth cessation-associated cellular senescence at this particular time of life.47 We found that stem/progenitor cells in the metaphysis of long bone are highly proliferative during early puberty but undergo progressive cell senescence during late puberty (Fig. 2b). Cellular senescence was defined by the presence of SA-βGal and p16INK4a and decreased expression of Ki67 and nestin. We also found that the senescence process is controlled by an epigenetic mechanism.47 Ezh2-H3K27me3 maintains the self-renewal and proliferative capacity of cells in primary spongiosa of fast-growing bones, whereas loss of Ezh2-H3K27me3 during late puberty leads to cell senescence. Bone cell senescence in late puberty should be a normal physiological event because it is restricted to a particular region of the long bone, follows a specific time course, and is programmed by a conserved mechanism. This senescence program during late puberty somewhat resembles the senescence process that has been recognized during embryogenesis, in which the SnCs are thought to play a beneficial role in establishing the initial formation of tissues/organs.64 Regarding the skeletal system, SnCs were found in embryonic limbs at the apical ectodermal ridge, which is the main region coordinating limb outgrowth and patterning. Loss of senescence by p21 knockout results in impaired pattern formation and proliferation of the mesenchyme cells,65 reflecting the beneficial role of senescence in embryonic skeletal development.64,65 Importantly, embryogenesis-associated senescence is also time and location restricted.64,79 It seems that, during development, SnCs can be automatically eliminated by apoptosis or immune cells.64,65,80 Therefore, the remodeling of developing structures can be accomplished by promoting immune-mediated cell clearance of particular cell populations or by modifying the tissue microenvironment.81 It is of interest to test whether immune cells are also involved in clearing the SnCs in bone metaphysis during late puberty. In future studies, it will also be important to determine whether SnCs are increased in the cortical bone or bone tissue in other locations (e.g., lumbar vertebrae, skull) during the postnatal period, when the rates of bone growth and mineral acquisition slow. Further uncovering of the detailed mechanisms that control skeletal (bone and cartilage) cell senescence during embryonic development and childhood growth will provide exciting new opportunities to understand the pathogenic mechanisms associated with congenital and childhood skeletal abnormalities.

Cell senescence in long bone during late puberty is a programmed physiological process. a Changes in bone mineral density (BMD)/bone acquisition across the lifespan. BMD/acquisition increases rapidly during childhood and early puberty periods, becomes slow during late puberty, and reaches a plateau in young adulthood. b SA-βGal staining images show increased senescent cells in primary spongiosa in mice during late puberty (8 weeks old) relative to early puberty (3 weeks old)
In children with genetic skeletal disorders or chronic disease, bone growth and mineral accrual are often compromised, leading to osteoporosis and high rates of bone fracture. During childhood, fractures are common, with an annual fracture incidence of 205 per 10 000 person-years in those younger than 16 years, with the highest rate during the rapid growth spurts of puberty.82,83 Up to 25% of children with a fracture history experience more than one fracture, often representing primary or secondary bone loss or osteoporosis.82 Moreover, bone mineral accrual during childhood and adolescence influences long-term bone health. Epidemiologic studies suggest that 60% of the risk for osteoporosis later in life can be explained by the bone mineral acquired by childhood and early adulthood. Pediatric osteoporosis is often caused by an underlying medical condition (i.e., secondary osteoporosis) or a genetic disorder. Glucocorticoid-induced osteoporosis is the most common form of secondary pediatric osteoporosis. Long-term glucocorticoid therapy has been used widely in the treatment of chronic inflammatory childhood illnesses. Although the use of glucocorticoids has led to improved outcomes and survival rates, it has major adverse effects on bone. Epidemiologic studies have shown a prevalence of up to 34% for vertebral fractures in children and youth who have undergone long-term glucocorticoid therapy.84 We recently found that glucocorticoid treatment in young mice induces vascular endothelial cell senescence in metaphysis of long bone, and that inhibition of endothelial cell senescence improves glucocorticoid-impaired bone angiogenesis with coupled osteogenesis.34 We identified ANG, a ribonuclease that is secreted by osteoclasts and that has pro-angiogenic activity, as a key factor for protecting the neighboring vascular cells against senescence. We found evidence that ANG secreted from metaphyseal osteoclasts is essential for maintaining the closely associated blood vessels in growing long bone through an ANG/PLXNB2-rRNA transcription signaling pathway (Fig. 3). Glucocorticoid treatment inhibits ANG production through suppression of osteoclast formation in metaphysis, leading to blood vessel cell senescence and impairment of angiogenesis with coupled osteogenesis. Our results reveal cellular senescence as a new line of mechanisms for the deleterious effects of glucocorticoids on the growing skeleton and suggest that the ANG/PLXNB2 axis functions as a molecular basis for the osteoclast-vascular interplay in maintaining bone blood vessels from senescence. An important future question is whether the decline or loss of ANG/PLXNB2 signaling in adult bone is also a key mechanism leading to cellular senescence during aging or under disease conditions.

Osteoclasts protect bone blood vessels against senescence through the ANG/PLXNB2 axis. a ANG secreted from metaphyseal osteoclasts maintains blood vessel homeostasis in growing long bone through an ANG/PLXNB2-rRNA transcription signaling. b Enlarged image for the black square in (a)
Osteoporosis with aging
Bone aging is a normal process caused by the disrupted balance between bone resorption and formation as a result of the changes in osteoblast and osteoclast activity, leading to bone loss. In fact, peak bone mass (PBM) occurs during the third decade of life, after which bone mass plateaus and then declines. Men and women have different rates of bone loss after PBM. Markedly accelerated bone loss in women occurs during perimenopause and the early postmenopausal period. In men, bone loss with aging is less profound but is maintained at a persistent lower rate.2,85,86 In humans and mice, bone loss differs between the trabecular and cortical compartments with age. Trabecular bone aging is associated with reduced trabecular number, increased spacing, and decreased thickness.87,88 In cortical bone, endocortical resorption and periosteal bone formation often occur during aging, leading to cortical thinning with marrow cavity expansion and increased cortical porosity.2,89,90 In addition to the reduction in bone quantity, bone quality and strength also diminish during aging, leading to increased fracture risk, especially from falls. Bone aging is mediated by complex interactions of cells at multiple levels with the combination of intrinsic changes and extrinsic factors. Intrinsic changes include alterations in hormone status, cell components, and gene expression, as well as local microenvironmental changes caused by the altered production of growth factors, cytokines, and inflammatory factors. Extrinsic factors include nutrition and comorbid medical conditions. At the molecular level, it has been proposed that genomic instability, epigenetic alterations, telomere attrition, loss of proteostasis, mitochondrial dysfunction, deregulated nutrient sensing, and cellular senescence gene activation may contribute to age-related dysfunction in bone.91,92,93
In the past few years, there is growing evidence to suggest that cellular senescence is a major player in the pathogenesis of osteoporosis, a prevalent age-related bone disorder characterized by deterioration of bone microarchitecture and low bone mineral density (BMD). SnC accumulation has been observed in bone/bone marrow, as shown by increased SA-βGal, SADS, telomere dysfunction-induced foci, p16INK4a, or p21CIP1 expression in many types of cells in aged mice, including bone marrow myeloid cells, T cells, B cells, MSCs, osteoprogenitors, osteoblasts, and osteocytes.42,89,94 These SnCs also acquire SASP and secrete various factors to affect their microenvironment, leading to the loss of bone mass and architecture. SASP secretion of bone marrow MSCs and osteoblastic cells mediates enhanced osteoclastogenesis and the development of osteoporosis.42,95 The study of senescence and the SASP of osteocytes, the longest-living bone cells embedded in the bone matrix, has drawn much attention. Senescent osteocyte accumulation has been found in the aged bone environment of mice and humans,42,89 and induction of osteocyte senescence is sufficient to stimulate receptor activator of nuclear factor kappa-B ligand production, leading to increased osteoclast formation and activity.96 The presence of SnCs and the corresponding SASP in osteoporotic bone has also been confirmed in elderly people.42 Importantly, the genetic elimination of the SnCs using INK-ATTAC mice or administration of senolytic drugs was able to inhibit production of SASP in the bone marrow microenvironment, leading to increased bone mass and microarchitecture in aged osteoporotic mice.97 Therefore, targeting cellular senescence may represent a new therapeutic strategy to prevent or treat osteoporosis in the elderly. However, activation of p16-3MR transgene failed to eliminate senescent osteocytes and alleviate age-associated bone loss,98 indicating that genetic approaches used for the elimination of p16‐expressing cells are generally tissue selective. Another interesting question is the role of cellular senescence in accelerated skeletal aging in response to sex steroid deficiency. Estrogen has a crucial effect on bone metabolism, and its decline is causal in the pathogenesis of osteoporosis. In one report, ovariectomy led to the generation of premature T-cell senescence in mice, and estrogen treatment reversed the phenotype.99 However, recent work by Farr et al.100 using multiple approaches in mice and humans suggests no evidence of changes in senescence biomarkers or SASP factors in mouse or human bone in response to estrogen deficiency. Moreover, elimination of SnCs during young adulthood does not restore the bone loss caused by estrogen deficiency. Therefore, it is likely that estrogen deficiency-induced bone loss occurs independently of cellular senescence.
