
Abstract
Extra copies of chromosome 1q21 (+1q: gain = 3 copies, amp >= 4 copies) are associated with worse outcomes in multiple myeloma (MM). This systematic review assesses the current reporting trends of +1q, the efficacy of existing regimens on +1q, and its prognostic implications in MM randomized controlled trials (RCTs). Pubmed, Embase and Cochrane Registry of RCTs were searched from January 2012 to December 2022. Only MM RCTs were included. A total of 124 RCTs were included, of which 29 (23%) studies reported on +1q. Among them, 10% defined thresholds for +1q, 14% reported survival data separately for gain and amp, and 79% considered +1q a high-risk cytogenetic abnormality. Amongst RCTs that met the primary endpoint showing improvement in progression free survival (PFS), lenalidomide maintenance (Myeloma XI), selinexor (BOSTON), and isatuximab (IKEMA and ICARIA) were shown to improve PFS for patients with evidence of +1q. Some additional RCT’s such as Myeloma XI+ (carfilzomib), ELOQUENT-3 (elotuzumab), and HOVON-65/GMMG-HD4 (bortezomib) met their endpoint showing improvement in PFS and also showed improvement in PFS in the +1q cohort, although the confidence interval crossed 1. All six studies that reported HR for +1q patients vs. without (across both arms) showed worse OS and PFS for +1q. There is considerable heterogeneity in the reporting of +1q. All interventions that have shown to be successful in RCTs and have clearly reported on the +1q subgroup have shown concordant direction of results and benefit of the applied intervention. A more standardized approach to reporting this abnormality is needed.
Similar content being viewed by others

Introduction
Despite advances in diagnostics and therapeutics, multiple myeloma (MM) remains associated with significant morbidity and mortality, with a broad range of patient survival observed. The cytogenetic alterations historically associated with high-risk disease include translocations (4;14), (14;16), (14;20), and deletion of chromosome 17p [1]. Recently, copy number gains of the long arm of chromosome 1 (+1q) have also been associated with worse outcomes [2].
Generally, +1q signifies the presence of one or more additional copies of a segment of chromosome 1q within the malignant plasma cells. Amongst MM with +1q, gain(1q) refers to those who possess only one extra copy of 1q, resulting in three total copies, while amp(1q) denotes patients exhibiting amplification of 1q, characterized by the presence of two or more additional copies, totaling four or more copies [2]. Through fluorescent in situ hybridization (FISH), around 32–40% of newly diagnosed MM is found to harbor +1q [3, 4]. A significant obstacle to understanding the role of +1q in MM is the need for more consistency in reporting and annotating cytogenetics [5]. However, the adverse prognostic implications of +1q have led to a clearer understanding of “double-hit” and “triplet-hit”, where an additive adverse prognostic effect of +1q is seen when combined with other high-risk abnormalities [6]. +1q has also been added to more recent myeloma staging systems, highlighting its prognostic significance [7, 8].
The introduction of immunomodulatory drugs (IMiDs), proteasome inhibitors (PIs), and anti-CD38 antibodies have significantly enhanced outcomes for patients with newly diagnosed MM. However, approximately 10–20% of patients have a 2-year PFS of ≤50% despite current treatment, which is categorized as high-risk MM [9,10,11]. Prior studies have reported amp(1q) to be linked to poor survival, which warrants recognition as a high-risk cytogenetic abnormality. Despite many studies indicating that gain(1q) is also high-risk, its influence seems less deleterious than amp(1q), though patients with gain(1q) still experience worse outcomes than those without it [2, 4, 12,13,14,15]. Moreover, the concomitant presence of other high-risk cytogenetic abnormalities or gene expression profiles could play a crucial role in determining the extent of additional risk conferred by this abnormality [2]. As an example, patients with both a high-risk status as determined by gene expression profiling (GEP70) and the presence of baseline gain(1q) had an especially poor response to daratumumab-based therapy [16].
To evaluate the current reporting trends of +1q in MM RCTs, we aimed to estimate the prevalence of +1q reporting, its impact on prognosis, how treatments impact outcomes for this subset of patients, and other characteristics of how it is reported through a systematic review of MM RCTs.
Methods
Direct patient information was not obtained, and the data was gathered from publicly available and deidentified sources; therefore, this study was considered exempt from approval of the institutional review board.
