
Abstract
Bone-produced fibroblast growth factor 23 (FGF23) increases in response to inflammation and iron deficiency and contributes to cardiovascular mortality in chronic kidney disease (CKD). Neutrophil gelatinase-associated lipocalin (NGAL or lipocalin 2; LCN2 the murine homolog) is a pro-inflammatory and iron-shuttling molecule that is secreted in response to kidney injury and may promote CKD progression. We investigated bone FGF23 regulation by circulating LCN2. At 23 weeks, Col4a3KO mice showed impaired kidney function, increased levels of kidney and serum LCN2, increased bone and serum FGF23, anemia, and left ventricular hypertrophy (LVH). Deletion of Lcn2 in CKD mice did not improve kidney function or anemia but prevented the development of LVH and improved survival in association with marked reductions in serum FGF23. Lcn2 deletion specifically prevented FGF23 elevations in response to inflammation, but not iron deficiency or phosphate, and administration of LCN2 increased serum FGF23 in healthy and CKD mice by stimulating Fgf23 transcription via activation of cAMP-mediated signaling in bone cells. These results show that kidney-produced LCN2 is an important mediator of increased FGF23 production by bone in response to inflammation and in CKD. LCN2 inhibition might represent a potential therapeutic approach to lower FGF23 and improve outcomes in CKD.
Similar content being viewed by others

Introduction
Bone production of fibroblast growth factor 23 (FGF23) is increased in patients and animals with chronic kidney disease (CKD)1,2,3 and is associated with the development of left ventricular hypertrophy (LVH), heart failure, and mortality.1,2,4,5,6,7 Excess circulating FGF23 is the first major perturbation of mineral metabolism that occurs in CKD, however, the complex mechanisms that trigger elevations of FGF23 in CKD remain incompletely understood. Among these, multiple studies showed contributions of inflammation,8 iron deficiency,9 anemia,2 and local osteocyte defects.1 Notably, circulating FGF23 levels increase as kidney disease progresses, suggesting that kidney-bone crosstalk may contribute to excessive production of FGF23 by bone in response to kidney injury.10,11
Lipocalin 2, (LCN2) also known as neutrophil gelatinase-associated lipocalin in humans (NGAL) is a 25 kD lipophilic glycoprotein member of the lipocalin superfamily12 involved in innate immunity. The established role of LCN2 is to limit bacterial growth by binding to bacterial siderophores, which are low molecular weight chelators of ferric iron that are produced by bacteria to scavenge iron from their surrounding environment. In addition, LCN2 functions as an iron transporter by binding mammalian siderophores,13,14 and stabilizes labile iron/siderophore complexes.15,16 LCN2 allows cells to tolerate supra-physiological iron concentrations by scavenging free iron17,18,19 and protects against labile iron-mediated cytotoxicity. LCN2 is secreted by various cell types and tissues, including but not limited to immune cells,20 bone,21 liver,22 intestines,23 heart24 and kidney,25 and its expression is regulated mainly by infection and inflammatory status.
In patients with acute and CKD, kidney production of NGAL/LCN2 increases and can be detected in the urine and plasma; elevated urinary NGAL/LCN2 is a biomarker of acute kidney injury (AKI).25 Increased kidney LCN2 expression in AKI is thought to be a component of the systemic inflammatory response to AKI that helps redirect iron to support repair of renal tubular cells.26 In CKD, kidney expression and urine and serum LCN2 levels are also elevated, presumably in response to chronic kidney injury, inflammation, and infiltrating cells.27,28,29,30 Studies in which Lcn2 genetic deletion delayed CKD progression in mice demonstrate that LCN2 is not only a biomarker but could be a potential driver of CKD progression.31 Despite the links between LCN2 regulation and iron homeostasis, inflammation, and kidney disease, each of which is also involved in FGF23 regulation, potential direct relationships between LCN2, FGF23 regulation, and FGF23-associated outcomes have not been studied.
In the present study, we propose a novel mechanism to explain coincident increases in LCN2 and FGF23 soon after kidney injury,32 and the strong independent association between elevated levels of FGF23 and inflammatory markers.33 We hypothesized that bone is a target of kidney-secreted LCN2 and that increased LCN2 stimulates bone production of FGF23 in CKD. To test our hypothesis, we investigated the role of LCN2 in FGF23 regulation in health and in CKD. We show that circulating levels of LCN2 increased and paralleled CKD progression in the Col4a3KO mouse model of CKD, and that kidney was the organ with the highest expression of Lcn2 in CKD. We further show that genetic deletion of LCN2 in mice that develop CKD prevented increases in bone and circulating levels of FGF23 and development of LVH, and improved lifespan, despite CKD and anemia of unchanged severity. Finally, we show that increased circulating LCN2 stimulates Fgf23 transcription through stimulation of cyclic AMP-mediated signaling in bone cells.
Results
Increased serum NGAL is associated with excess FGF23 in patients with CKD
NGAL/LCN2 is a secreted pro-inflammatory and iron shuttling glycoprotein which might contribute to the progression of CKD.31 Given that FGF23 production is increased in response to inflammation and iron deficiency, we investigated whether FGF23 levels correlate with circulating NGAL. In serum collected from healthy volunteers and patients with CKD, we found that NGAL, cFGF23, and iFGF23 levels increased as kidney function declined (Fig. 1a–c). We further found that log(cFGF23) and log(iFGF23) strongly correlated with NGAL levels (log cFGF23 R2 = 0.79; log iFGF23 R2 = 0.73) and significantly associated with ascending NGAL levels (Fig. 1d, e). These associations remained significant after adjusting for kidney function [partial correlation log cFGF23 R2 = 0.57; partial correlation log iFGF23 R2 = 0.54] (Fig. 1f, g). Linear regression models showed that NGAL, independently of eGFR, was strongly associated with excess total and intact FGF23 (Supplementary Tables 1 and 2). Taken together, the strong eGFR-independent associations of NGAL/LCN2 with FGF23 levels suggest that NGAL/LCN2 might regulate FGF23 production in CKD.