Osteoporosis in diabetes mellitus
Osteoporosis, bone fractures, and bone-related disorders are more common in people with diabetes mellitus (DM) than in the general population. DM can cause osteopenia and osteoporosis and aggravates both conditions. Pathologic bone changes differ between those with type 1 DM (T1DM) and type 2 DM (T2DM). Patients with T1DM often develop osteoporosis with reduced BMD at an early age, whereas bone in T2DM exhibits normal or high BMD. However, patients with T2DM have a higher rate of fractures, mainly because of compromised bone quality and microarchitecture.101,102 Eckhardt et al.103 examined cellular senescence in a T2DM mouse model, in which accelerated accumulation of senescent osteocytes with SASP was detected in bone while bone microarchitecture and biomechanical strength were compromised. The finding suggests that the SnCs and corresponding SASP are likely involved in skeletal fragility in T2DM. It remains unclear whether cellular senescence and SASP are involved in the pathogenesis of T1DM-related bone loss. Future studies are needed to determine whether other cell types in the bone/bone marrow microenvironment also undergo cellular senescence with DM and whether they are factors in the disease pathogenesis. SnCs accumulate in adipose tissue of humans and mice with T2DM, obesity, and age-related metabolic dysfunction.104 Given that bone mass/quality is negatively correlated with many of these metabolic disorders, and more fat cells are present in the bone marrow in these conditions, it has long been proposed that increased marrow adipocyte differentiation from mesenchymal stem/progenitor cells accounts for age- and DM-associated bone loss. However, a recent study showed that increased marrow adipogenesis did not contribute to age‐ and DM-associated appendicular bone loss in female mice,105 and provides a compelling argument against the above hypothesis. Additional studies are still needed to clarify whether bone marrow adipocytes have a senescence phenotype and secrete SASP factors during DM to regulate the function of surrounding bone cells.
Cellular senescence in osteoarthritis development
Cellular senescence in the pathogenesis of osteoarthritis
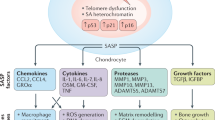
The articular cartilage in human joints is subject to extreme biologic and biomechanical demands in allowing movement and function. The term “articular cartilage” typically refers to hyaline cartilage, which must be maintained within a narrow range of biochemical composition and morphologic architecture to maintain its integrity, and the intrinsic healing potential of articular cartilage is very limited. Therefore, any insult, such as injury, aging, or metabolic dysregulation, can initiate a cascade of events that lead to degradation and erosion of articular cartilage and the development of osteoarthritis. Osteoarthritis is a common chronic musculoskeletal disease and a leading cause of disability with no effective disease-modified therapy.41,106,107 Increasing evidence in the past several years has shown that SnCs accumulate in mouse and human osteoarthritic joint tissue, and SASP has been implicated in osteoarthritis progression.108,109,110,111 SnCs typically develop in response to cellular stress, preventing the proliferation of damaged or dysfunctional cells, thereby protecting organisms against diseases. Initially, SnCs are a crucial component of the wound-healing process; however, if they are not cleared in a timely manner, their persistence can lead to tissue deterioration that causes aging, as well as many chronic conditions and age-related diseases.56,58,112 Senescent chondrocytes, for example, have been linked to post-traumatic osteoarthritis (PTOA) and age-related osteoarthritis since they were identified in the cartilage tissue isolated from patients with osteoarthritis undergoing joint replacement surgery.11,108,113
As osteoarthritis progresses, the chondrocyte phenotype is skewed toward matrix degradation accompanied by SnC-associated SASP factors, specifically matrix metalloproteinases (MMP)-13 and aggrecan-degrading ADAMTS-5.114,115 In addition, oxidative stress promotes chondrocyte senescence, mainly by activating p38 MAPK and PI3K/Akt signaling, leading to SASP factor production and increased inflammation and tissue deterioration.116 Macrophages, in response to the SASP, are recruited to the articular space, secrete proinflammatory factors interleukin (IL)-1β, IL-6, and tumor necrosis factor α (TNFα), as well as growth factors TGFβ and BMPs, which activate synovial fibroblasts to upregulate MMPs and ADAMTS and promote osteophyte formation and drive osteoarthritis progression.117,118,119 More recently, we have shown that damage to the joint (anterior cruciate ligament transection [ACLT]) results in increased cellular senescence (p16+), upregulated SnC-associated proinflammatory factors, as well as a Th17-type immune signature in the joint and draining lymph nodes, and ultimately osteoarthritis development.108,110 Historically, osteoarthritis has been defined as a primarily localized disease, but through in vitro and in vivo studies we have identified a systemic Th17-SnC feedback where Th17 T cells induce senescence in healthy fibroblasts and SnCs skew naive T cells toward a Th17 phenotype in the presence of TGFβ.110 Thus, with this new understanding of the systemic immunological feedback that exacerbates cartilage degeneration leading to osteoarthritis, more effective therapeutics can be developed to combat not only the pathological SnC, but also the greater effects of their SASP. The immune system initiates and conducts critical responses to tissue damage. SnCs, through their SASP, secrete several cytokines that influence the immune response orchestrating the balance between a proinflammatory and pro-regenerative response to injury or trauma.56,112,120 Trauma induces a cascade of local and systemic immune events that recruit various cells to the injured site to protect the tissue from infection and promote tissue repair. Cartilage, however, because of its avascular composition, has limited self-repair capacity. Therefore, SnC accumulation and SASP signaling leading to chronic inflammation in the joint after trauma not only results in the inability for cartilage repair and osteoarthritis development but also induces systemic immune changes, possibly exacerbating responses to other injuries or diseases and promoting further tissue deterioration. The canonical proinflammatory molecules associated with SnC accumulation and their SASP in osteoarthritis are cytokines (IL-1β, IL-6, TNFα), chemokines (CCL2, CCL4), proteases (MMPs, ADAMTS), and growth factors (TGFβ, IGFBP).109 The identification of the SnCs in the osteoarthritic joints and their contribution to the disease development have also been discussed in recent comprehensive review articles.109,121
Osteoarthritis has heterogeneous causes that lead to accelerated structural damage and has long been considered as the unique consequence of a “wear and tear” process leading to cartilage degradation. Indeed, direct joint injury and mechanical overloading are important contributors to the development of osteoarthritis.122,123 However, this initial paradigm has been modified during the past decade because of critical findings from epidemiologic and prospective clinical studies. Specifically, only 12% of the overall prevalence of symptomatic osteoarthritis is attributable to PTOA of the hip, knee, or ankle,124 indicating that mechanisms other than biomechanical factors are involved in osteoarthritis development. Older age is the greatest risk factor for osteoarthritis, and age-related changes could accelerate the development of its pathologic process. Furthermore, metabolic osteoarthritis has now been considered as a subtype of osteoarthritis defined by the presence of individual metabolic syndrome (MetS) components or MetS as a whole.125 Epidemiologic studies have shown that osteoarthritis is strongly associated with cardiovascular disease,126,127,128,129 and the rate of cardiovascular mortality is directly proportional to the extent of radiographic evidence of osteoarthritis. Furthermore, osteoarthritis patients have a higher prevalence of cardiovascular risk factors, including dyslipidemia, obesity, hypertension, and DM,130,131,132,133 all of which are aspects of MetS. Particularly, 59% of patients with osteoarthritis had MetS compared with 23% of the general population, and the middle-aged population with osteoarthritis has more than a five-fold increase in the risk of MetS compared with the population without osteoarthritis.134 Although aging remains the most important risk factor for osteoarthritis, the metabolic phenotype has become the second most frequent subtype of osteoarthritis among patients enrolled in clinical studies.125,135,136,137 However, the pathogenic mechanisms of osteoarthritis associated with MetS remain unclear. It will be important to know whether cellular senescence is also involved in the pathogenesis of MetS-associated osteoarthritis. Understanding the role of cellular senescence and the SASP in the development of different subtypes of osteoarthritis will undoubtedly inform the development of targeted treatment approaches to stop or slow osteoarthritis progression.