A previously published systematic review and search strategy was utilized for this study [17]. We performed a search of three databases: (MEDLINE/PubMed, Embase, and Cochrane Registry of RCTs). The search terms used are highlighted in Supplementary Table 1. Two independent reviewers (KAK and GRM) screened all studies, and any conflict was resolved through mutual discussion. This systematic review was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) recommendations. Our search strategy was restricted to MM RCTs published in peer-reviewed journals from January 2012 to December 2022. This time period was chosen to ascertain the presumed increased reporting of +1q over time. All other studies were excluded, including editorials, case reports, case series, review articles, case-control, retrospective/prospective cohort, and single-arm studies. The search strategy was not restricted to language. Abstracts from conference proceedings that were captured on these databases via our search strategy, such as those on Embase, were included for final analysis in our study. This study was not registered on PROSPERO. Two authors (KN and GGF) performed and verified all data extraction. Extracted data was tabulated using Microsoft Excel (Microsoft, Redmond, Washington, United States). We identified the following characteristics of studies: name of the RCT, year of publication, number of participants, location of study (enrollment in one country versus multi-national), therapeutic agent under study, whether +1q was reported or not as a high-risk cytogenetic alteration, definition of +1q with respect to the percentage of cells with this abnormality detected, documentation of distinction between gain(1q) and amp(1q) in analysis, the prevalence of +1q in enrolled population, and the outcomes of patients [overall survival (OS) and progression-free survival (PFS)] in patients with +1q in the experimental versus control arm and in patients with or without +1q.
The primary outcome of this study was to determine the prevalence of +1q in MM RCTs. A key secondary outcome was to understand the prognostic significance of +1q in RCTs.
Results
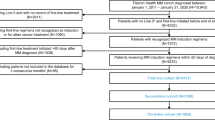
After excluding duplicate trials and trials that did not meet the inclusion criteria above, 124 discrete RCTs were identified (Fig. 1 highlights the study selection strategy). Table 1 highlights the characteristics of the included studies.

Flow diagram depicting our search strategy and study inclusion.
Reporting of +1q
Among these trials, 29 (23%) studies reported data on +1q, including 26 studies that reported data in the primary manuscript and three studies that reported in separate publications. These RCTs reported 2754 patients with +1q representing 25% of all enrolled patients. Out of 29 RCTs, three trials (10%) specified the criteria for categorizing patients as +1q (for example, in IKEMA and IFM-99: the presence of at least three copies in at least 30% of analyzed plasma cells was required, and in ELOQUENT-2 positivity for 1q was assigned based on identifying at least one abnormal cell) [18,19,20]. Only four trials (14%) reported survival data on gain and amp separately [21,22,23,24], and the remaining 25 (86%) studies reported for gain or did not specify gain versus amp. One study reported three or more copies as amp(1q) in its original publication, although it did report separately for four or more copies in a follow-up publication [21, 25]. Only one (4%) RCT reported survival outcomes for patients with isolated +1q but no other high-risk cytogenetics [26]. Among the RCTs that reported +1q, 23 (79%) considered this a high-risk cytogenetic abnormality, with the remaining studies reporting on +1q, but not including it within the high-risk category. Reporting of +1q is summarized in Fig. 2.

Bar graph depicting percentages of multiple myeloma trials reporting on +1q.
Reporting of +1q has been increasing recently, with +1q being reported by 58.3% of MM RCTs published between 2020–2022 vs. 18.3% of MM RCTs published between 2016–2019 and 9.8% of MM RCTs published between 2012–2015. Most of these studies (86%) were from outside the US, and 71% included frontline or consolidation/maintenance therapy (Table 1).
Survival outcomes
Amongst RCTs that met their primary endpoint showing improvement in PFS and clearly reported on +1q, the following drugs/regimens also improved PFS for those with +1q (when comparing HR for intervention versus control arm in the +1q subgroup): selinexor in BOSTON (HR 0.63, 95% CI 0.34–1.17, p = 0.07) [25], lenalidomide (len) maintenance in Myeloma XI (HR 1.5, 95% CI 0.9–2.7, p = 0.02) [26], isatuximab in IKEMA (HR 0.582, 95% CI 0.368–0.932) [18] and ICARIA (HR 0.41, 95% CI 0.2–0.7, p = 0.137) [27]. Although len maintenance improved PFS after autologous transplant as maintenance in Myeloma XI for patients with +1q overall (including those with other concurrent high-risk abnormalities), it did not appear to improve PFS for patients with isolated +1q (with no other concurrent genetic abnormalities) [26].