FGF23 and NGAL levels increase in patients with CKD. Levels of serum (a) NGAL, (b) total FGF23 (cFGF23), and (c) intact FGF23 (iFGF23) increase with the progression of kidney disease. NGAL correlates with both (d) and (f) cFGF23 (R2 = 0.79, partial correlation R2 = 0.57) and (e) and (g) iFGF23 (R2 = 0.73, partial correlation R2 = 0.54) levels, (d) and (e) unadjusted variables or (f) and (g) adjusted by eGFR. P values were determined by a two-sided, paired t-test. Values are mean ± SE, n ≥ 12/group, P < 0.05 vs. *Healthy, $Stage 2–4
Col4a3KO mouse model of CKD with increased Lipocalin 2
We reported that C57Bl6–Col4a3KO mice experience progressive declines in renal function and develop LVH.1,34 Here, we report that circulating levels of LCN2 increased in an age-dependent manner in WT and Col4a3KO mice, and were higher in Col4a3KO mice with CKD than in WT mice (Fig. 2a). Lcn2 is one of the leading upregulated genes in kidneys from Col4a3KO mice.30 In our current study, expression of Lcn2 was highly elevated in the kidney, and to a lower extent in the hearts, but not in bone or bone marrow of 23-week-old Col4a3KO mice with advanced CKD compared to WT mice (Fig. 2b). This suggests that LCN2 is secreted into the circulation mainly by the injured kidneys.

Increased production of lipocalin 2 does not contribute to impaired kidney function in the Col4a3KO mouse model of chronic kidney disease. (a) Levels of serum lipocalin 2 (LCN2) measured in 4–23-week-old wild-type (WT) and Col4a3KO mice. (b) Levels of Lcn2 mRNA expression (reported to Rpl19 expression, and set at 1 in WT kidneys) in kidney, heart, bone, and bone marrow from 23-week-old WT and Col4a3KO mice. (c) Body weight, (d) blood urea nitrogen (BUN) levels, (e) 24 h urine albumin levels and (f) bright-field microscopy of hematoxylin & eosin (H&E) and picrosirius red (PSR, scale bar = 75 µm) stainings of kidneys from 23-week-old WT, Lcn2KO, Col4a3KO, and Col4a3KO/Lcn2KO (CPD) mice. P values were determined by 2-sided, paired t-test. Data are presented as mean ± SE, n ≥ 5 per group, P < 0.05 vs.*WT, $Lcn2KO
Lipocalin 2 deletion does not improve renal function in Col4a3KO mice
To assess the contribution of LCN2 to CKD progression and CKD-associated outcomes, we deleted Lcn2 from WT and Col4a3KO mice and studied the phenotype of WT, Lcn2KO, Col4a3KO, and mice with compound deletion of Lcn2 and Col4a3 (CPD: Lcn2KO/Col4a3KO) littermates. Col4a3KO and CPD mice showed reduced body weight compared to WT and Lcn2KO mice (Fig. 2c) but BUN and albuminuria were similar in Col4a3KO and CPD mice (Fig. 2d, e). Similar degrees of glomerulosclerosis, tubular atrophy, and interstitial fibrosis were also recorded in Col4a3KO and CPD mice (Fig. 2f). These results suggest that Lcn2 deletion in CKD does not protect against loss of kidney function in the Col4a3KO model.
Lipocalin 2 deficiency does not prevent anemia of CKD
We next tested the hypothesis that LCN2 contributes to disturbed iron metabolism and erythropoiesis in CKD,35,36 given that one of the major functions of LCN2 is to transport iron. At 23 weeks, Lcn2KO mice did not display changes in circulating iron or hematological parameters (Fig. 3a–h). Col4a3KO animals showed decreased circulating iron, ferritin (as a measure of iron stores), hemoglobin, red blood cell number, hematocrit, and mean corpuscular volume consistent with the development of microcytic anemia (Fig. 3a–h).2 Deletion of Lcn2 in Col4a3KO mice partially corrected circulating iron, transferrin saturation, and ferritin levels, suggesting that increased levels of LCN2 contribute to disordered iron metabolism in CKD (Fig. 3a–c). Serum EPO levels were inappropriately low in both Col4a3KO and CPD mice (Fig. 3d), and despite increases in circulating iron and iron stores, CPD mice showed a similar degree of microcytic anemia as Col4a3KO, assessed by reduced hemoglobin, red blood cell number, hematocrit and mean corpuscular volume (Fig. 3f–h).

Increased lipocalin 2 contributes to the elevation of FGF23 in Col4a3KO mice with advanced chronic kidney disease. Levels of (a) serum iron, (b) serum transferrin saturation, (c) serum ferritin, (d) serum erythropoietin, (e) hemoglobin (HB), (f) red blood cells (RBC), (g) hematocrit (HCT) and (h) mean corpuscular volume (MCV) measured in 23-week-old WT, Lcn2KO, Col4a3KO, and Col4a3KO/Lcn2KO (CPD) mice. Levels of (i), (n) serum total FGF23 (cFGF23), (j), (o) serum intact FGF23 (iFGF23), (k) urine phosphate (Pi), (l) serum Pi, and (m) bone Fgf23 mRNA expression, measured in baseline (a)–(l) and control-treated (m)–(o) 23-week-old WT, Lcn2KO, Col4a3KO, and Col4a3KO/Lcn2KO (CPD) mice, and in mouse recombinant lipocalin 2 (mrLCN2, 50 ng·g−1 per day)–treated Col4a3KO and CPD mice (m)–(o). Data are presented as mean ± SE, n ≥ 3 per group, P < 0.05 vs.*WT, $Lcn2KO, &Col4a3KO, #CPD, @Col4a3KO + mrLCN2
Deletion of Lipocalin 2 reduces FGF23 production in CKD
Total cFGF23 and iFGF23 levels were similar in Lcn2KO and WT mice (Fig. 3i, j) at 23 weeks of age. This was accompanied by normal urine Pi excretion and serum phosphate (Fig. 3k, l). As previously reported, serum cFGF23 and iFGF23 levels were highly increased in Col4a3KO mice with advanced CKD (Fig. 3i, j), which also showed hyperphosphatemia with increased urine Pi excretion (Fig. 3k, l). These changes were markedly attenuated by Lcn2 deletion in Col4a3KO mice, which demonstrated 60% reductions in serum cFGF23 and iFGF23, 80% reduction in bone Fgf23 mRNA expression (Fig. 3i, j, m), and significantly reduced urine Pi excretion in CPD mice (Fig. 3k) and serum phosphate levels also decreased in CPD mice (Fig. 3l).
To further confirm that LCN2 directly regulates FGF23 in CKD, we administered murine recombinant LCN2 to Col4a3KO and CPD mice for 8 weeks, from 16 to 23 weeks, using osmotic minipumps. Administration of LCN2 to Col4a3KO mice did not further increase bone Fgf23 mRNA or serum cFGF23 levels (Fig. 3m, n), but resulted in higher levels of serum iFGF23 (Fig. 3o). More importantly, LCN2 administration to CPD mice increased bone Fgf23 mRNA and both serum cFGF23 and iFGF23 levels to similar levels observed in control Col4a3KO mice (Fig. 3m–o), showing that increased LCN2 levels contribute to increased FGF23 production in CKD.
