
Abstract
As key developmental regulators, HOX cluster genes have varied and context-specific roles in normal and malignant hematopoiesis. A complex interaction of transcription factors, epigenetic regulators, long non-coding RNAs and chromatin structural changes orchestrate HOX expression in leukemia cells. In this review we summarize molecular mechanisms underlying HOX regulation in clinical subsets of AML, with a focus on NPM1 mutated (NPM1mut) AML comprising a third of all AML patients. While the leukemia initiating function of the NPM1 mutation is clearly dependent on HOX activity, the favorable treatment responses in these patients with upregulation of HOX cluster genes is a poorly understood paradoxical observation. Recent data confirm FOXM1 as a suppressor of HOX activity and a well-known binding partner of NPM suggesting that FOXM1 inactivation may mediate the effect of cytoplasmic NPM on HOX upregulation. Conversely the residual nuclear fraction of mutant NPM has also been recently shown to have chromatin modifying effects permissive to HOX expression. Recent identification of the menin-MLL interaction as a critical vulnerability of HOX-dependent AML has fueled the development of menin inhibitors that are clinically active in NPM1 and MLL rearranged AML despite inconsistent suppression of the HOX locus. Insights into context-specific regulation of HOX in AML may provide a solid foundation for targeting this common vulnerability across several major AML subtypes.
Similar content being viewed by others
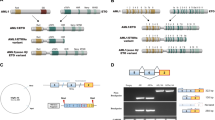
Introduction
Acute myeloid leukemia (AML), an aggressive heterogenous hematological malignancy [1], is driven by recurrent cytogenetic abnormalities and hotspot mutations that suppress normal myeloid differentiation and prime malignant transformation [2,3,4]. AML is characterized by a hijacking of normal transcriptional networks [5]. The clustered homeobox (HOX) gene family of transcription factors have an established role in normal hematopoiesis and their deregulation has been linked to pathways critical for leukemic stem cell activity. Overexpression of several HOX family members in experimental models alters self-renewal and differentiation properties of hematopoietic stem and progenitor cells and increased levels of certain HOX A genes has been linked to unfavorable prognosis in patients with AML [6, 7].
The HOX genes contain a highly conserved nucleotide sequence called homeodomain and encode a large family of transcription factors. In humans, 39 HOX genes are organized into four genomic clusters with shared enhancers namely the HOX A, B, C and D clusters, located on chromosomes 7, 17, 12 and 2 respectively [8, 9]. These clusters allow for spatial and temporal regulation of transcription needed for development [3, 4]. HOX A and HOX B families, are critical for hematopoietic lineage development [10, 11]. While HOX 1–4 genes are highly expressed in stem and early hematopoietic progenitor cells, HOX 7–11 expression is maximal in lineage committee progenitors and there is progressive down regulation with terminal differentiation. The selective DNA binding of HOX proteins is heavily dependent on cofactors such as pre-B-cell leukemia (PBX) [12] and myeloid ecotropic insertion site (MEIS) families [13]. Given their fundamental developmental role, dysregulation of HOX genes through altered expression or mutation is strongly implicated in cancer biology [14]. Depending on cellular context, HOX genes can be proto-oncogenes or tumor suppressors. In the context of hematopoiesis [15, 16], induced overexpression of several HOX genes leads to progenitor cell expansion, differentiation arrest and in the case of HOX A9 [17] and A10 [18], development of AML.
Numerous studies have shown that HOX genes can promote the development of AML by forming chimeric fusions with other genes [19, 20], or overexpression due to altered upstream regulators such as CDX proteins [21]. In this review we attempt to reconcile recent findings with known paradigms of HOX regulation focusing on HOX-dependent subsets of AML (Fig. 1).

A Major mechanisms of regulation of expression of HOX A/B genes in human leukemia cells include (i) cytogenetic rearrangements resulting in novel fusion proteins, (ii) long non-coding RNA association with the HOX gene locus and (iii) binding of upstream protein such as CDX proteins. B Regulation of HOX A in the context of NPM1mut AML includes two discrete mechanisms whereby (i) residual nuclear mutant NPM1 directly binds to chromatin and hijacks the transcriptional complex (RNA Pol II, MLL-Menin and super elongation complex) and (ii) the bulk of mutant NPM1 sequesters and relocalizes to the cytoplasm several repressors of HOX gene expression including FOXM1, and PU.1. * Additional cytogenetic arrangements inducing HOX expression include NUP98-NSD1, SET -NUP214, DEK-NUP214.