Several RCTs met their endpoint and showed improvement in PFS in the +1q cohort in the same direction as the overall study results but did not reach statistical significance. These included carfilzomib, len, dex, and cyclophosphamide vs. len, dexamethasone (dex), and cyclophosphamide (or thalidomide, dex, and cyclophosphamide in Myeloma XI+ (HR 0.63, 95% CI 0.38–1.06, p = 0.89) [28], the addition of elotuzumab to pomalidomide and dex (HR 0.56, 95% CI = 0.29–1.09) in ELOQUENT-3 [29], and bortezomib-based treatment before and after autologous stem cell transplantation vs. no bortezomib (HR 0.76, 95% CI 0.48–1.18, p = 0.22) in HOVON-65/GMMG-HD4 [23]. Although not powered for PFS, the addition of daratumumab to len and dex (HR 0.42, 95% CI 0.14–1.27) in GRIFFIN demonstrated a trend towards improvement in PFS for the +1q cohort [30].
Six RCTs, including ENDURANCE [22], IFM-99 [19], HOVON87/NMSG18 [31], HOVON-65/GMMG-HD4 [23], FORTE [4], and Myeloma IX [32] reported HR for patients with +1q in the trial (across both arms) compared to those without. Worse outcomes were seen in PFS and OS for those with +1q versus those without +1q in all these studies (Table 2). FORTE reported worse OS and PFS in patients with amp(1q) compared to gain(1q) (OS: HR 3.13, 95% CI 1.73–5.68, p < 0.001, and PFS: HR 1.84, 95% CI 1.21–2.81, p = 0.004) [4].
Among the RCTs that reported PFS and OS for the +1q group, three trials, SWOG-1211 [11], Myeloma XI [33] and, ENDURANCE [22] did not meet their primary endpoints. In Myeloma XI [33], no benefit was seen in the +1q group with the addition of vorinostat, and in SWOG-1211 [11], there was no statistically significant difference in PFS or OS in patients with +1q in patients treated with bort, len, and dex with or without elotuzumab. Though primary endpoint was not reached in ENDURANCE trial, the addition of carfilzomib vs. bortezomib to len and dex showed benefit in gain(1q) with HR 0.75, 95% CI 0.49–1.14, p = 0.17 but not in amp(1q) with HR 1.46, 95% CI 0.73–2.92, p = 0.281 [34].
The gap in the evidence
Important interventions for which subgroup analysis of +1q was not presented in trial results, and hence conclusions about the efficacy of the drugs specifically for patients with +1q cannot be ascertained, include pomalidomide and ixazomib. Although GRIFFIN (a Phase 2) study has shown benefit of daratumumab in patients with +1q, other publications of daratumumab randomized phase 3 trials have not reported outcomes of the effect of daratumumab on +1q [30]. Two recent contemporary RCTs that isolated the effect of autologous transplant (DETERMINATION and IFM-2009) did not report +1q [35, 36]. However, in the FORTE trial, the adverse prognostic implications of +1q were not seen in the arm receiving carfilzomib, len, dex, and autologous transplant, indicating a possible role of carfilzomib and autologous transplant in ameliorating the adverse prognostic implications of +1q [24]. We did not find any RCT that enrolled patients exclusively with +1q.
Discussion
In this systematic review of MM RCTs looking at +1q, we find three key findings. Firstly, in all studies that report outcomes in patients with +1q compared to those without, outcomes are poorer in those with +1q. Secondly, all interventions that have shown to be successful in RCTs also demonstrate benefit in those with +1q, with no specific evidence of any one particular therapy being uniquely effective in +1q. Third, we find considerable heterogeneity in the reporting of +1q in the literature and +1q to be inconsistently classified as a poor prognostic factor in subgroup analysis of randomized MM RCTs. Although various other narrative reviews have been conducted on this topic, our systematic review is the first attempt at categorizing reporting of +1q in all MM RCTs, through a systematic search [1, 2, 37, 38], and the knowledge gained from our review can serve as an impetus to standardize reporting in future RCTs.
Our results show that for interventions that were successful in meeting their endpoint in RCTs, the benefit was also seen in the +1q group. Conversely, for RCTs that were negative and did not meet their endpoint, no benefit was seen in the +1q group either for the intervention [33]. Given that such a substantial fraction of patients with MM have +1q, it is only logical that interventions that generally benefit patients with MM also benefit patients with +1q. It is claimed that drugs such as isatuximab may be uniquely efficacious in patients with +1q [39]. Whereas data on the efficacy of daratumumab specific for +1q was not reported in its trials other than the GRIFFIN trial, our overall results indicate that a drug that is generally effective in MM (such as daratumumab), is also likely to be effective in +1q. Further reporting on +1q in trials that have evaluated daratumumab in a randomized fashion would help clarify this. Targeted therapies specific to this subgroup could be an area of further interest, as we found no RCTs exclusively enrolling patients with +1q.