Deletion of Lipocalin 2 prevents LVH and improves survival in CKD
In CKD, elevated FGF23 is associated with the development of LVH, heart failure, and death, and administration of FGF23 to WT mice induces LVH.4 As previously reported,1,34 C57Bl6-Col4a3KO mice show a shortened lifespan and die at an average of 23 weeks of age (Fig. 4a). Development of LVH in C57Bl6-Col4a3KO mice with advanced CKD may contribute to a shortened lifespan, as an increase in whole heart and left ventricular mass in Col4a3KO mice eventually leads to impaired cardiac function (Fig. 4b–f), and rescue of LVH is associated with extended lifespan in this model.1 Despite a similar degree of CKD and anemia, Lcn2 deletion improved survival, as CPD mice lived on average 3 weeks longer than Col4a3KO mice (Fig. 4a). Indeed, histology and echocardiography analyses showed a lower heart weight to tibia length ratio, reduced LV mass, and posterior wall thickness in CPD vs. Col4a3KO mice with advanced CKD, demonstrating that Lcn2 deletion prevents the development of LVH in CKD (Fig. 4b–e). In addition, CPD mice showed higher ejection fraction (EF) than Col4a3KO mice with CKD, suggesting that Lcn2 deletion also preserves cardiac function (Fig. 4f), perhaps by limiting FGF23 production.

Lipocalin 2 deletion in Col4a3KO mice improves survival and prevents the development of left ventricular hypertrophy. (a) Kaplan–Meier cumulative proportion of mice surviving. Both Col4a3KO (red) and Col4a3KO/Lcn2KO (coral) mice show reduced lifespan (P < 0.05 vs. WT), but Col4a3KO/Lcn2KO survive longer than Col4a3KO mice (P < 0.05 vs. Col4a3KO). (b) Heart weight to tibia length (HW/TL) ratio, and (c) left ventricular (LV) mass, (d) LV posterior wall (LVPW) thickness, (e) bright-field microscopy of hematoxylin & eosin staining (H&E) of heart cross-sections (scale bar = 1 mm), and M-mode echocardiography, and (f) ejection fraction (EF) measured in 23-week-old WT, Lcn2KO, Col4a3KO, and Col4a3KO/Lcn2KO (CPD) mice. c, d, f are calculated from echocardiography analysis. Data are presented as mean ± SE, n ≥ 10 per group, P < 0.05 vs.*WT, $Lcn2KO
Lipocalin 2 regulates bone FGF23 production in response to inflammation, but not to phosphate or iron deficiency
Hyperphosphatemia, iron deficiency/anemia, and inflammation are potent stimuli of bone FGF23 production during CKD progression.2,8 We tested whether Lcn2 deletion would alter FGF23 regulation by phosphate, iron, or inflammatory stimuli induced by dietary Pi loading, iron restriction, and acute IL1β injections, respectively. As expected, WT animals fed a high phosphate diet displayed higher levels of both serum cFGF23 and iFGF23 levels, and Lcn2KO mice showed similar elevations of cFGF23 and iFGF23 on the high phosphate diet, suggesting that phosphate regulation of FGF23 is independent of LCN2 (Fig. 5a, b). As previously shown,8 WT mice fed a low iron diet also showed increased cFGF23 and mildly elevated iFGF23 levels. Despite the role of LCN2 as an iron transporter, Lcn2KO mice showed similar increases in both serum cFGF23 and iFGF23 levels to WT mice in response to a low iron diet (Fig. 5c, d), suggesting that LCN2 regulation of FGF23 is iron-independent. Finally, consistent with previous reports,8 acute inflammation induced by administration of IL-1β to WT mice dramatically increased cFGF23 levels and, to a much lower extent, iFGF23 levels. However, Lcn2KO mice showed a blunted response to IL-1β, as cFGF23 and iFGF23 elevations were reduced by ~50% in IL-1β-treated Lcn2KO mice (Fig. 5e, f). This demonstrates that LCN2 partially mediates FGF23 production during inflammation (Fig. 5g).

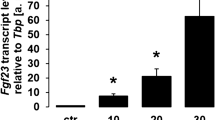
Lipocalin 2 mediates inflammation-induced FGF23 elevation. Levels of serum total FGF23 (cFGF23) and serum intact FGF23 (iFGF23) inWT and Lcn2KO mice fed (a), (b) a 0.8% (Ctr) or a 2% high phosphate (HP) diet from 6 to 12 weeks, (c), (d) a 48 ppm (Ctr) or a 3 ppm iron deficient (ID) diet from 3 to 6 weeks, and (e), (f) 6 h post-injection of a single dose of saline (Ctr) or interleukin 1β (IL-1β, 250 ng·g−1) in 6-week old mice. (g) Serum levels of LCN2 in WT mice submitted to the same challenges. Data are presented as mean ± SE, n ≥ 5 per group, P < 0.05 vs.*Ctr-WT, $Ctr-Lcn2KO, &IL-1β-WT. Levels of (h), (k) bone Fgf23 mRNA, (i), (l) serum total FGF23 (cFGF23) and (j), (m) serum intact FGF23 (iFGF23) in WT mice treated continuously for 4 weeks with 0 (0.9%NaCl), 5 and 50 ng·g−1 of mouse recombinant LCN2 (mrLCN2) (h)–(j) or injected daily for 4 d with 0 (0.9%NaCl) and 50 ng·g−1 of mrLCN2 (k)–(m). Data are presented as mean ± SE, n ≥ 5 per group, P < 0.05 vs. *non treated, &5 ng·g−1 mrLCN2. Fgf23 mRNA levels (n) in primary osteoblast and (o) in MC3T3-E1 osteoblasts cultures treated with 0–50 osteoblasts cultures treated with 0–50 ng·mL–1 of mrLCN2. (p) Fgf23 promoter activity in Fgf23 promoter-reporter MC3T3-E1 osteoblast cultures treated with 0–50 ng·mL−1 of mrLCN2. Data are presented as mean ± SE, n ≥ 3 per group, P < 0.05 vs. *non treated, &5 ng·mL−1, #25 ng·mL−1 mrLCN2
Lipocalin 2 directly regulates bone FGF23 production
To further understand whether LCN2 only potentiates the effects of inflammatory stimuli on FGF23 production in CKD or inflammation or whether LCN2 directly stimulates FGF23 production, we administered LCN2 to 6-week old WT mice either for 4 weeks using osmotic minipumps or during 4 days of repeated LCN2 injections. Continuous administration of LCN2 at low (5 ng·g–1 per day) or high doses (50 ng·g–1 per day), dose-dependently increased bone Fgf23 mRNA expression, serum cFGF23 levels, and led to a mild increase in iFGF23 levels (Fig. 5h–j). Similarly, short-term intermittent administration of 50 ng/g/day LCN2 to C57Bl6 mice resulted in increased osseous Fgf23 mRNA, higher circulating cFGF23 levels, and a mild increase in iFGF23 levels (Fig. 5k–m). In addition, 24 h of LCN2 treatment increased Fgf23 transcription in cultured bone marrow stromal cells (BMSC) or MC3T3-E1 cell lines (Fig. 5n–o), and dose-dependently increased Fgf23 promoter activity in MC3T3-E1 Fgf23-promoter-reporter cells (Fig. 5p). These combined results demonstrate that LCN2 increases FGF23 production in bone by stimulating Fgf23 expression in osteoblasts and osteocytes.