Seno-therapy to treat osteoarthritis
Despite of the intensive investigations in the past two decades on the pathogenesis of osteoarthritis, currently there are still no FDA-approved medications to slow or stop the disease progression. Cellular senescence and the SASP have causal roles in mediating many age-associated chronic diseases. Therapeutically directly eliminating SnCs or targeting the effects of SnCs hold promise to prevent, delay, or alleviate these conditions. To address the recent findings of the effect of SnC and the SASP in osteoarthritis, researchers have been investigating the efficacy of two families of senescent-related medications: senolytics and senomorphics. Senolytics preferentially induce apoptosis in SnCs. Senomorphics, also referred to as senostatics or SASP inhibitors, selectively block SASP factors that cause further inflammatory paracrine signaling and tissue damage.138
In initial studies exploring the role of senescence in age-associated abnormalities, mice that had undergone clearance of p16-expressing cells had an increased lifespan and delayed onset of age-related diseases.139,140 When a similar inducible p16-clearance model was used in osteoarthritic mice, the development of osteoarthritis was attenuated and cartilage repair and development increased.108 Senolytics typically induce preferential or ideally selective apoptosis of SnCs via targeting pro-survival signals and pathways that are activated in SnCs but not healthy cells (Fig. 4a). Timing of delivery, however, is critical with senolytic therapies because SnCs are crucial to the initial tissue injury response; therefore, senolytics are typically administered after the initial inflammatory phase, when tissue repair must be promoted. Navitoclax, a BCL-2 and BCL-Xl inhibitor, clears senescent, p16+ stem cells in the bone marrow and muscle, thus promoting regeneration when delivered systemically.141,142,143 Other pro-survival targets constitute several tyrosine kinases, such as BCR-ABL, SRC, c-KIT, ephrin A receptor, and PDGF-β-receptor tyrosine kinases. Dasatinib is a senolytic drug that targets all of the aforementioned tyrosine kinase targets and has been used in combination with quercetin, which inhibits PI3K and serpins.144 The dual treatment of dasatinib and quercetin eliminates SnCs in the cartilage and bone and promotes tissue regeneration.97,145,146

Senolytic and senomorphic therapeutic strategies. a Senolytic strategies seek to preferentially induce apoptosis in senescent cells and target molecules or signaling pathways that are increased in senescent cells such as p53, BCL-XL and BCL-2, and survival signaling through receptor tyrosine kinases (RTKs). b Senomorphic strategies target SASP factors that can cause further inflammatory paracrine signaling and tissue damage. CL82198, a selective MMP13 inhibitor, and rapamycin, an mTOR inhibitor, are both senomorphics under assessment. Blocking antibodies as well as neutralizing antibodies for several SASP factors (IL-1, IL-6, TNF) are being tested for efficacy against further tissue damage. Neutralizing IL-17a/f monoclonal antibodies (nIL-17 mAb) are under evaluation to disrupt the feedforward cycle of TH17 skewing and TH17-induced senescence. Ub Ubiquitin
UBX0101, via local intra-articular injection, is a senolytic that is currently being assessed in clinical trials for its efficacy in osteoarthritis treatment (ClinicalTrials.gov identifiers NCT03513016, NCT04229225, and NCT04349956). UBX0101 inhibits p53 ubiquitination and degradation by preventing MDM2, an E3 ubiquitin protein ligase, binding and has shown promise by eliminating SnCs and relieving osteoarthritis-related articular cartilage degradation and proteoglycan loss via local delivery.108 Another study sought to determine whether local and/or systemic senolytic delivery promotes cartilage repair.110 ACLT was performed in young and aged mice to create a model of PTOA. Mice were then treated with local UBX0101, systemic navitoclax, or both. Young mice responded well to solo treatment with either UBX0101 or navitoclax, showing improved tissue structure, decreased osteophytes, and potential tissue repair, whereas aged mice experienced significant improvement only with both local and systemic delivery.110 Faust et al.110 concluded that systemic senolysis is required in aged animals to restore a wound-healing and tissue-regeneration capacity. Furthermore, sirtuin-1 (SIRT1) is a crucial enzyme to ECM homeostasis and chondrocyte survival,147 and SIRT1 cleavage has been correlated with chondrosenescence and osteoarthritis.148 In another study, ACLT in mice induced SIRT1 cleavage, and systemic navitoclax delivery combined with intra-articular UBX0101 delivery decreased the amount of circulating cleaved SIRT1.148 Another senolytic that is currently being assessed in clinical trials is fisetin (NCT04210986). Fisetin, a flavonoid that activates sirtuins such as SIRT1, has been suggested to increase longevity and inhibit IL-1β–propagated inflammation in osteoarthritis.149,150 Via high-throughput drug screening against senescence in chondrocyte cell lines, new senolytics can be identified for further investigation.
Senomorphic strategies target SASP factors that can influence the surrounding tissue microenvironment by inflammatory paracrine signaling (Fig. 4b). SASP factors fall into two main classes: MMPs and cytokines. Of MMPs, the most highly expressed is MMP13, which can efficiently degrade type-II collagen in articular cartilage.151 Patients with osteoarthritis express higher levels of MMP13 in their chondrocytes than do healthy patients,152 and MMP13 has been identified as a primary driver of osteoarthritis.153 CL82198, an MMP13 inhibitor, decreased osteoarthritis-related degeneration and pain, decreased chondrocyte cell death, and increased type-II collagen levels.154 Therefore, MMP13 inhibitors could be a promising therapy for osteoarthritis patients.