HOX regulation in NPM1 mut AML
Mutations of the nucleophosmin 1 (NPM1) gene are the most highly prevalent genomic alteration present in nearly a third of adult AML patients [22,23,24,25,26,27]. NPM1, a ubiquitously expressed protein while mainly located in the nucleolus, continuously shuttles between nucleus and cytoplasm. [28, 29]. It is involved in multiple cellular functions including histone assembly, ribosome biogenesis and export, centrosome duplication, maintenance of genome stability, transcriptional regulation, and DNA repair [30]. NPM1 mutations are always heterozygous and mainly restricted to exon 12 [24, 31, 32] and result in the insertion of a new C-terminal nuclear export signal on the protein [24] whereby mutated NPM heterodimerizes with wild type NPM and is translocated from the nucleus to cytoplasm with the help of nuclear exporter CRM1 [33,34,35]. HOX gene (specially HOXA/B) upregulation is a pathognomonic feature of NPM1mut AML [15, 36,37,38,39,40,41] and a key consequence of this mutation. Nuclear relocalization of the mutant NPM protein by inactivating the nuclear export signal (NES) or targeted deletion of NPM1mut by CRISPR-Cas9 genome editing technology results in rapid loss of HOX gene expression, abrogates the undifferentiated state and induces growth arrest [42, 43]. Thus, NPM acts as a direct upstream activation factor of HOX gene expression to maintain the growth and undifferentiated state of NPM1mut AML cells. The mechanism underlying NPM1mut regulation of HOX has been the subject of many recent studies.
A recently described mechanism links mutant NPM1 to the regulation of the HOX A/B gene clusters and the oncogenic state of AML cells via cytoplasmic export of the Forkhead box M1 (FOXM1) transcription factor. FOXM1, a member of the Forkhead family transcription factors, promotes cell proliferation, survival, tissue invasion, metastasis, angiogenesis, therapy resistance, and stem cell self-renewal [44,45,46,47,48,49,50]. Its overexpression is strong predictor of poor outcomes across many cancers including AML [45, 49, 51, 52]. Wild-type NPM binds FOXM1 through the heterodimerization domain of NPM (residues 187–259) and regulates its intracellular localization and stability [53]. This interaction was confirmed in AML cell lines and primary samples with mutant and wild type NPM1 [51]. In AML cells expressing NPM1mut, FOXM1 is inactivated and localized in the cytoplasm together with mutant cytoplasmic NPM (NPM1c). Stable knockdown of FOXM1 in AML cell lines expressing wild-type NPM1 resulted in significant upregulation of HOX A cluster genes in RNA-seq analysis recapitulating the phenotype of NPM1mut cells [54] and conferred sensitivity to current therapies including cytarabine and venetoclax. Therefore, FOXM1 may be a key intermediate in the NPM1mut / HOX axis whereby transcriptional inactivation of FOXM1 by cytoplasmic sequestration allows de-repression of HOX cluster genes. Haploinsufficiency of FOXM1 was recently shown to accelerate leukemogenesis by enhancing self-renewal in short term HSC [55] and DNA-damage, although a downstream effect on HOX expression was not examined. A role for FOXM1 as a tumor suppressor was demonstrated in a breast cancer model where FOXM1 was shown to interact with Rb and caused transcriptional suppression of the mammary alveolar differentiation program [56]. Recent RNA-seq analysis from the BEAT AML study showed that FOXM1 is transcriptionally downregulated and several HOX genes are significantly upregulated in NPM1mut AML patients [57] strengthening the evidence for negative correlation of FOXM1 transcriptional activity and HOX expression in primary samples. RNA inhibition of FOXM1 in AML cells resulted in increased expression of HOXA genes suggesting FOXM1 is sufficient to regulate HOX expression, independent of NPM molecular status [54]. These data suggest that the oncogenicity and chemosensitivity of NPM1 mutant AML cells can be simultaneously reconciled by transcriptional inactivation of FOXM1 and consequent de-repression of HOX A/B cluster genes.