Lipocalin 2 stimulates bone FGF23 production through a cAMP-dependent mechanism
To understand the primary mechanisms leading to increased bone production of FGF23 in response to elevated LCN2, we performed RNA sequencing of cortical bone isolated from 12-week old WT and Lcn2KO mice. We found that 614 genes were differentially expressed in Lcn2KO compared to WT mice (cutoffs: P < 0.05, absolute fold change (FC) of 2). Of these, 369 genes were downregulated and 245 genes were upregulated. The top 50 most upregulated and downregulated genes are shown in Table 1. Of note, we found a net (−2-fold) albeit non-significant reduction in Fgf23 (P = 0.12). Using ingenuity pathway analysis (IPA, QIAgen), we identified the canonical signaling pathways predicted to be significantly changed in the cortical bone of Lcn2KO mice. Among these, the top five pathways predicted to be downregulated or upregulated, cAMP-mediated signaling was predicted to be the most inhibited pathway (Fig. 6a), based on the z-score activation of multiple genes. A total of ten genes in this pathway were dysregulated in Lcn2KO mice (P < 0.05, FC2), and selective enrichment of this pathway (P < 0.1, FC2) identified five additional genes of cAMP signaling in the entire dataset (Fig. 6b). To test whether LCN2 regulates FGF23 production through activation of cAMP-mediated signaling, we first verified that forskolin (FSK), a known inducer of cAMP and cAMP-sensitive pathways, stimulated Fgf23 promoter activity in MC3T3-E1 Fgf23-promoter-reporter cells (Fig. 6c). Then, we compared the effects of FSK and LCN2 treatment on cAMP activation and found that both FSK and LCN2 treatment increased intracellular cAMP levels (Fig. 6d) and phosphorylation of the cAMP response element-binding protein (CREB) in MC3T3-E1 osteoblast-like cells 6 h post-stimulation (Fig. 6e, f). Co-treatment of LCN2 stimulated cells with KT5720, an inhibitor of cAMP activation blocked the effects of LCN2 on CREB phosphorylation, suggesting that cAMP mediates the effects of LCN2. Finally, both FSK and LCN2 increased Fgf23 mRNA (Fig. 6g) and Fgf23 promoter activity (Fig. 6h) at 6 h, and their effects were partially blocked by co-treatment with the KT5720. These data demonstrate that stimulation of FGF23 production by LCN2 in osteoblasts is mediated at least in part by cAMP signaling and that LCN2 and FGF23 are part of a vicious cycle in CKD (Fig. 6i).

cAMP signaling mediates lipocalin 2-induced bone stimulation of FGF23 production in osteoblasts. RNA sequencing of WT and Lcn2KO cortical bone identified 614 differentially regulated genes in Lcn2KO compared to WT (cutoffs: P < 0.05 and absolute fold change of 2). (a) Top 5 canonical pathways identified by ingenuity pathway analysis from the 614 genes dataset. (b) Normalized expression of dataset genes involved in the cAMP pathway with a twofold cutoff and P < 0.05 with the exception of *which indicates P < 0.1. (c) Fgf23 promoter activity in Control (Ctr) or Forskolin (FSK) -treated Fgf23 promoter-reporter MC3T3-E1 osteoblast cultures. (d) cAMP levels in Ctr, FSK, and LCN2 -treated MC3T3-E1 osteoblast cultures. (e) Representative micrograph and quantification (f) of western blotting detection of phosphorylated CREB (p-CREB) and total CREB, normalized to β-actin, in protein extracts from MC3T3-E1 osteoblasts treated with Ctr, FSK, and LCN2, and co-treated with LCN2 and cAMP inhibitor KT5720. Effects of Ctr, FSK, and LCN2 -treatment and KT5720 co-treatment on Fgf23 mRNA in MC3T3 osteoblasts (g) and promoter activity (h) in Fgf23 promoter-reporter MC3T3-E1 osteoblast cultures. Data are presented as mean ± SE, n ≥ 3 per group, P < 0.05 vs.* 6 h-Ctr, α 24 h-Ctr, and 6 h-FSK, £ 6 h-FSK + KT5720, β 6 h-LCN2. (i) Progressive alterations in kidney morphology and function induce inflammation-dependent lipocalin secretion leading to increased circulating LCN2. In bone, LCN2 increases FGF23 production through a cAMP/PKA/CREB-dependent mechanism, which contributes to excess FGF23 in CKD. Elevated FGF23 exerts pro-inflammatory effects, aggravating the inflammatory status in CKD. Excess FGF23 also targets the heart and contributes to the development of cardiac disease and mortality
Discussion
Excess FGF23 is associated with adverse outcomes in patients with CKD.6,37,38 There is a tight correlation between progression of CKD and FGF23, as FGF23 increases as kidney function declines, suggesting that molecules secreted by the kidney might regulate FGF23 production in CKD. Here, we identified LCN2, as a kidney messenger that targets the bone cells to increase FGF23 production. We show that excess LCN2 in CKD contributes to excess circulating FGF23, development of cardiac disease, and premature death, and that genetic ablation of Lcn2 in CKD partially reduces Fgf23 bone mRNA expression, circulating FGF23 levels and results in marked improvement in cardiac function and lifespan. We also found that LCN2 mediates the expression of Fgf23 in response to inflammation and that LCN2 directly stimulates FGF23 in bone cells via activation of cAMP-mediated signaling.