TNF and IL-1β are proinflammatory cytokines that promote MMP production and secretion, preventing tissue regeneration and repair. Unfortunately, targeting these two cytokines has produced little clinical improvement in osteoarthritis trials, with little or no effects on pain and tissue repair.155,156 A more recent study, however, indicated a lower incidence of hip and knee replacement in a group treated with anti-IL-1β antibody compared with the placebo group.157 Clinically approved for treatment of rheumatoid arthritis, an anti-IL-6-receptor, tocilizumab, is being assessed for clinical efficacy for osteoarthritis of the hand (NCT02477059). One study reported that IL-6 knockout mice experience more severe osteoarthritis than wild-type mice158 so targeting certain immunologic mediators involved in the SASP is not straightforward. More recently, however, IL-17 was linked to the SASP profile, causing inflammation-induced SnCs.110,142 In aged animals, co-administration of local and systemic senolytics attenuated tissue degeneration after ACLT and corresponded to decreased IL-17 expression in the joint and draining lymph nodes.110 In vitro, SnCs skewed naive T cells toward a proinflammatory Th17 or Th1 type, while Th17 T cells were also able to induce senescence in fibroblasts, indicating a feedback loop that perpetuates senescence development and tissue damage.110 Faust et al.110 assessed the efficacy of an IL-17 neutralizing antibody against further osteoarthritic damage after ACLT and found that intra-articular injection of an anti-IL-17a neutralizing antibody decreased MMP13 and p21 expression, significantly improved tissue structure, and relieved SnC burden and the pathologic effect of cartilage injury. Therefore, future studies should explore the connection between IL-17 and senescence and the efficacy of IL-17-blocking senomorphics to promote cartilage regeneration.
Cellular senescence in the pathogenesis of sarcopenia
Cellular senescence in sarcopenia
Skeletal muscle attaches to bone via strong connective tissues and therefore is key to controlling locomotion. Skeletal muscle is also a critical factor in whole-body metabolism and energy expenditure and is essential for maintaining quality of life.159,160,161,162 Skeletal muscle contains several cell populations, including myocytes, skeletal stem cells, fibroblasts, nerve fibers, and blood vessel cells. Adult muscle stem cells are referred to as satellite cells, which are responsible for muscle maintenance, repair, and regeneration in aging and injured muscles.160,163 Loss of lean muscle mass begins at a slow rate around the fifth decade of life and accelerates over time, resulting in 30%–50% of lean mass reduction by age 70.164,165 Muscle strength deficiency precedes muscle loss and declines much faster than muscle mass. Age-related decline in muscle mass and associated muscle weakness are referred to as sarcopenia.166
Preliminary studies have linked increased senescence, not only of the muscle cells themselves, but also of their SASP, to the characteristic muscle-fiber thinning of sarcopenia. A recent study sought to determine whether SnC-implanted adjacent to skeletal muscle would affect healthy surrounding muscle cells via the “bystander effect” and senescent-like signaling.167 SnC transplantation adjacent to skeletal muscle induced an increase in senescent markers in the muscle, as well as muscle-fiber thinning, indicative of sarcopenia.167 SASP factors have also been linked to the induction of several factors, such as sarcolipin, that have been shown to promote skeletal muscle fibrosis and, ultimately, sarcopenia.168
Other factors, such as endothelin-1, TRIM32, and GSK-3α have been shown to induce muscle stem cell senescence and suppress autophagy in mouse models. Knockout of each of these factors results in muscle degeneration and sarcopenia development.169,170,171 In addition, aberrant p38α/β ΜΑPK signaling has also been observed in aged muscle stem cells, leading to reduced regenerative capacity and functional decline.172 Pharmacologic inhibition of this p38α/β signaling in preclinical studies enhances aged muscle stem cell proliferation, restores aged muscle stem cell regenerative capacity, and provides long-term functionality and increased strength. Furthermore, in murine muscle stem cells, a recent study reported reduced expression of the transcription factor Slug and identified Slug as a transcriptional repressor of senescence-initiating gene p16Ink4a.173 The authors proposed to develop therapeutics to overexpress Slug as a potential inhibitor of age-related skeletal muscle degeneration and even sarcopenia development. Wnt-3 is another therapeutic target against skeletal muscle senescence and deterioration. Wnt-3 was increased in the serum of elderly people, and Wnt-3 expression induces CCN1 expression.174 Any of these molecules could serve as promising potential targets for combatting age-related muscle degeneration and promote muscle regeneration in cases of sarcopenic development.
Autophagy is the organized degradation and recycling of dysfunctional cellular components. Typically, in muscle stem cells, autophagy activity declines with age, resulting in accumulation of damaged cell components and a transition from quiescence to senescence.175 Maintenance of basal autophagy in muscle stem cells is critical for preventing senescence and maintaining regenerative capacity.176 Therefore, promotion of basal autophagy in aged muscle is a promising therapy to reverse senescence in muscle stem cells in elderly populations.175 Some disorders in which autophagy is impaired lead to sarcopenia via senescence induction. For example, hyperphosphatemia, an electrolyte disorder in which circulating phosphate levels are higher than normal, induces myoblast senescence via autophagy impairment and integrin-linked kinase overexpression, which could propagate muscle degeneration and sarcopenia.177
Recent studies have begun investigating immunogenic changes that can be influenced by SnCs in the development of sarcopenia. Preliminary studies have shown that bone marrow transplants from young mice into old mice prevent sarcopenia.178,179 Macrophages present in muscle repair have been shown to secrete nicotinamide phosphoribosyltransferase (NAMPT) to stimulate muscle stem cells and promote myoblast proliferation.180 Because NAMPT has been shown to inhibit senescence in endothelial progenitor cells via SIRT1 regulation,181 the study suggests that macrophages, by producing NAMPT, may prevent/inhibit cellular senescence and the corresponding SASP and therefore positively regulate muscle regeneration.
Metabolic irregularities, which disrupt typical metabolic homeostasis, can lead to systemic inflammation and senescence. Aberrant metabolic signaling in skeletal muscle, a critical metabolic tissue, contributes to senescence and the SASP, which accelerates sarcopenia.182,183 Oxidative stress induces senescence in mesenchymal progenitor cells, which typically support myogenic cell function but can further differentiate into fibrous/adipose tissue that is detrimental to skeletal muscle support.184 These senescent mesenchymal progenitor cells, when cultured with myoblasts, prevented myotube formation and accelerated sarcopenia.185,186 Several metabolic enzymes are involved in metabolic sarcopenia. Peroxiredoxin 6, an antioxidant enzyme, prevents SIRT1 cleavage, as well as forkhead box O1 (FoxO1) expression, and when absent leads to muscle degeneration associated with sarcopenia.187 Increased FoxO expression increases proteolysis, leads to increased senescence, and accelerates age- and obesity-induced muscle degeneration.188 The role of perimuscular adipose tissue accumulation around muscle has also been explored in the development or sarcopenia. Perimuscular adipose tissue transplantation in mice increased activation and nuclear translocation of FoxO transcription factors.188 Optic atrophy 1 (OPA1), another general metabolic regulator, is a mitochondrial protein in which decreased levels have been associated with sedentary but not active people experiencing muscle loss.189 Via FGF21, OPA1 regulates muscle stem cell activity and senescence, and OPA1 abrogation leads to endoplasmic reticulum stress and induction of FoxO transcription factors, leading to muscle catabolism and loss.189
These studies indicate that senescence plays a pivotal role in sarcopenia development, and a deeper understanding of senescence induction, the SASP, and sarcopenia progression would provide insight into regenerative therapeutics for muscle loss.