Most RCTs reviewed did not specify the percent of cells needed to harbor +1q to be classified as such. A prospective, non-randomized clinical trial investigated the prognostic implications of the size of the clones of +1q. They divided the patients according to the percentage of monoclonal plasma cells with the mentioned cytogenic alteration (20–50% vs. >50%). This trial did not find a statistically significant difference in PFS or OS between these groups [40]. However, the discovery of a small percentage of cells carrying +1q needs to be interpreted cautiously. This could be due to two different reason. Firstly, this could be a result of test being performed in a non CD138 enriched sample leading to an erroneous result not reflective of a new clone in malignant plasma cells. Secondly, this could represent the emergence of a new non-dominant sub-clone, a development that may be of future clinical significance over time.
Furthermore, we found that +1q was inconsistently reported as a high-risk cytogenetic abnormality. Some trials may report a higher overall proportion of patients with high-risk disease by including all patients +1q [24], whereas others may have a lower proportion of high-risk disease by only including those with other high-risk abnormalities such as deletion 17p or t(4;14). This is particularly noteworthy when comparing outcomes for high-risk diseases across trials and would benefit from standardization.
Data from Emory in a large cohort of 1000 patients treated with triplet induction (bortezomib/lenalidomide/dexamethasone), autologous transplant, and maintenance showed no adverse impact of gain(1q) without concurrent high-risk cytogenetics, but an adverse impact of four or more copies, or when present in conjunction with other cytogenetic features [12]. However, more recent data indicates a more likely direct prognostic impact of gain(1q). This is highlighted by a patient-level meta-analysis of 2,596 patients from three trials (German-speaking Myeloma Multicenter Group (GMMG), GMMG MM5 trial, EudraCT 2010-019173-16) and the Myeloma XI trial) that showed adverse prognostic impact of gain(1q) as well, with no discernible difference from amp(1q) in terms of prognosis [14]. Another meta-analysis of patients from the Myeloma IX and the XI trials also showed similar results- that gain(1q) was significantly associated with shorter PFS and OS compared to normal 1q copy number status. Amp(1q) was also linked to shorter PFS and OS compared to normal copy numbers of 1q, but there was no significant difference compared to gain(1q) [41]. Thus, while amp(1q) has been consistently shown to have an adverse effect on the prognosis as evidenced by the statistically significant worse HR for patients with amp(1q) in ENDURANCE [22, 34], HOVON-65/GMMG-HD4 [23] and FORTE [4], the effect of gain(1q) on prognosis is not clear. Although previous data from Mayo Clinic did not show a difference in median OS in patients with or without +1q [42], recent data shows that the presence of +1q was associated with high tumor burden, an advanced stage of disease and decreased overall survival on a multivariate analysis [43]. A summary of the data to date increasingly demonstrates an adverse prognostic impact of +1q, although the signal on whether the number of abnormal copies of +1q has a further effect on prognosis remains controversial, given conflicting results from the studies mentioned above.
Our results show that most RCTs do not provide granular detail on patients with +1q. As the data indicates, which concurrent cytogenetic abnormalities exist with +1q has a great impact on prognosis. As an example, in an analysis of 737 patients from Spain, +1q by itself did not improve the predictive value of the Revised International Staging System. However, the co-existence of hyperdiploidy with +1q improved the prognosis of patients undergoing stem cell transplant (10-year OS of 62.5% versus 96%) [44].
We recognize that unlike current knowledge that allows us to propose suggestions for future reporting of +1q, authors in earlier trials lacked the benefit of hindsight when studying this cytogenetic abnormality. Early issues with reporting were anticipated, and the diverse reporting approaches we found on our review do not cast a critical light on the efforts of other authors. Based on the evolving knowledge on this topic, we provide recommendations to standardize reporting of +1q in clinical trials. Firstly, the demographic information should clearly document the percentage of patients for whom testing for +1q was performed, and for what proportion of those patients was +1q detected. A clear description of the copy numbers of the 1q chromosome and consistent usage of the words amp to denote 4 or more copies or gain to denote just three copies is paramount. Description of the concurrent cytogenetic abnormalities (hyperdiploidies, or high-risk features) and the proportion of patients should be clearly reported in the demographics table. These subgroups of patients (+1q overall, those with only three copies, those with 4 or more copies, those with isolated +1q with no other abnormalities, those with +1q and other hyperdiploidies, and those with +1q and other high-risk cytogenetic features) represent distinct biology, and pre-planned subgroup analysis of these patients should be reported, allowing future patient-level meta-analysis of granular data. Furthermore, clear definitions of what threshold of cells for which +1q is found will aid in consistent analysis in the future. A recent analysis of 2596 patients from the German group had a cut-off of 10%, which would be a reasonable threshold to utilize for future studies [14]. The HARMONY alliance from Europe, which recently contributed to the development of the Second Revision of the International Staging System (R-2 ISS) of Myeloma represents another pivotal step forward in further understanding the prognostic implications of +1q [7]. Given that new staging systems now incorporate +1q, implementation of these staging systems will lead to improved reporting and understanding of +1q. As our understanding of cytogenetic abnormalities evolves, it is increasingly clear that there is a cumulative risk imparted by having +1q in addition to other cytogenetic abnormalities, and it is encouraging to see recent trials such as the IsKia study of isatuximab, carfilzomib, lenalidomide, dexamethasone vs carfilzomib, lenalidomide, dexamethasone presented at the American Society of Hematology 2023 meeting clearly demarcate outcomes based on the number of high-risk cytogenetic features [45]. Such analysis have been performed for other studies done recently [46, 47].