Additional nuclear binding partners of NPM including myeloid transcription factor PU.1 and the architectural protein CCCTC binding factor CTCF, and both are required for differentiation and physiological HOX downregulation. Mutant NPM directly binds and translocates these putative factors from the nucleus to cytoplasm [58, 59]. The self-renewal and undifferentiation phenotype of PU.1-null myeloid precursor is significantly abrogated by reintroduction of nuclear PU.1 [58]. Deletion of CTCF in AML cells induced HOXA9 gene expression [60]. The data obtained from these investigations suggest that mutant nucleophosmin acts as a derepressor of HOX gene expression indirectly by translocating HOX repressors such as PU.1 and CTCF from the nucleus to cytoplasm. These data collectively support the premise that cytoplasmic relocalization of NPM and its binding partners is critical to the oncogenic function of this mutation.
Previous studies reported that mutant NPM1 could directly regulate HOX gene expression through an unknown but potentially chromatin-associated mechanism [42, 61, 62]. Recent studies elegantly demonstrated that mutant nucleophosmin (NPM1c) binds a subset of gene promoters and transcriptionally upregulates HOXA/B cluster genes through distinct mechanisms. The mutant NPM1c protein was shown to bind to chromatin at MLL-menin binding sites amplifying the function of this histone methyltransferase [63] while also inhibiting the activity of histone deacetylases [64] to maintain active transcription This chromatin bound mutant NPM directly modulates the active nascent transcription of HOXA/B cluster genes and cofactor MEIS1 by controlling the overall transcriptional activity mediated by transcriptional complexes such as RNA polymerase II, MLL-Menin complex and super elongation complex [63, 64].
Long non-coding RNAs (lncRNAs), a major transcribed product of the genome, play a role in chromatin remodeling or directing transcription factors to their target genomic sequences It was recently shown that lncRNA HOTTIP drives aberrant posterior HOXA gene expression through alteration of topologically associated domain (TADs) in the AML genome without affecting the anterior HOXA locus [65]. HOTTIP lncRNA is aberrantly expressed in the subset of AML patients with NPM1mut and MLL rearrangements [65] where it regulates the recruitment of WDR5/MLL complex to coordinate active chromatin modifications and HOXA gene expression [66]. Loss of HOTTIP lncRNA leads to reduced leukemic burden in an MLL rearranged mouse model and leukemogenesis by HOX A TADs is restored by reactivation of HOTTIP.
A more recent study [67] introduced the novel concept of long non coding RNA from the HOX B locus-HOXBLINC as a critical downstream mediator of NPM1c + -associated leukemic transcription program and leukemogenesis.
In summary, we describe three complimentary mechanisms convergently regulating the HOX A/B locus in NPM1 mutant AML, namely (i) cytoplasmic delocalization of repressors, (ii) chromatin modification by NPM1c directly or (iii) indirectly through long non coding RNA. Collectively this data underscores the critical oncogenic dependency on HOX A/B expression in this genomic subset of AML. Earlier work has shown that high HOX expression maintains the leukemic state in NPM1-mutant AML [42] and we postulate this results in potentially redundant mechanisms to sustain expression of this key transcription factor family.
HOX regulation in AML with recurrent cytogenetic abnormalities
One of the earliest defined subsets of AML is characterized by chromosomal translocations which generate leukomogenic fusion proteins that often act as aberrant transcription factors [68]. Recurrent chromosomal translocations in AML influencing HOX include direct fusion of HOX with nucleoporin genes such as NUP98 and rearrangements that involve upstream regulators, such as the mixed lineage leukemia gene (MLL) [69, 70].