Senolytic and senomorphic treatments have also been used against SnCs and the SASP that contribute to sarcopenia.190 FoxO4 expression, a senolytic target, is elevated in SnCs and has been identified as a key molecule in maintaining SnC viability.191 A small molecule peptide senolytic, FoxO4-DRI, blocks FoxO4-p53 interaction, shows the ability to preferentially target SnCs, and should be assessed for efficacy against sarcopenic muscle loss.191 In addition, MABp1 administration, a senomorphic, efficiently targets and prevents sarcopenia progression by neutralizing anti-human IL-1α.192 Chung et al.142 explored the influence of IL-17-regulated senescence in the foreign body response after volumetric muscle loss and found that SnC development was abrogated in IL-17A–/– and IL-17RA–/– mice, indicating that SnC induction is dependent on IL-17. This finding could indicate that a senomorphic IL-17-blocking therapeutic, as discussed in the cartilage repair section, could be a promising strategy to attenuate further senescence/SASP–related damage and promote muscle repair and regeneration. In addition, treatment with an IL-6 neutralizing antibody showed decreased senescent-induced Il17a expression, indicating that IL-6-targeted senomorphic therapies may also slow or even prevent sarcopenia progression.142 Finally, when navitoclax was systemically administered to mice 4 weeks after volumetric muscle loss, it cleared SnCs, reduced the SASP (particularly IL-17-related), and prevented further tissue damage.142 This study highlighted the efficacy of combination therapies of senolytics and senomorphics (navitoclax + anti-IL-17a/f neutralizing antibodies), illustrating that, when these treatments are combined, the SASP is even more diminished and less tissue damage occurs.142 These combination therapies should be assessed in the context of tissue regeneration, not only in the muscle, but also in age-related/senescence-associated bone and cartilage abnormalities.
Other treatments seek to target the factors that induce senescence. Tocotrienol-rich fraction treatment of patients with sarcopenia has been shown to reverse myoblast aging and senescence, providing muscle cells with the regenerative capacity needed to reverse muscle loss and promote repair.193,194,195 More recently, researchers have targeted microRNAs that are related to key factors that induce satellite cell senescence, such as Igfbp5, Wnt-3α signaling, and dysregulation of AMPK/SIRT1.196,197,198 Although these microRNA-targeted therapies have shown promise in preclinical trials, further studies and characterization are needed to assess the efficacy in preventing pathological senescence, reversing sarcopenic muscle loss, and promoting regeneration.
Summary and perspective
With aging, various mechanistic changes occur in the MSK, which result in structural degeneration, mechanical pain, decreased mobility, and limited function. The degeneration of bones, joints, and skeletal muscles accelerates frailty and makes older people prone to osteoporosis, osteoarthritis, and sarcopenia. During the past two decades, studies using mouse models have established that SnCs accumulate in different components of the MSK with aging, and elimination of SnCs results in attenuation/deceleration of disease progression. However, several issues need to be resolved before interventions to remove SnCs are tested in humans to treat age-associated disorders in the MSK. First, it is important to understand the spatio-temporal distribution of SnCs in human musculoskeletal tissues, under diverse conditions and during all stages of the lifespan. Second, we must explore the heterogeneity of SASP in different cell types in the complex MSK microenvironment. Third, although excessive accumulation of SnCs is associated with increased susceptibility to chronic musculoskeletal diseases, SnCs also have beneficial physiologic functions during embryonic development, late pubertal bone growth cessation, and tissue remodeling in adulthood. Therefore, it is imperative to better understand the mechanisms that distinguish beneficial from deleterious senescence and the heterogeneity underlying SnC states. Mapping the landscape of cellular senescence under physiologic or pathologic conditions will undoubtedly aid the targeted development of therapeutic approaches for SnCs. Transformational advances in the combined use of single-cell technologies and in situ image-based approaches would allow us to generate a molecular atlas of SnC types in vivo while mapping their spatial and functional organization.
References
Roberts, S. et al. Ageing in the musculoskeletal system. Acta Orthopaedica 87, 15–25 (2016).
Goltzman, D. The aging skeleton. Adv. Exp. Med. Biol. 1164, 153–160 (2019).
Patel, J. Economic implications of osteoporotic fractures in postmenopausal women. Am. J. Manag. Care 26, S311–S318 (2020).
Ma, V. Y., Chan, L. & Carruthers, K. J. Incidence, prevalence, costs, and impact on disability of common conditions requiring rehabilitation in the United States: stroke, spinal cord injury, traumatic brain injury, multiple sclerosis, osteoarthritis, rheumatoid arthritis, limb loss, and back pain. Arch. Phys. Med. Rehabil. 95, 986–995.e1 (2014).
von Haehling, S., Morley, J. E. & Anker, S. D. An overview of sarcopenia: facts and numbers on prevalence and clinical impact. J. Cachexia Sarcopenia Muscle 1, 129–133 (2010).
Guo, Y., Phillips, B. E., Atherton, P. J. & Piasecki, M. Molecular and neural adaptations to neuromuscular electrical stimulation; Implications for ageing muscle. Mech. Ageing Dev. 193, 111402 (2021).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37, 614–636 (1965).
Wiley, C. D. et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314 (2016).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Childs, B. G., Durik, M., Baker, D. J. & van Deursen, J. M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med. 21, 1424–1435 (2015).
Childs, B. G., Li, H. & van Deursen, J. M. Senescent cells: a therapeutic target for cardiovascular disease. J. Clin. Investig. 128, 1217–1228 (2018).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011 (2017).
Ito, Y., Hoare, M. & Narita, M. Spatial and temporal control of senescence. Trends Cell Biol. 27, 820–832 (2017).
Harley, C. B., Futcher, A. B. & Greider, C. W. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 (1990).
Levy, M. Z., Allsopp, R. C., Futcher, A. B., Greider, C. W. & Harley, C. B. Telomere end-replication problem and cell aging. J. Mol. Biol. 225, 951–960 (1992).
Marcand, S., Gilson, E. & Shore, D. A protein-counting mechanism for telomere length regulation in yeast. Science 275, 986–990 (1997).
Bartkova, J. et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006).
Di Micco, R. et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642 (2006).
Mallette, F. A., Gaumont-Leclerc, M. F. & Ferbeyre, G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 21, 43–48 (2007).
Webley, K. et al. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol. Cell. Biol. 20, 2803–2808 (2000).
Takahashi, A. et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 8, 1291–1297 (2006).
Imai, Y. et al. Crosstalk between the Rb pathway and AKT signaling forms a quiescence-senescence switch. Cell Rep. 7, 194–207 (2014).
Terzi, M. Y., Izmirli, M. & Gogebakan, B. The cell fate: senescence or quiescence. Mol. Biol. Rep. 43, 1213–1220 (2016).
Watanabe, S., Kawamoto, S., Ohtani, N. & Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 108, 563–569 (2017).
Bracken, A. P. et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525–530 (2007).
Gil, J., Bernard, D., Martínez, D. & Beach, D. Polycomb CBX7 has a unifying role in cellular lifespan. Nat. Cell Biol. 6, 67–72 (2004).
Jacobs, J. J., Kieboom, K., Marino, S., DePinho, R. A. & van Lohuizen, M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397, 164–168 (1999).
Childs, B. G. et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 16, 718–735 (2017).
Nishimura, K. et al. Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep. 10, 1310–1323 (2015).
Lessard, F. et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat. Cell Biol. 20, 789–799 (2018).
Del Toro, N. et al. Ribosomal protein RPL22/eL22 regulates the cell cycle by acting as an inhibitor of the CDK4-cyclin D complex. Cell Cycle 18, 759–770 (2019).
Pantazi, A. et al. Inhibition of the 60S ribosome biogenesis GTPase LSG1 causes endoplasmic reticular disruption and cellular senescence. Aging Cell 18, e12981 (2019).
Liu, X. et al. Osteoclasts protect bone blood vessels against senescence through the angiogenin/plexin-B2 axis. Nat. Commun. 12, 1832 (2021).
Hernandez-Segura, A., Rubingh, R. & Demaria, M. Identification of stable senescence-associated reference genes. Aging Cell 18, e12911 (2019).
Dimri, G. P. et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA. 92, 9363–9367 (1995).
Hall, B. M. et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 9, 1867–1884 (2017).
Odgren, P. R. et al. False-positive beta-galactosidase staining in osteoclasts by endogenous enzyme: studies in neonatal and month-old wild-type mice. Connect. Tissue Res. 47, 229–234 (2006).