LCN2 is primarily a bacteriostatic agent,39 that binds to hydrophobic ligands such as iron siderophores produced by bacteria.40,41 LCN2 excess is observed in multiple aseptic pathologies of inflammatory nature,42,43 and as such, LCN2 has become increasingly relevant in recent years as a potential clinical biomarker in inflammatory diseases.44,45,46 Most importantly, blood and urinary levels of LCN2 have been extensively studied as potential biomarkers for an early diagnosis of AKI and for monitoring of CKD severity. Transcriptome and proteomic studies identified LCN2 to be one of the most upregulated genes and one of the most highly induced proteins in the kidney in animal models of AKI47,48 and CKD.30 Beyond its status as a biomarker and early predictor of AKI, data also suggested that LCN2 could serve as a biomarker in CKD.27 In a previous study, LCN2 was shown to lead to progressive renal failure31 and Lcn2 genetic deletion protected against CKD progression in 3/4 nephrectomized mice or jck mice with polycystic kidney disease. In contrast, we report that the severity of kidney disease was similar in the Col4a3KO mouse model of progressive CKD, with and without global deletion of Lcn2. It is possible that Lcn2 deletion might delay the onset of CKD, but at the advanced stage of CKD in our model, differences in kidney function and morphology following Lcn2 deletion would be barely perceptible. In line with our results, Lcn2 deletion in the severe acute tubular necrosis model did not rescue kidney morphology and function.49
Deletion of LCN2 leads to increased circulating iron in Col4a3KO mice. Prior studies showed that LCN2 was a key factor in the regulation of erythrocyte growth due to its ability to inhibit the maturation of bone marrow erythroid precursors50 or inhibition of erythropoiesis through induction of apoptosis.50,51 However, we found that the role of LCN2 in anemia of CKD was limited. Indeed, hemoglobin and hematocrit were similar in Col4a3KO and in CPD mice, which is consistent with studies showing no significant correlations between LCN2 and the levels of hemoglobin, hematocrit, and erythrocytes in patients on hemodialysis.52
Iron deficiency, hyperphosphatemia, and inflammation contribute to FGF23 production during CKD.1,8,9 Whether the effects of Lcn2 deletion on FGF23 production in CKD are partially mediated by the increase in circulating iron or decrease in circulating phosphate that we observed in CPD mice requires further study, but the lack of effects of LCN2 deletion on FGF23 regulation in diet-induced iron deficiency strongly suggests an iron independent effect. Similarly, the lack of effects of LCN2 on FGF23 in a model of diet-induced hyperphosphatemia suggests that LCN2 does not play a role in phosphate-induced FGF23 production. Alternatively, LCN2 may constitute a mediator of the stimulatory effects of inflammation on FGF23 production. Lcn2 deletion in unchallenged WT mice only resulted in a trend toward reduced osseous Fgf23 mRNA both by PCR and RNAseq without significant reduction in FGF23 levels. However, Lcn2 deletion attenuated the increase in serum FGF23 levels induced by inflammatory stimuli. We previously showed that inflammation stimulates Fgf23 transcription in the bone which results in increased serum cFGF23 levels with only mild elevations in circulating levels of biologically active iFGF23 levels due to coupled activation of FGF23 cleavage mechanisms.8 Consistent with these effects,8 LCN2 administration mainly increased Fgf23 transcription and secretion of cFGF23 and had a very modest impact on serum iFGF23 levels in the non-CKD models in the present study. However, LCN2 administration showed more pronounced effects on iFGF23 levels in Col4a3KO mice with CKD, confirming that impaired FGF23 cleavage contributes to elevated serum iFGF23 levels in CKD.8,53,54
Interestingly, LCN2 directly targets bone cells and induces activation of cAMP/PKA/CREB signaling in osteoblasts. Previous studies have shown that LCN2 also activates cAMP signaling in the brain and spermatozoa,21,55 supporting cAMP signaling as a specific mediator of LCN2 effects. In bone, cAMP is also an important mediator of PTH effects56,57 and we confirm that cAMP-mediated signaling is a potent regulator of FGF23 transcription.58 The fact that cAMP signaling is regulated by both PTH and LCN2 upstream of FGF23 production suggests that cAMP-mediated signaling may play a central role in the regulation of FGF23 in CKD. Nevertheless, the overall impact of LCN2 on FGF23 circulating levels is highlighted by the independent relationship between FGF23 and NGAL in patients with CKD.
Excess FGF23 during CKD progression is associated with cardiovascular mortality via direct and reversible effects of FGF23 on cardiac myocytes that lead to the development of LVH.4,5,59,60,61 In the present study, inhibition of FGF23 production by genetic deletion of Lcn2 may have prevented the development of LVH and increased survival in Col4a3KO mice with CKD. We previously showed in Col4a3KO mice with CKD that overexpression of a bone matrix protein, DMP1, or treatment with ferric citrate, an iron-based phosphate binder, reduces FGF23 production and cardiovascular disease. This is the third study from our group showing that FGF23 reduction in CKD using different approaches, consistently leads to improvement of cardiac morphology and function despite persistent kidney disease.1,2 Ours study also suggests that LCN2 may be involved in secondary outcomes associated with progressive CKD, including the development of heart disease. Whether these effects are only mediated by FGF23 or whether there may also be some contribution from other known and possibly independent cardiac effects of LCN2,24,62 warrants additional studies which were beyond the scope of the present manuscript. Nonetheless, taken together our results emphasize the need for future studies focusing on FGF23 reduction in CKD.
To conclude, we showed that increased circulating LCN2 minimally contributes to iron deficiency in CKD, and does not have a determinant role on anemia of CKD, but mediates the stimulatory effects of inflammation on FGF23 production in bone by activating cAMP-mediated signaling, and contributes to the development of cardiac hypertrophy and mortality, likely through stimulation of FGF23 production. Since FGF23 also exerts pro-inflammatory effects,30,63 and strong correlations are observed in CKD between FGF23 and inflammatory markers,33 this represents a feed-forward loop, in which the effects of inflammation and FGF23 are fueling one another (Fig. 6i), offering one possible explanation for the maladaptive and exponential increases in FGF23 levels that occur in advanced CKD. Therefore, serum LCN2 emerges as a major kidney-bone crosstalk molecule that links the inflammation of kidney disease to FGF23 secretion from the bone and the development of cardiac disease during CKD.
Materials and methods
Human subjects
We used stored serum samples from 12 healthy volunteers and 36 patients with CKD stages 2–5 who participated in previous IRB-approved physiologic studies. All participants provided written informed consent to have their samples stored for future use for analysis of biomarkers related to kidney function and mineral metabolism. We measured NGAL using the NGAL ELISA assay (Abcam, Cambridge, MA, USA). We used a human intact FGF23 (iFGF23) enzyme-linked immunosorbent assay (ELISA) to measure the active iFGF23 protein and a C-terminal FGF23 (cFGF23) ELISA that recognizes the full-length protein and its C-terminal cleavage fragments to measure total FGF23 (both from Immutopics, Carlsbad, CA, USA).