NUP98
It has been reported that the recurring translocations in AML fuse NUP98 directly to HOXA9 and HOXD13, as well as to histone methyltransferase nuclear receptor-binding SET domain protein 1 (NSD1) [71,72,73,74,75,76]. NUP98-NSD1 fusion protein has been reported to induce AML in vivo, maintain self-renewal of myeloid stem cells in vitro and enforce expression of proto-oncogenes such as HOXA (A7, A9 and A10) and MEIS1. NUP98-NSD1 directly activates HOXA gene expression by binding to chromatin regions marked by histone H3 Lys 36 methylation (H3K36me) and prevents transcriptional repression of HOXA locus genes mediated by binding of EZH2-complexes to the HOXA gene locus [77]. A study using mouse embryonic stem cells showed that NUP89-HOX9A is significantly recruited to the HOX cluster region with the help of CRM1 and activates expression of HOX genes [78]. NUP98-homebox fusions have greater effect on self-renewal and aberrant gene expression than NUP98-nonhomebox fusions [79]. NUP98 fusion proteins such as NUP98-HOXA9, NUP98-HOXD13, and NUP98-NSD1 interact with non-specific lethal (NSL) and MLL1 histone-modifying complexes to drive HOX gene expression [75]. Analysis of chromatin immunoprecipitation sequencing data showed that NUP98-HOXA9 and MLL1 are recruited to HOXA and HOXB cluster gene loci marked by H3K4me3 and H4K16ac. Inactivation of MLL1 significantly reduces NUP98-HOXA9-induced gene expression as well as growth and survival of NUP98-HOXA9 driven leukemogenesis in vitro and in vivo [75]. These results demonstrate that NUP98 fusion protein mediates leukemogenesis and elevated HOX gene expression by controlling the gene transcriptional activity of histone modifying complex. In addition to direct transcriptional activation through the homeodomain, the NUP98 moiety also plays an important role in transformation by inhibiting CRM1-mediated nuclear export with resultant retention and enhanced activity of oncogenic transcription factors NFAT and NFκB [80].
NUP214
NUP214 is another nucleoporin involved in translocations with two chromatin remodeling proteins SET and DEK resulting in fusion proteins that influence HOX expression [70, 74, 76, 81, 82]. SET-NUP214 is associated with both acute lymphoblastic leukemia (ALL) as well as AML, while DEK-NUP214 is exclusively involved in AML. The SET-NUP214 fusion protein is recruited to the HOX cluster region with help of CRM1 where it recruits active RNA polymerase II leading to aberrant transcriptional activity of HOX genes [78]. DEK-NUP214 fusion protein also causes upregulation of HOX genes, specially HOXA9 in AML [83].
MLL
It has previously been reported that histone methyltransferase KMT2A (MLL1- mixed-lineage leukemia1) is genetically rearranged in upto 10% of AML with more than 64 translocation partner genes [84] and transformation of murine bone marrow by MLL fusion proteins is Hox gene dependent [85]. MLL directly binds to and utilizes its highly conserved SET domain H3 (Lys4) methyltransferase to regulate HOX promoters. Leukemia associated MLL-fusion proteins have deletion of the SET domain that may compromise stage-specific down regulation of HOX transcription [86] .
Upstream mechanisms of HOX regulation in AML
CDX
The CDX gene, a member of the ParaHox gene family has been shown to influence organogenesis and hematopoiesis through regulation of HOX gene expression and Cdx proteins can act directly on Hox gene regulatory elements [87, 88]. CDX2 is aberrantly expressed in leukemic cells of 90% AML patients [21]. Three CDX homologs CDX1, CDX2, and CDX4 have consensus binding sites in the promoters of multiple HOX genes [87]. Both in vivo and in vitro experiments showed that CDX2 and CDX4 expression led to dysregulation of HOX expression. Overexpression of CDX2 leads to aberrant HOX gene expression in murine hematopoietic progenitors and this link was confirmed in AML primary cells [21, 89]. CDX2 acts as a positive upstream regulator of several HOX genes such as HOXA6/10 and HOXB8, and the proliferation of AML cell lines was inhibited by siRNA targeting of CDX2 [89].
In addition, overexpression of CDX4 can transform hematopoietic cells in cooperation with MEIS1 leading to AML in mouse models [90]. CDX4 and HOXA10 were shown to regulate one in a positive feedback loop [91], with the HOXA10 promoter having a CDX4-binding cis element and vice versa. β-catenin also activates CDX4 transcription and is upregulated as a consequence of HOX effects on FGF2 [92, 93]. Thus, there is cross regulation at several levels between HOX, HOX target genes such as FGF2, β-catenin and CDX.
Our review highlights a confluence of different mechanisms regulating HOX expression. Hox-binding cis elements often occur in tandem in genes (like the CYBB, NCF2 and ARIH2 genes) [94] with differential binding affinity for each of the “cassettes” that bind Hox proteins. This tremendous flexibility in the regulation of target gene expression is germane to the developmental role of HOX transcription factors.