Yang, N. C. & Hu, M. L. The limitations and validities of senescence associated-beta-galactosidase activity as an aging marker for human foreskin fibroblast Hs68 cells. Exp. Gerontol. 40, 813–819 (2005).
Alcorta, D. A. et al. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl Acad. Sci. USA. 93, 13742–13747 (1996).
Veronese, N. et al. Pain increases the risk of developing frailty in older adults with osteoarthritis. Pain Med. 18, 414–427 (2017).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 31, 1920–1929 (2016).
Swanson, E. C., Manning, B., Zhang, H. & Lawrence, J. B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 203, 929–942 (2013).
Criscione, S. W. et al. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2, e1500882 (2016).
Hewitt, G. et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708 (2012).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453 (2018).
Li, C. et al. Programmed cell senescence in skeleton during late puberty. Nat. Commun. 8, 1312 (2017).
Su, J. et al. Cellular senescence mediates the detrimental effect of prenatal dexamethasone exposure on postnatal long bone growth in mouse offspring. Stem Cell Res. Ther. 11, 270 (2020).
Hermann, A. et al. Age-dependent neuroectodermal differentiation capacity of human mesenchymal stromal cells: limitations for autologous cell replacement strategies. Cytotherapy 12, 17–30 (2010).
Wang, A. S., Ong, P. F., Chojnowski, A., Clavel, C. & Dreesen, O. Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin. Sci. Rep. 7, 15678 (2017).
Davalos, A. R. et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629 (2013).
Zhang, Y. et al. Nuclear Nestin deficiency drives tumor senescence via lamin A/C-dependent nuclear deformation. Nat. Commun. 9, 3613 (2018).
Hernandez-Segura, A. et al. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol. 27, 2652–2660.e2654 (2017).
Basisty, N. et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 18, e3000599 (2020).
Kohli, J. et al. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat. Protoc. 16, 2471–2498 (2021).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Laberge, R. M. et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 (2015).
Wiley, C. D. et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab. 33, 1124–1136.e5 (2021).
Kuilman, T. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Liu, X. & Wan, M. A tale of the good and bad: cell senescence in bone homeostasis and disease. Int. Rev. Cell Mol. Biol. 346, 97–128 (2019).
Munoz-Espin, D. et al. Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118 (2013).
Storer, M. et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130 (2013).
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007).
Iannello, A., Thompson, T. W., Ardolino, M., Lowe, S. W. & Raulet, D. H. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med. 210, 2057–2069 (2013).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P. Y. & Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl Acad. Sci. USA. 98, 12072–12077 (2001).
Kirkland, J. L. & Tchkonia, T. Cellular senescence: a translational perspective. EBioMedicine 21, 21–28 (2017).
Song, S., Lam, E. W., Tchkonia, T., Kirkland, J. L. & Sun, Y. Senescent cells: emerging targets for human aging and age-related diseases. Trends Biochem. Sci. 45, 578–592 (2020).
Borodkina, A. V., Deryabin, P. I., Giukova, A. A. & Nikolsky, N. N. “Social life” of senescent cells: what is SASP and why study it? Acta Nat. 10, 4–14 (2018).
Kumari, R. & Jat, P. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 9, 645593 (2021).
Cuollo, L., Antonangeli, F., Santoni, A. & Soriani, A. The senescence-associated secretory phenotype (SASP) in the challenging future of cancer therapy and age-related diseases. Biology 9, 485 (2020).
Birch, J. & Gil, J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 34, 1565–1576 (2020).
Payea, M. J., Anerillas, C., Tharakan, R. & Gorospe, M. Translational control during cellular senescence. Mol. Cell. Biol. 41, e00512–e00520 (2021).
Tanaka, Y. & Takahashi, A. Senescence-associated extracellular vesicle (SA-EV) release plays a role in senescence-associated secretory phenotype (SASP) in age-associated diseases. J. Biochem. 169, 147–153 (2021).
Rauch, F. The dynamics of bone structure development during pubertal growth. J. Musculoskelet. Neuronal Interact. 12, 1–6 (2012).
Yakar, S. & Isaksson, O. Regulation of skeletal growth and mineral acquisition by the GH/IGF-1 axis: lessons from mouse models. Growth Horm. IGF Res. 28, 26–42 (2016).
Muñoz-Espín, D. & Serrano, M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014).
Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742–758 (2011).
Triana-Martinez, F., Pedraza-Vazquez, G., Maciel-Baron, L. A. & Konigsberg, M. Reflections on the role of senescence during development and aging. Arch. Biochem. Biophys. 598, 40–49 (2016).
Marrani, E., Giani, T., Simonini, G. & Cimaz, R. Pediatric osteoporosis: diagnosis and treatment considerations. Drugs 77, 679–695 (2017).
Nellans, K. W., Kowalski, E. & Chung, K. C. The epidemiology of distal radius fractures. Hand Clin. 28, 113–125 (2012).
Huber, A. M. et al. Prevalent vertebral fractures among children initiating glucocorticoid therapy for the treatment of rheumatic disorders. Arthritis Care Res. 62, 516–526 (2010).
Berger, C. et al. Change in bone mineral density as a function of age in women and men and association with the use of antiresorptive agents. CMAJ 178, 1660–1668 (2008).
Glatt, V., Canalis, E., Stadmeyer, L. & Bouxsein, M. L. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J. Bone Miner. Res. 22, 1197–1207 (2007).
Ding, M. & Hvid, I. Quantification of age-related changes in the structure model type and trabecular thickness of human tibial cancellous bone. Bone 26, 291–295 (2000).
Javaheri, B. & Pitsillides, A. A. Aging and mechanoadaptive responsiveness of bone. Curr. Osteoporos. Rep. 17, 560–569 (2019).
Piemontese, M. et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight 2, e93771 (2017).
Pignolo, R. J., Samsonraj, R. M., Law, S. F., Wang, H. & Chandra, A. Targeting cell senescence for the treatment of age-related bone loss. Curr. Osteoporos. Rep. 17, 70–85 (2019).
Farr, J. N. & Khosla, S. Cellular senescence in bone. Bone 121, 121–133 (2019).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Manolagas, S. C. The quest for osteoporosis mechanisms and rational therapies: how far we’ve come, how much further we need to go. J. Bone Miner. Res. 33, 371–385 (2018).
Kim, H. N. et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 16, 693–703 (2017).
Chen, Q. et al. DNA damage drives accelerated bone aging via an NF-κB-dependent mechanism. J. Bone Miner. Res. 28, 1214–1228 (2013).
Kim, H. N. et al. Osteocyte RANKL is required for cortical bone loss with age and is induced by senescence. JCI Insight 5, e138815 (2020).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Kim, H. N. et al. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 18, e12923 (2019).
Tyagi, A. M. et al. Premature T cell senescence in Ovx mice is inhibited by repletion of estrogen and medicarpin: a possible mechanism for alleviating bone loss. Osteoporos. Int. 23, 1151–1161 (2012).
Farr, J. N. et al. Independent roles of estrogen deficiency and cellular senescence in the pathogenesis of osteoporosis: evidence in young adult mice and older humans. J. Bone Miner. Res. 34, 1407–1418 (2019).
Costantini, S. & Conte, C. Bone health in diabetes and prediabetes. World J. Diabetes 10, 421–445 (2019).
Valderrábano, R. J. & Linares, M. I. Diabetes mellitus and bone health: epidemiology, etiology and implications for fracture risk stratification. Clin. Diabetes Endocrinol. 4, 9 (2018).
Eckhardt, B. A. et al. Accelerated osteocyte senescence and skeletal fragility in mice with type 2 diabetes. JCI Insight 5, e135236 (2020).