Animals
Heterozygous C57Bl6/J-LCN+/−, expressing a neo-cassette targeting a 1.9 kb fragment in exons 2–4,39 obtained from Dr. Shizuo Akira at Osaka University were crossed to generate LCN2+/+ (WT) and LCN2−/− (Lcn2KO) mice. To assess the effects of acute inflammation, 12-week-old WT and Lcn2KO mice have injected ip with 250 ng·g−1 of recombinant murine interleukin 1 beta (IL-1β, Cell signaling). All diets were manufactured by Teklad (Envigo, Dever, CO, USA). Mice were maintained on a standard diet (Teklad 7012) except when otherwise specified. True iron deficiency was induced by feeding 3-week-old mice an iron deficient diet (TD.80396) or a mineral adjusted control diet (TD.80394) for 3 weeks. WT and Lcn2KO mice were fed a 2% high phosphate diet (TD.160039) or a mineral adjusted control diet (TD.80394) from 6 to 12 weeks of age. Six-week-old C57Bl6/J WT mice have injected ip during four days with murine recombinant LCN2 (50 ng·g−1 per day, 1857-LC-050, R&D systems, Minneapolis, MN, USA). Six-week-old C57Bl6/J WT mice were implanted ip with Alzet osmotic minipumps (Model 1004, Alzet, Cupertino, CA, USA) for 4 weeks to deliver murine recombinant LCN2 (5 or 50 ng·g−1 per day).
Heterozygous C57Bl6/J-Col4a3tm1Dec mice were crossed to LCN2+/− 39 mice to generate C57Bl6/J- Col4a3+/+LCN2+/+ (WT), Col4a3+/+LCN2−/− (Lcn2KO), Col4a3−/−LCN2+/+ (Col4a3KO) and Col4a3−/−LCN2−/− (CPD). We harvested samples on a set of 23-week-old male littermates. We recorded body weight at sacrifice. In a separate set of animals, we recorded the age of death on Col4a3KO and CPD littermates to assess effects on lifespan. Col4a3KO and CPD mice have implanted subcutaneously with Alzet osmotic minipumps for 8 weeks to deliver 50 ng·g−1 per day of murine recombinant LCN2. All studies were approved by Institutional Animal Care and Use Committee at Northwestern University.
Biochemistry of mouse samples
We collected overnight urine samples from fasted animals housed overnight in metabolic cages and serum samples by intracardiac exsanguination. We used a murine intact FGF23 (iFGF23) enzyme-linked immunosorbent assay (ELISA) to measure the active iFGF23 protein and a C-terminal FGF23 (cFGF23) ELISA that recognizes the full-length protein and its C-terminal cleavage fragments to measure total FGF23 (both from Immutopics, Carlsbad, CA, USA). Phosphate, blood urea nitrogen (BUN), albumin, iron, and transferrin saturation were measured using colorimetric assays (Pointe Scientific, Canton, MI, USA). We measured serum ferritin using mouse ELISA assays (Alpco, Salem, NH, USA), circulating LCN2 using LCN2 mouse ELISA assay (Abcam, Cambridge, MA, USA), and erythropoietin using mouse EPO quantikine ELISA kit (R&D Systems, Minneapolis, MN, USA).
RNA isolation, RT-PCR, and RNA sequencing
We isolated total RNA from tissues at sacrifice and from cell cultures using TRI reagent (Waltham, MA, USA) and purified RNA using RNeasy kit (Qiagen, Germantown, MD, USA).
For RNA sequencing, the total RNA library for each individual tibia was prepared using the TruSeq Total RNA-Seq Library Preparation Kit (Illumina, San Diego, CA), and the bar-coded cDNA libraries were sequenced for 75 bp single reads on one lane of Illumina NexSeq to generate a minimum of 100 million reads/library. Reads from each library were mapped to the mouse transcriptome and genome (UCSC mm10), filtered using StrandNGS software suite (Strand Life Sciences, Bangalore, Karnataka, India), and following Strand alignment and filtering pipelines. Reads were normalized using DESeq and we used baseline transformation to the median for each sample. FC and P value were calculated using moderated T-test and data were used for subsequent downstream pathway analyzes using the Ingenuity Pathway Analysis platform (IPA, Qiagen).
For RT-PCR, we synthesized first-strand cDNA (iScript cDNA Synthesis Kit, Bio-Rad Laboratories, Hercules, CA) and used the iCycler iQ real-time PCR detection system, iQ SYBR Green supermix (Bio-Rad Laboratories, Hercules, CA), and adequate primer pairs for real-time quantitative PCR analysis. The threshold of detection of each gene expression was set at optimal reaction efficiency. The expression was plotted against a standard dilution curve of relative concentration, normalized to 60S ribosomal protein L19 (Rpl19) expression in the same sample, and expressed as fold change versus respective controls.
Echocardiography
We performed echocardiography under isoflurane anesthesia 1 week prior to sacrifice (at 22 weeks of age) using a Vevo 770 High-Resolution Imaging System (VisualSonics, Toronto, ON, Canada). We used the parasternal short- and long-axis views to obtain two-dimensional and M-mode images as previously described.1,2,34
Hematologic analysis
Hematologic parameters were acquired in whole blood using the HEMAVET 950 hematology system (Drew Scientific Inc., Oxford, CT, USA) and analyzed with multispecies software using mouse settings as previously described.2
Histology
Heart weight and tibia length were measured post sacrifice. Kidneys and hearts were collected at sacrifice, fixed in 100% ethanol, and embedded in paraffin. We collected 5-µm-thick sections using a rotary microtome. For analysis of the cardiac phenotype, we used cross-sections from the mid-chamber of the heart. We stained the sections with hematoxylin and eosin (H&E) to determine renal and cardiac morphology, picrosirius red (PSR) to determine kidney fibrosis. Images were acquired using light microscopy (Leica Microsystems, Buffalo Grove, IL, USA).
Cell cultures and assays
We cultured MC3T3-E1 osteoblastic cell lines (ATCC) according to American type culture collection guidelines. We prepared BMSCs from 6-week-old mice according to a previously described protocol.64 We maintained MC3T3-E1 and BMSCs in α-MEM containing 10% FBS, 10 U·mL–1 penicillin, and 100 μg·mL–1 streptomycin. For all experimental conditions, we plated MC3T3-E1 at 3 × 104 cells per well and BMSCs at 10 × 104 cells per well and cultured for 3 weeks in osteoblast-differentiating medium (α-minimal essential medium, 10% fetal bovine serum, 10 U·mL–1 penicillin, 100 μg·mL–1 streptomycin, 10 mmol·L–1 β-glycerophosphate, and 50 μg·mL–1 ascorbic acid; Sigma–Aldrich, St Louis, MO) prior to treatment and collection. To assess Fgf23 promoter activity, MC3T3-E1 cells were stably transfected with the pLuc-Fgf23 promoter plasmid carrying a secreted luciferase expression cassette under the control of the proximal Fgf23 promoter, a secreted alkaline phosphatase (SEALP) under the control of the CMV promoter, and a puromycin resistance cassette (Genecopoeia, Rockville, MD) as previously described.1,8 At day 21, cells were treated with recombinant murine 5–50 ng·mL–1 LCN2 (R&D systems), 10 μmol·L–1 FSK (Sigma Aldrich, St. Louis, MO, USA) and 12 μmol·L–1 KT5720 (Abcam). For promoter activity assays, Optimem medium containing 1% FBS 10 U•mL–1 penicillin and 100 μg·mL–1 streptomycin, supplemented with LCN2, FSK, and/or KT5720 was used for the last 24 h of culture. Promoter activity is represented by a relative luciferase unit normalized to pSEALP-CMV control. We conducted all experiments in triplicate.