Challenges in targeting HOX in leukemia and future directions
Using a CRISPR/Cas9 genome editing screen [95], the histone modifier MLL1 was shown to be critical factor regulating HOX expression in NPM1mut AML and the menin binding site of MLL was proven to be a potent target in NPM1mut AML. Early therapeutic development focused on DOT1L, the histone methyltransferase that specifically targets nucleosomal H3K79 aberrantly recruited to MLL target genes including HOXA9 but these inhibitors lacked specificity and clinical trials of EPZ-5676 showed variable effects on histone methylation and HOXA9 inhibition not necessarily correlated with clinical outcomes [96]. Recent therapeutic development of menin inhibitors in HOX-addicted AML has been very encouraging leading to ongoing review by the FDA for a first-in-class menin inhibitor in AML [97]. However the inhibitory activity of these compounds is predominantly at the co-factors MEIS and PBX3 with inconsistent suppression of the HOXA locus [98]
Context-specific knowledge of transcriptional regulation of HOX expression has given impetus to therapeutic targeting in genomic subsets of AML. In addition, recent data on direct effects of mutant NPM1c on chromatin structure may uncover novel targets. There is increasing interest in cross regulation of HOX proteins by each other and other homeodomain proteins such as CDXs as well as potential targets downstream of HOX such as FGF2. There is an ongoing clinical trial with nintedanib (FGFR inhibitor) in HOXA overexpressing leukemia (NCT03513484).
While enhanced oncogenic activity is a functional property of HOX overexpression that is being aggressively targeted, the relationship of HOX overexpression to increased sensitivity to chemotherapy and bcl2 inhibitors [37, 99] remains elusive. It is still to be determined whether high HOXA expression in chemosensitive subsets of AML indicates an alternate functional role of HOXA as an effector of treatment responses or a biomarker for loss of FOXM1 or additional mediators of chemoresistance.
References
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–98.
Moore MA. Converging pathways in leukemogenesis and stem cell self-renewal. Exp Hematol. 2005;33:719–37.
Wiseman DH, Greystoke BF, Somervaille TC. The variety of leukemic stem cells in myeloid malignancy. Oncogene. 2014;33:3091–8.
Assi SA, Imperato MR, Coleman DJL, Pickin A, Potluri S, Ptasinska A, et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat Genet. 2019;51:151–62.
Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16:186–95.
Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–7.
Duboule D, Dolle P. The structural and functional organization of the murine HOX gene family resembles that of Drosophila homeotic genes. EMBO J. 1989;8:1497–505.
Scott MP, Tamkun JW, Hartzell GW 3rd. The structure and function of the homeodomain. Biochim Biophys Acta. 1989;989:25–48.
Abramovich C, Humphries RK. Hox regulation of normal and leukemic hematopoietic stem cells. Curr Opin Hematol. 2005;12:210–6.
Sauvageau G, Lansdorp PM, Eaves CJ, Hogge DE, Dragowska WH, Reid DS, et al. Differential expression of homeobox genes in functionally distinct CD34+ subpopulations of human bone marrow cells. Proc Natl Acad Sci USA. 1994;91:12223–7.
Nakamura T, Largaespada DA, Shaughnessy JD, Jenkins NA, Copeland NG. Cooperative activation of Hoxa and Pbx1-related genes in murine myeloid leukaemias. Nat Genet. 1996;12:149–53.
Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998;17:3714–25.
Grier DG, Thompson A, Kwasniewska A, McGonigle GJ, Halliday HL, Lappin TR. The pathophysiology of HOX genes and their role in cancer. J Pathol. 2005;205:154–71.
Argiropoulos B, Humphries RK. Hox genes in hematopoiesis and leukemogenesis. Oncogene. 2007;26:6766–76.
Wheadon H, Ramsey JM, Dobbin E, Dickson GJ, Corrigan PM, Freeburn RW, et al. Differential Hox expression in murine embryonic stem cell models of normal and malignant hematopoiesis. Stem Cells Dev. 2011;20:1465–76.
Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99:121–9.
Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Lansdorp PM, Lawrence HJ, et al. Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol Cell Biol. 1997;17:495–505.
Borrow J, Shearman AM, Stanton VP, Becher R, Collins T, Williams AJ, et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP96 and class I homeoprotein HOXA9. Nat Genet. 1996;12:159–67.
Hatano M, Roberts CWM, Minden M, Crist WM, Korsmeyer SJ. Deregulation of a Homeobox Gene, HOX11, by the t(10;14) in T Cell Leukemia. Science. 1991;253:79–82.
Scholl C, Bansal D, Döhner K, Eiwen K, Huntly BJP, Lee BH, et al. The homeobox gene CDX2 is aberrantly expressed in most cases of acute myeloid leukemia and promotes leukemogenesis. J Clin Investig. 2007;117:1037–48.