Khosla, S., Farr, J. N., Tchkonia, T. & Kirkland, J. L. The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol. 16, 263–275 (2020).
Almeida, M. et al. Increased marrow adipogenesis does not contribute to age-dependent appendicular bone loss in female mice. Aging Cell 19, e13247 (2020).
Cleveland, R. J. & Callahan, L. F. Can osteoarthritis predict mortality? North Carol. Med. J. 78, 322–325 (2017).
Misra, D. et al. Knee osteoarthritis and frailty: findings from the Multicenter Osteoarthritis Study and Osteoarthritis Initiative. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 70, 339–344 (2015).
Jeon, O. H. et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 23, 775–781 (2017).
Jeon, O. H., David, N., Campisi, J. & Elisseeff, J. H. Senescent cells and osteoarthritis: a painful connection. J. Clin. Investig. 128, 1229–1237 (2018).
Faust, H. J. et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J. Clin. Investig. 130, 5493–5507 (2020).
Jeon, O. H. et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 4, e125019 (2019).
van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
Martin, J. A., Brown, T. D., Heiner, A. D. & Buckwalter, J. A. Chondrocyte senescence, joint loading and osteoarthritis. Clin. Orthop. Rel. Res. S96–S103 (2004).
Goldring, M. B. & Marcu, K. B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 11, 224 (2009).
Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J. P. & Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42 (2011).
Yu, S. M. & Kim, S. J. Thymoquinone-induced reactive oxygen species causes apoptosis of chondrocytes via PI3K/Akt and p38kinase pathway. Exp. Biol. Med. 238, 811–820 (2013).
Benito, M. J., Veale, D. J., FitzGerald, O., van den Berg, W. B. & Bresnihan, B. Synovial tissue inflammation in early and late osteoarthritis. Ann. Rheum. Dis. 64, 1263–1267 (2005).
Blom, A. B. et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage 12, 627–635 (2004).
van Lent, P. L. et al. Crucial role of synovial lining macrophages in the promotion of transforming growth factor beta-mediated osteophyte formation. Arthritis Rheum. 50, 103–111 (2004).
Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 75, 685–705 (2013).
Coryell, P. R., Diekman, B. O. & Loeser, R. F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 17, 47–57 (2021).
Heijink, A. et al. Biomechanical considerations in the pathogenesis of osteoarthritis of the knee. Knee Surg. Sports Traumatol. 20, 423–435 (2012).
Zhang, Y. & Jordan, J. M. Epidemiology of osteoarthritis. Clin. Geriatr. Med. 26, 355–369 (2010).
Brown, T. D., Johnston, R. C., Saltzman, C. L., Marsh, J. L. & Buckwalter, J. A. Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J. Orthop. Trauma 20, 739–744 (2006).
Zhuo, Q., Yang, W., Chen, J. & Wang, Y. Metabolic syndrome meets osteoarthritis. Nat. Rev. Rheumatol. 8, 729–737 (2012).
Philbin, E. F. et al. Osteoarthritis as a determinant of an adverse coronary heart disease risk profile. J. Cardiovascular Risk 3, 529–533 (1996).
Cerhan, J. R., Wallace, R. B., el-Khoury, G. Y., Moore, T. E. & Long, C. R. Decreased survival with increasing prevalence of full-body, radiographically defined osteoarthritis in women. Am. J. Epidemiol. 141, 225–234 (1995).
Haara, M. M. et al. Osteoarthritis of finger joints in Finns aged 30 or over: prevalence, determinants, and association with mortality. Ann. Rheum. Dis. 62, 151–158 (2003).
Saleh, A. S. et al. Arterial stiffness and hand osteoarthritis: a novel relationship? Osteoarthr. Cartil. 15, 357–361 (2007).
Francisco, V. et al. Biomechanics, obesity, and osteoarthritis. The role of adipokines: when the levee breaks. J. Orthop. Res. 36, 594–604 (2018).
Piva, S. R. et al. Links between osteoarthritis and diabetes: implications for management from a physical activity perspective. Clin. Geriatr. Med. 31, 67–87, viii (2015).
Baudart, P., Louati, K., Marcelli, C., Berenbaum, F. & Sellam, J. Association between osteoarthritis and dyslipidaemia: a systematic literature review and meta-analysis. RMD Open 3, e000442 (2017).
Vinatier, C., Dominguez, E., Guicheux, J. & Carames, B. Role of the inflammation-autophagy-senescence integrative network in osteoarthritis. Front. Physiol. 9, 706 (2018).
Puenpatom, R. A. & Victor, T. W. Increased prevalence of metabolic syndrome in individuals with osteoarthritis: an analysis of NHANES III data. Postgrad. Med. 121, 9–20 (2009).
Chadha, R. Revealed aspect of metabolic osteoarthritis. J. Orthop. 13, 347–351 (2016).
Courties, A., Sellam, J. & Berenbaum, F. Metabolic syndrome-associated osteoarthritis. Curr. Opin. Rheumatol. 29, 214–222 (2017).
Farnaghi, S., Crawford, R., Xiao, Y. & Prasadam, I. Cholesterol metabolism in pathogenesis of osteoarthritis disease. Int. J. Rheum. Dis. 20, 131–140 (2017).
Kang, C. Senolytics and senostatics: a two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol. Cells 42, 821–827 (2019).
Baker, D. J. et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Chung, L. et al. Interleukin 17 and senescent cells regulate the foreign body response to synthetic material implants in mice and humans. Sci. Transl. Med. 12, eaax3799 (2020).
Sessions, G. A. et al. Controlled induction and targeted elimination of p16(INK4a)-expressing chondrocytes in cartilage explant culture. FASEB J. 33, 12364–12373 (2019).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Hickson, L. J. et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456 (2019).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Matsuzaki, T. et al. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 73, 1397–1404 (2014).
Batshon, G. et al. Serum NT/CT SIRT1 ratio reflects early osteoarthritis and chondrosenescence. Ann. Rheum. Dis. 79, 1370–1380 (2020).
Yousefzadeh, M. J. et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018).
Zheng, W. et al. Fisetin inhibits IL-1β-induced inflammatory response in human osteoarthritis chondrocytes through activating SIRT1 and attenuates the progression of osteoarthritis in mice. Int. Immunopharmacol. 45, 135–147 (2017).
Shiomi, T., Lemaître, V., D’Armiento, J. & Okada, Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol. Int. 60, 477–496 (2010).
Roach, H. I. et al. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 52, 3110–3124 (2005).
Neuhold, L. A. et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 107, 35–44 (2001).
Wang, M. et al. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 15, R5 (2013).
Fleischmann, R. M. et al. A phase II trial of lutikizumab, an anti-interleukin-1α/β dual variable domain immunoglobulin, in knee osteoarthritis patients with synovitis. Arthritis Rheumatol. 71, 1056–1069 (2019).
Kloppenburg, M. et al. Etanercept in patients with inflammatory hand osteoarthritis (EHOA): a multicentre, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 77, 1757–1764 (2018).
Schieker, M. et al. Effects of interleukin-1β inhibition on incident hip and knee replacement: exploratory analyses from a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 173, 509–515 (2020).
de Hooge, A. S. et al. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthr. Cartil. 13, 66–73 (2005).
Baskin, K. K., Winders, B. R. & Olson, E. N. Muscle as a “mediator” of systemic metabolism. Cell Metab. 21, 237–248 (2015).
Manickam, R., Duszka, K. & Wahli, W. PPARs and microbiota in skeletal muscle health and wasting. Int. J. Mol. Sci. 21, 8056 (2020).
Periasamy, M., Herrera, J. L. & Reis, F. C. G. Skeletal muscle thermogenesis and its role in whole body energy metabolism. Diabetes Metab. J. 41, 327–336 (2017).
Sartori, R., Romanello, V. & Sandri, M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat. Commun. 12, 330 (2021).