Protein assays and SDS-Page
MC3T3/E1 osteoblast cells grown in the osteogenic medium were treated for 6 h with FSK, LCN2, or LCN2 + KT5720 in Opti-MEM with 1% FBS. Protein extracts were prepared using T-Per lysis buffer (Thermo Fisher Scientific, MA, USA) containing protease inhibitors cocktail and immunoblots were performed as previously described.2,8 For SDS-Page, 100 µg of total proteins were loaded on a 4-to-12% Bis-Tris (midi) Gel (Invitrogen). CREB expression was detected using rabbit antibodies against phosphorylated CREB (1:1 000; 9198) and CREB (1:1 000; 9197) (Cell Signaling Technology, MA, USA) and goat anti-β Actin (1:1 000, 8229) (Abcam, MA, USA). Cyclic AMP accumulation in cells was measured using a cAMP direct immunoassay (Abcam) from protein extracts in 100 µL of 0.1 mol·L–1 HCl.
Statistics
Data are presented as mean ± SEM. Univariate and multiple regression, ANOVA followed by Fisher and two-sided, paired t tests were used for statistical inference using Statistica software (Statsoft, OK, USA). P values < 0.05 were considered statistically significant.
Study approval
All human participants studies provided written informed consent to have their samples stored for future use for analysis of biomarkers related to kidney function and mineral metabolism and were enrolled in IRB-approved physiologic studies. All animal studies were approved by Institutional Animal Care and Use Committee at Northwestern University.
Data availability
All data associated with this study are present in the paper. Materials and protocols are available upon demand.
References
Dussold, C. et al. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone Res. 7, 12 (2019).
Francis, C. et al. Ferric citrate reduces fibroblast growth factor 23 levels and improves renal and cardiac function in a mouse model of chronic kidney disease. Kidney Int. 96, 1346–1358 (2019).
Pereira, R. C. et al. Patterns of FGF-23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone 45, 1161–1168 (2009).
Faul, C. et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 121, 4393–4408 (2011).
Grabner, A. et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 22, 1020–1032 (2015).
Isakova, T. et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 305, 2432–2439 (2011).
Gutierrez, O. M. et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 359, 584–592 (2008).
David, V. et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 89, 135–146 (2016).
Farrow, E. G. et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl Acad. Sci. USA 108, E1146–E1155 (2011).
Grabner, A. & Wolf, M. Kidney to bone via bedside to bench…and back? J. Clin. Investig. 130, 1106–1108 (2020).
Simic, P. et al. Glycerol-3-phosphate is a FGF23 regulator derived from the injured kidney. TJ. Clin. Investig. 130, 1513–1526 (2020).
Li, J. Y. et al. Detection of intracellular iron by its regulatory effect. Am. J. Physiol. Cell Physiol. 287, C1547–C1559 (2004).
Devireddy, L. R., Hart, D. O., Goetz, D. H. & Green, M. R. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell 141, 1006–1017 (2010).
Bao, G. et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat. Chem. Biol. 6, 602–609 (2010).
Cherayil, B. J. The role of iron in the immune response to bacterial infection. Immunol. Res. 50, 1–9 (2011).
Correnti, C. & Strong, R. K. Mammalian siderophores, siderophore-binding lipocalins, and the labile iron pool. J. Biol. Chem. 287, 13524–13531 (2012).
Roudkenar, M. H. et al. Neutrophil gelatinase-associated lipocalin: a new antioxidant that exerts its cytoprotective effect independent on Heme Oxygenase-1. Free Radic. Res. 45, 810–819 (2011).
Roudkenar, M. H. et al. Neutrophil gelatinase-associated lipocalin acts as a protective factor against H(2)O(2) toxicity. Arch. Med. Res. 39, 560–566 (2008).
Hu, L. et al. NGAL decreases E-cadherin-mediated cell-cell adhesion and increases cell motility and invasion through Rac1 in colon carcinoma cells. Lab. Investig. 89, 531–548 (2009).
Kjeldsen, L., Johnsen, A. H., Sengelov, H. & Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 268, 10425–10432 (1993).
Mosialou, I. et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543, 385–390 (2017).
Xu, M. J. et al. Liver is the major source of elevated serum lipocalin-2 levels after bacterial infection or partial hepatectomy: a critical role for IL-6/STAT3. Hepatology 61, 692–702 (2015).
Chassaing, B. et al. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 7, e44328 (2012).
Xu, G. et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J. Biol. Chem. 287, 4808–4817 (2012).
Mishra, J. et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 365, 1231–1238 (2005).
Mori, K. et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J. Clin. Investig. 115, 610–621 (2005).
Nickolas, T. L. et al. NGAL (Lcn2) monomer is associated with tubulointerstitial damage in chronic kidney disease. Kidney Int. 82, 718–722 (2012).
Bolignano, D. et al. Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin. J. Am. Soc. Nephrol. 4, 337–344 (2009).
Bhavsar, N. A., Kottgen, A., Coresh, J. & Astor, B. C. Neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule 1 (KIM-1) as predictors of incident CKD stage 3: the atherosclerosis risk in communities (ARIC) study. Am. J. Kidney Dis. 60, 233–240 (2012).
Dai, B. et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS One 7, e44161 (2012).
Viau, A. et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J. Clin. Investig. 120, 4065–4076 (2010).
Christov, M. et al. Plasma FGF23 levels increase rapidly after acute kidney injury.Kidney Int. 84, 776–785 (2013).
Munoz Mendoza, J. et al. Fibroblast growth factor 23 and Inflammation in CKD. Clin. J. Am. Soc. Nephrol. 7, 1155–1162 (2012).
Neuburg, S. et al. Genetic background influences cardiac phenotype in murine chronic kidney disease. Nephrol. Dial. Transpl. 33, 1129–1137 (2018).