Boissel N, Renneville A, Biggio V, Philippe N, Thomas X, Cayuela JM, et al. Prevalence, clinical profile, and prognosis of NPM mutations in AML with normal karyotype. Blood. 2005;106:3618–20.
Dohner K, Schlenk RF, Habdank M, Scholl C, Rucker FG, Corbacioglu A, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–6.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl J Med. 2005;352:254–66.
Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–9.
Suzuki T, Kiyoi H, Ozeki K, Tomita A, Yamaji S, Suzuki R, et al. Clinical characteristics and prognostic implications of NPM1 mutations in acute myeloid leukemia. Blood. 2005;106:2854–61.
Verhaak RG, Goudswaard CS, van Putten W, Bijl MA, Sanders MA, Hugens W, et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood. 2005;106:3747–54.
Borer RA, Lehner CF, Eppenberger HM, Nigg EA. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56:379–90.
Cordell JL, Pulford KA, Bigerna B, Roncador G, Banham A, Colombo E, et al. Detection of normal and chimeric nucleophosmin in human cells. Blood. 1999;93:632–42.
Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493–505.
Falini B, Brunetti L, Sportoletti P, Martelli MP. NPM1-mutated acute myeloid leukemia: from bench to bedside. Blood. 2020;136:1707–21.
Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109:874–85.
Bolli N, Nicoletti I, De Marco MF, Bigerna B, Pucciarini A, Mannucci R, et al. Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res. 2007;67:6230–7.
Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S, et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia. 2009;23:1731–43.
Falini B, Martelli MP, Bolli N, Bonasso R, Ghia E, Pallotta MT, et al. Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood. 2006;108:1999–2005.
Alcalay M, Tiacci E, Bergomas R, Bigerna B, Venturini E, Minardi SP, et al. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood. 2005;106:899–902.
Kontro M, Kumar A, Majumder MM, Eldfors S, Parsons A, Pemovska T, et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia. 2017;31:301–9.
Mullighan CG, Kennedy A, Zhou X, Radtke I, Phillips LA, Shurtleff SA, et al. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia. 2007;21:2000–9.
Spencer DH, Young MA, Lamprecht TL, Helton NM, Fulton R, O’Laughlin M, et al. Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia. 2015;29:1279–89.
Uckelmann HJ, Kim SM, Wong EM, Hatton C, Giovinazzo H, Gadrey JY, et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science. 2020;367:586–90.
Vassiliou GS, Cooper JL, Rad R, Li J, Rice S, Uren A, et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet. 2011;43:470–5.
Brunetti L, Gundry MC, Sorcini D, Guzman AG, Huang YH, Ramabadran R, et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell. 2018;34:499–512.e9.
Dovey OM, Cooper JL, Mupo A, Grove CS, Lynn C, Conte N, et al. Molecular synergy underlies the co-occurrence patterns and phenotype of NPM1-mutant acute myeloid leukemia. Blood. 2017;130:1911–22.
Borhani S, Gartel AL. FOXM1: a potential therapeutic target in human solid cancers. Expert Opin Ther Targets. 2020;24:205–17.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45.
Halasi M, Gartel AL. FOX(M1) news—it is cancer. Mol Cancer Ther. 2013;12:245–54.
Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–36.
Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7:847–59.
Nakamura S, Hirano I, Okinaka K, Takemura T, Yokota D, Ono T, et al. The FOXM1 transcriptional factor promotes the proliferation of leukemia cells through modulation of cell cycle progression in acute myeloid leukemia. Carcinogenesis. 2010;31:2012–21.
Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–94.
Khan I, Halasi M, Zia MF, Gann P, Gaitonde S, Mahmud N, et al. Nuclear FOXM1 drives chemoresistance in AML. Leukemia. 2017;31:251–5.
Khan I, Patel AA, Halasi M, Schultz R, Chen Y-H, Aardsma N, et al. Validation of FOXM1 as a therapeutic target in acute myeloid leukemia. Blood 2017;130:300.
Bhat UG, Jagadeeswaran R, Halasi M, Gartel AL. Nucleophosmin interacts with FOXM1 and modulates the level and localization of FOXM1 in human cancer cells. J Biol Chem. 2011;286:41425–33.