Yamakawa, H., Kusumoto, D., Hashimoto, H. & Yuasa, S. Stem cell aging in skeletal muscle regeneration and disease. Int. J. Mol. Sci. 21, 1830 (2020).
Oikawa, S. Y., Holloway, T. M. & Phillips, S. M. The impact of step reduction on muscle health in aging: protein and exercise as countermeasures. Front. Nutr. 6, 75 (2019).
Romanello, V. The interplay between mitochondrial morphology and myomitokines in aging sarcopenia. Int. J. Mol. Sci. 22, 91 (2020).
Morley, J. E., Anker, S. D. & von Haehling, S. Prevalence, incidence, and clinical impact of sarcopenia: facts, numbers, and epidemiology-update 2014. J. Cachexia Sarcopenia Muscle 5, 253–259 (2014).
da Silva, P. F. L. et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 18, e12848 (2019).
Chen, Q. N. et al. Effect of sarcolipin-mediated cell transdifferentiation in sarcopenia-associated skeletal muscle fibrosis. Exp. Cell Res. 389, 111890 (2020).
Alcalde-Estévez, E. et al. Endothelin-1 induces cellular senescence and fibrosis in cultured myoblasts. A potential mechanism of aging-related sarcopenia. Aging 12, 11200–11223 (2020).
Kudryashova, E., Kramerova, I. & Spencer, M. J. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J. Clin. Investig. 122, 1764–1776 (2012).
Zhou, J. et al. GSK-3α is a central regulator of age-related pathologies in mice. J. Clin. Investig. 123, 1821–1832 (2013).
Cosgrove, B. D. et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 20, 255–264 (2014).
Zhu, P. et al. The transcription factor Slug represses p16(Ink4a) and regulates murine muscle stem cell aging. Nat. Commun. 10, 2568 (2019).
Du, J. et al. Aging increases CCN1 expression leading to muscle senescence. Am. J. Physiol. Cell Physiol. 306, C28–C36 (2014).
Sousa-Victor, P. et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321 (2014).
García-Prat, L. et al. Autophagy maintains stemness by preventing senescence. Nature 529, 37–42 (2016).
Sosa, P. et al. Hyperphosphatemia promotes senescence of myoblasts by impairing autophagy through Ilk overexpression, a possible mechanism involved in sarcopenia. Aging Dis. 9, 769–784 (2018).
Wang, Y. et al. Aging of the immune system causes reductions in muscle stem cell populations, promotes their shift to a fibrogenic phenotype, and modulates sarcopenia. FASEB J. 33, 1415–1427 (2019).
Wang, Y., Wehling-Henricks, M., Samengo, G. & Tidball, J. G. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 14, 678–688 (2015).
Ratnayake, D. et al. Macrophages provide a transient muscle stem cell niche via NAMPT secretion. Nature 591, 281–287 (2021).
Ming, G. F., Wu, K., Hu, K., Chen, Y. & Xiao, J. NAMPT regulates senescence, proliferation, and migration of endothelial progenitor cells through the SIRT1 AS lncRNA/miR-22/SIRT1 pathway. Biochem. Biophys. Res. Commun. 478, 1382–1388 (2016).
Baraibar, M. et al. Impaired metabolism of senescent muscle satellite cells is associated with oxidative modifications of glycolytic enzymes. Free Radic. Biol. Med. 75 (Suppl 1), S23 (2014).
Baraibar, M. A. et al. Impaired energy metabolism of senescent muscle satellite cells is associated with oxidative modifications of glycolytic enzymes. Aging 8, 3375–3389 (2016).
Dennison, E. M., Sayer, A. A. & Cooper, C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat. Rev. Rheumatol. 13, 340–347 (2017).
Alsharidah, M. et al. Primary human muscle precursor cells obtained from young and old donors produce similar proliferative, differentiation and senescent profiles in culture. Aging cell 12, 333–344 (2013).
Sugihara, H., Teramoto, N., Yamanouchi, K., Matsuwaki, T. & Nishihara, M. Oxidative stress-mediated senescence in mesenchymal progenitor cells causes the loss of their fibro/adipogenic potential and abrogates myoblast fusion. Aging 10, 747–763 (2018).
Pacifici, F. et al. Prdx6 plays a main role in the crosstalk between aging and metabolic sarcopenia. Antioxidants (Basel) 9, 329 (2020).
Zhu, S. et al. Aging- and obesity-related peri-muscular adipose tissue accelerates muscle atrophy. PLoS One 14, e0221366 (2019).
Tezze, C. et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 25, 1374–1389.e1376 (2017).
von Kobbe, C. Targeting senescent cells: approaches, opportunities, challenges. Aging 11, 12844–12861 (2019).
Baar, M. P. et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147.e116 (2017) .
O’Sullivan Coyne, G. & Burotto, M. MABp1 for the treatment of colorectal cancer. Expert Opin. Biol. Ther. 17, 1155–1161 (2017).
Khor, S. C., Wan Ngah, W. Z., Mohd Yusof, Y. A., Abdul Karim, N. & Makpol, S. Tocotrienol-rich fraction ameliorates antioxidant defense mechanisms and improves replicative senescence-associated oxidative stress in human myoblasts. Oxid. Med. Cell. Longev. 2017, 3868305 (2017).
Lim, J. J., Ngah, W. Z., Mouly, V. & Abdul Karim, N. Reversal of myoblast aging by tocotrienol rich fraction posttreatment. Oxid. Med. Cell. Longev. 2013, 978101 (2013).
Lim, J. J., Wan Zurinah, W. N., Mouly, V. & Norwahidah, A. K. Tocotrienol-rich fraction (TRF) treatment promotes proliferation capacity of stress-induced premature senescence myoblasts and modulates the renewal of satellite cells: microarray analysis. Oxid. Med. Cell. Longev. 2019, 9141343 (2019).
Hu, Z. et al. MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging 6, 160–175 (2014).
Soriano-Arroquia, A., McCormick, R., Molloy, A. P., McArdle, A. & Goljanek-Whysall, K. Age-related changes in miR-143-3p:Igfbp5 interactions affect muscle regeneration. Aging Cell 15, 361–369 (2016).
Zheng, Y., Kong, J., Li, Q., Wang, Y. & Li, J. Role of miRNAs in skeletal muscle aging. Clin. Interv. Aging 13, 2407–2419 (2018).
Acknowledgements
For their editorial assistance, we thank Jenni Weems, MS, Kerry Kennedy, BA, and Rachel Box, MS, in the Editorial Services group of The Johns Hopkins Department of Orthopaedic Surgery. This work was supported by the National Institutes of Health grant R01 AG068226 and R01AG072090 to M. W. and P01AG066603 to X. C.
Author information
Authors and Affiliations
Contributions
M. W. and E. F. G.-G. drafted the manuscript and J. H. E. proofread the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wan, M., Gray-Gaillard, E.F. & Elisseeff, J.H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res 9, 41 (2021). https://doi.org/10.1038/s41413-021-00164-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-021-00164-y
This article is cited by
-
Age-related decline in melatonin contributes to enhanced osteoclastogenesis via disruption of redox homeostasis
Molecular Medicine (2024)
-
Age-related secretion of grancalcin by macrophages induces skeletal stem/progenitor cell senescence during fracture healing
Bone Research (2024)
-
Senescent-like macrophages mediate angiogenesis for endplate sclerosis via IL-10 secretion in male mice
Nature Communications (2024)
-
Interleukin-19 in Bone Marrow Contributes to Bone Loss Via Suppressing Osteogenic Differentiation Potential of BMSCs in Old Mice
Stem Cell Reviews and Reports (2024)
-
Fighting age-related orthopedic diseases: focusing on ferroptosis
Bone Research (2023)