Kim, I. Y. et al. Plasma neutrophil gelatinase-associated lipocalin is associated with iron status in anemic patients with pre-dialysis chronic kidney disease. Clin. Exp. Nephrol. 22, 28–34 (2018).
Yazdani, M. et al. Association between neutrophil geletinase-associated lipocalin and iron deficiency anemia in children on chronic dialysis. J. Res. Med. Sci. 19, 624–628 (2014).
Kestenbaum, B. et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 16, 520–528 (2005).
Levin, A. et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am. J. Kidney Dis. 34, 125–134 (1999).
Flo, T. H. et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432, 917–921 (2004).
Goetz, D. H. et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 10, 1033–1043 (2002).
Holmes, M. A., Paulsene, W., Jide, X., Ratledge, C. & Strong, R. K. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 13, 29–41 (2005).
Guo, H., Jin, D. & Chen, X. Lipocalin 2 is a regulator of macrophage polarization and NF-kappaB/STAT3 pathway activation. Mol. Endocrinol. 28, 1616–1628 (2014).
Yang, J. & Moses, M. A. Lipocalin 2: a multifaceted modulator of human cancer. Cell Cycle 8, 2347–2352 (2009).
Wilson, R., Belluoccio, D., Little, C. B., Fosang, A. J. & Bateman, J. F. Proteomic characterization of mouse cartilage degradation in vitro. Arthritis Rheum. 58, 3120–3131 (2008).
Chakraborty, S., Kaur, S., Guha, S. & Batra, S. K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys. Acta 1826, 129–169 (2012).
Lindberg, S. et al. Plasma neutrophil gelatinase-associated lipocalinin in the general population: association with inflammation and prognosis. Arterioscler. Thromb. Vasc. Biol. 34, 2135–2142 (2014).
Mishra, J. et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol. 14, 2534–2543 (2003).
Yuen, P. S., Jo, S. K., Holly, M. K., Hu, X. & Star, R. A. Ischemic and nephrotoxic acute renal failure are distinguished by their broad transcriptomic responses. Physiol. Genom. 25, 375–386 (2006).
Berger, T. et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc. Natl Acad. Sci. USA 103, 1834–1839 (2006).
Miharada, K. et al. Lipocalin 2-mediated growth suppression is evident in human erythroid and monocyte/macrophage lineage cells. J. Cell. Physiol. 215, 526–537 (2008).
Miharada, K., Hiroyama, T., Sudo, K., Nagasawa, T. & Nakamura, Y. Lipocalin 2 functions as a negative regulator of red blood cell production in an autocrine fashion. FASEB J. 19, 1881–1883 (2005).
Bolignano, D. et al. Neutrophil gelatinase-associated lipocalin (NGAL) reflects iron status in haemodialysis patients. Nephrol. Dial. Transpl. 24, 3398–3403 (2009).
Shimada, T. et al. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J. Clin. Endocrinol. Metab. 95, 578–585 (2010).
Stubbs, J. R. et al. Longitudinal evaluation of FGF23 changes and mineral metabolism abnormalities in a mouse model of chronic kidney disease. J. Bone Miner. Res. 27, 38–46 (2012).
Watanabe, H. et al. Lipocalin 2 binds to membrane phosphatidylethanolamine to induce lipid raft movement in a PKA-dependent manner and modulates sperm maturation. Development 141, 2157–2164 (2014).
Meir, T. et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 86, 1106–1115 (2014).
Lavi-Moshayoff, V., Wasserman, G., Meir, T., Silver, J. & Naveh-Many, T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimetal kidney failure: a bone parathyroid feedback loop. Am J. Physiol. Ren. Physiol. 299, F882–F889 (2010).
Han, X., Xiao, Z. & Quarles, L. D. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF-23. J. Biol. Chem. 290, 10447–10459 (2015).
Gutierrez, O. M. et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 119, 2545–2552 (2009).
Mitsnefes, M. M. et al. FGF23 and left ventricular hypertrophy in children with CKD. Clin. J. Am. Soc. Nephrol. 13, 45–52 (2018).
Leifheit-Nestler, M. et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol. Dial. Transpl. 31, 1088–1099 (2016).
Marques, F. Z. et al. Experimental and human evidence for lipocalin-2 (neutrophil gelatinase-associated lipocalin [NGAL]) in the development of cardiac hypertrophyand heart failure. J. Am. Heart Assoc. 6, e005971 (2017).
Singh, S. et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 90, 985–996 (2016).
Martin, A. et al. Degradation of MEPE, DMP1, and release of SIBLING ASARM-peptides (minhibins): ASARM-peptide(s) are directly responsible for defective mineralization in HYP. Endocrinology 149, 1757–1772 (2008).
Acknowledgements
This study was supported by grants from the National Institute of Health to V.D. (R01DK102815, R01DK114158) and A.M. (R01DK101730).
Author information
Authors and Affiliations
Contributions
V.D. designed the study. G.C., A.M., and V.D. drafted the manuscript. All authors contributed to data acquisition and data interpretation. All authors revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
V.D. receives research funding from Akebia and has received research funding from Vifor Pharma and consulting honoraria from Keryx Biopharmaceuticals, Vifor Pharma, Luitpold, and Amgen outside of submitted work. M.W. has received personal fees from Akebia, Amag, Amgen, Ardelyx, Diasorin, Keryx, Lutipold, and Sanofi, and grants from the Shire, outside the submitted work. T.I. received personal fees from Kirin and Guidepoint Global, outside the submitted work. J.L.B. has an ownership interest in Ferrumax Pharmaceuticals and has received consulting fees from Keryx Biopharmaceuticals and Disc Medicine outside of submitted work. All other authors have nothing to disclose.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Courbon, G., Francis, C., Gerber, C. et al. Lipocalin 2 stimulates bone fibroblast growth factor 23 production in chronic kidney disease. Bone Res 9, 35 (2021). https://doi.org/10.1038/s41413-021-00154-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-021-00154-0
This article is cited by
-
FGF23 and klotho at the intersection of kidney and cardiovascular disease
Nature Reviews Cardiology (2024)
-
Regulation of FGF23 Production in Osteocytes
Current Osteoporosis Reports (2024)
-
Mechanisms of Rostellularia procumbens (L.) Nees on treating chronic glomerulonephritis explored by network pharmacology, RNA-seq, and in vitro experiments
BMC Complementary Medicine and Therapies (2023)
-
Regulation of FGF23 production and phosphate metabolism by bone–kidney interactions
Nature Reviews Nephrology (2023)
-
Messages from the Mineral: How Bone Cells Communicate with Other Tissues
Calcified Tissue International (2023)