Chesnokov MS, Borhani S, Halasi M, Arbieva Z, Khan I, Gartel AL. FOXM1-AKT positive regulation loop provides venetoclax resistance in AML. Front Oncol. 2021;11:696532.
Yu C, Sheng Y, Yu F, Ni H, Qiu A, Huang Y, et al. Foxm1 haploinsufficiency drives clonal hematopoiesis and promotes a stress-related transition to hematologic malignancy in mice. J Clin Invest. 2023;133:e163911.
Kopanja D, Chand V, O’Brien E, Mukhopadhyay NK, Zappia MP, Islam A, et al. Transcriptional repression by FoxM1 suppresses tumor differentiation and promotes metastasis of breast cancer. Cancer Res. 2022;82:2458–71.
Khan I, Kaempf A, Raghuwanshi S, Chesnokov M, Zhang X, Wang Z, et al. Favorable outcomes of NPM1(mut) AML patients are due to transcriptional inactivation of FOXM1, presenting a new target to overcome chemoresistance. Blood Cancer J. 2023;13:128.
Gu X, Ebrahem Q, Mahfouz RZ, Hasipek M, Enane F, Radivoyevitch T, et al. Leukemogenic nucleophosmin mutation disrupts the transcription factor hub that regulates granulomonocytic fates. J Clin Invest. 2018;128:4260–79.
Wang AJ, Han Y, Jia N, Chen P, Minden MD. NPM1c impedes CTCF functions through cytoplasmic mislocalization in acute myeloid leukemia. Leukemia. 2020;34:1278–90.
Luo H, Wang F, Zha J, Li H, Yan B, Du Q, et al. CTCF boundary remodels chromatin domain and drives aberrant HOX gene transcription in acute myeloid leukemia. Blood. 2018;132:837–48.
Oka M, Mura S, Otani M, Miyamoto Y, Nogami J, Maehara K, et al. Chromatin-bound CRM1 recruits SET-Nup214 and NPM1c onto HOX clusters causing aberrant HOX expression in leukemia cells. Elife. 2019;8:e46667.
Zhang W, Zhao C, Zhao J, Zhu Y, Weng X, Chen Q, et al. Inactivation of PBX3 and HOXA9 by down-regulating H3K79 methylation represses NPM1-mutated leukemic cell survival. Theranostics. 2018;8:4359–71.
Uckelmann HJ, Haarer EL, Takeda R, Wong EM, Hatton C, Marinaccio C, et al. Mutant NPM1 directly regulates oncogenic transcription in acute myeloid leukemia. Cancer Discov. 2023;13:746–65.
Wang XQD, Fan D, Han Q, Liu Y, Miao H, Wang X, et al. Mutant NPM1 hijacks transcriptional hubs to maintain pathogenic gene programs in acute myeloid leukemia. Cancer Discov. 2023;13:724–45.
Luo H, Zhu G, Xu J, Lai Q, Yan B, Guo Y, et al. HOTTIP lncRNA promotes hematopoietic stem cell self-renewal leading to AML-like disease in mice. Cancer Cell. 2019;36:645–59.e8.
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4.
Zhu G, Luo H, Feng Y, Guryanova OA, Xu J, Chen S, et al. HOXBLINC long non-coding RNA activation promotes leukemogenesis in NPM1-mutant acute myeloid leukemia. Nat Commun. 2021;12:1956.
Rowley JD. The critical role of chromosome translocations in human leukemias. Annu Rev Genet. 1998;32:495–519.
Dorrance AM, Liu S, Yuan W, Becknell B, Arnoczky KJ, Guimond M, et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J Clin Invest. 2006;116:2707–16.
Xu S, Powers MA. Nuclear pore proteins and cancer. Semin Cell Dev Biol. 2009;20:620–30.
Borrow J, Shearman AM, Stanton VP Jr., Becher R, Collins T, Williams AJ, et al. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet. 1996;12:159–67.
Nakamura T, Largaespada DA, Lee MP, Johnson LA, Ohyashiki K, Toyama K, et al. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7;11)(p15;p15) in human myeloid leukaemia. Nat Genet. 1996;12:154–8.
Cerveira N, Correia C, Doria S, Bizarro S, Rocha P, Gomes P, et al. Frequency of NUP98-NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia. 2003;17:2244–7.
Zhou MH, Yang QM. NUP214 fusion genes in acute leukemia (Review). Oncol Lett. 2014;8:959–62.
Xu H, Valerio DG, Eisold ME, Sinha A, Koche RP, Hu W, et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer Cell. 2016;30:863–78.
Mendes A, Fahrenkrog B. NUP214 in leukemia: it’s more than transport. Cells. 2019;8:76.
Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol. 2007;9:804–12.
Oka M, Mura S, Yamada K, Sangel P, Hirata S, Maehara K, et al. Chromatin-prebound Crm1 recruits Nup98-HoxA9 fusion to induce aberrant expression of Hox cluster genes. Elife. 2016;5:e09540.
Saw J, Curtis DJ, Hussey DJ, Dobrovic A, Aplan PD, Slape CI. The fusion partner specifies the oncogenic potential of NUP98 fusion proteins. Leuk Res. 2013;37:1668–73.
Takeda A, Sarma NJ, Abdul-Nabi AM, Yaseen NR. Inhibition of CRM1-mediated nuclear export of transcription factors by leukemogenic NUP98 fusion proteins. J Biol Chem. 2010;285:16248–57.
Ageberg M, Drott K, Olofsson T, Gullberg U, Lindmark A. Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes Chromosomes Cancer. 2008;47:276–87.
Takeda A, Yaseen NR. Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol. 2014;27:3–10.
Ikeda D, Chi S, Uchiyama S, Nakamura H, Guo YM, Yamauchi N, et al. Molecular classification and overcoming therapy resistance for acute myeloid leukemia with adverse genetic factors. Int J Mol Sci. 2022;23:5950.
Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–9.
Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17:2298–307.
Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–17.
Charité J, de Graaff W, Consten D, Reijnen MJ, Korving J, Deschamps J. Transducing positional information to the Hox genes: critical interaction of cdx gene products with position-sensitive regulatory elements. Development. 1998;125:4349–58.
Subramanian V, Meyer BI, Gruss P. Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell. 1995;83:641–53.
Rawat VP, Thoene S, Naidu VM, Arseni N, Heilmeier B, Metzeler K, et al. Overexpression of CDX2 perturbs HOX gene expression in murine progenitors depending on its N-terminal domain and is closely correlated with deregulated HOX gene expression in human acute myeloid leukemia. Blood. 2008;111:309–19.
Bansal D, Scholl C, Frohling S, McDowell E, Lee BH, Dohner K, et al. Cdx4 dysregulates Hox gene expression and generates acute myeloid leukemia alone and in cooperation with Meis1a in a murine model. Proc Natl Acad Sci USA. 2006;103:16924–9.
Bei L, Huang W, Wang H, Shah C, Horvath E, Eklund E. HoxA10 activates CDX4 transcription and Cdx4 activates HOXA10 transcription in myeloid cells. J Biol Chem. 2011;286:19047–64.
Shah CA, Bei L, Wang H, Platanias LC, Eklund EA. HoxA10 protein regulates transcription of gene encoding fibroblast growth factor 2 (FGF2) in myeloid cells*. J Biol Chem. 2012;287:18230–48.
Bei L, Shah C, Wang H, Huang W, Roy R, Eklund EA. β-Catenin activates the HOXA10 and CDX4 genes in myeloid progenitor cells. J Biol Chem. 2012;287:39589–601.
Wang H, Bei L, Shah CA, Horvath E, Eklund EA. HoxA10 influences protein ubiquitination by activating transcription of ARIH2, the gene encoding Triad1. J Biol Chem. 2011;286:16832–45.
Kuhn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 2016;6:1166–81.
Stein EM, Garcia-Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131:2661–9.
Issa GC, Aldoss I, DiPersio J, Cuglievan B, Stone R, Arellano M, et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature. 2023;615:920–4.
Gundry MC, Goodell MA, Brunetti L. It’s all about MEis: Menin-MLl inhibition eradicates NPM1-mutated and MLL-rearranged acute leukemias in mice. Cancer Cell. 2020;37:267–9.
DiNardo CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135:791–803.
Author information
Authors and Affiliations
Contributions
ALG conceptualized and edited the manuscript; IK and MAA wrote the manuscript; EAE provided supervision and critical input.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, I., Amin, M.A., Eklund, E.A. et al. Regulation of HOX gene expression in AML. Blood Cancer J. 14, 42 (2024). https://doi.org/10.1038/s41408-024-01004-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-024-01004-y