Although our analysis focused on the prognostic implications of +1q and not deletion 1p, abnormalities of deletion 1p may co-exist with +1q, and may compound the inferior prognostic implications of +1q [48]. Furthermore, biallelic deletion of 1p has been shown to be a particularly poor prognostic factor, based on a retrospective study of 2551 patients [49]. It is crucial that these cytogenetic abnormalities be clearly listed in future studies, and we believe that these abnormalities should be demarcated and their prognostic impact isolated rather than lumped as “chromosome 1 abnormalities”.
Limitations of our study include that some relevant or recent studies may not have been picked up despite searching three datasets. Furthermore, our study also carries the limitations inherited with systemic reviews, such as publication bias (studies with positive results are more likely to be published compared to those with negative results), selection bias, and selective outcome reporting bias.
Conclusion
This systematic review finds significant heterogeneity in +1q reporting in recent RCTs, and an association of +1q with poor outcomes when found. We find that interventions that generally work for patients with MM, also demonstrate efficacy in the +1q subgroup. We propose standardization of +1q reporting to better understand the implications of this abnormality.
References
Hanamura I. Multiple myeloma with high-risk cytogenetics and its treatment approach. Int J Hematol. 2022;115:762–77.
Schmidt TM, Fonseca R, Usmani SZ. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 2021;11:83.
Hanamura I, Stewart JP, Huang Y, Zhan F, Santra M, Sawyer JR, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108:1724–32.
D’Agostino M, Ruggeri M, Aquino S, Giuliani N, Arigoni M, Gentile M, et al. Impact of gain and amplification of 1q in newly diagnosed multiple myeloma patients receiving carfilzomib-based treatment in the forte trial. Blood. 2020;136:38–40.
Yu Y, Brown Wade N, Hwang AE, Nooka AK, Fiala MA, Mohrbacher A, et al. Variability in cytogenetic testing for multiple myeloma: a comprehensive analysis from across the United States. JCO Oncol Pract. 2020;16:e1169–80.
Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33:159–70.
D’agostino M, Cairns DA, Lahuerta JJ, Wester R, Bertsch U, Waage A, et al. Second revision of the International Staging System (R2-ISS) for overall survival in multiple myeloma: a European Myeloma Network (EMN) report within the HARMONY project. J Clin Oncol. 2022;40:3406–18.
Abdallah NH, Binder M, Rajkumar SV, Greipp PT, Kapoor P, Dispenzieri A, et al. A simple additive staging system for newly diagnosed multiple myeloma. Blood Cancer J. 2022;12:21.
Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95:548–67.
Sonneveld P, Avet-Loiseau H, Lonial S, Usmani S, Siegel D, Anderson KC, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127:2955–62.
Usmani SZ, Hoering A, Ailawadhi S, Sexton R, Lipe B, Hita SF, et al. Bortezomib, lenalidomide, and dexamethasone with or without elotuzumab in patients with untreated, high-risk multiple myeloma (SWOG-1211): primary analysis of a randomised, phase 2 trial. Lancet Haematol. 2021;8:e45–e54.
Schmidt TM, Barwick BG, Joseph N, Heffner LT, Hofmeister CC, Bernal L, et al. Gain of Chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J. 2019;9:94.
Costa LJ, Chhabra S, Medvedova E, Dholaria BR, Schmidt TM, Godby KN, et al. Daratumumab, carfilzomib, lenalidomide, and dexamethasone with minimal residual disease response-adapted therapy in newly diagnosed multiple myeloma. J Clin Oncol. 2022;40:2901–12.
Weinhold N, Salwender HJ, Cairns DA, Raab MS, Waldron G, Blau IW, et al. Chromosome 1q21 abnormalities refine outcome prediction in patients with multiple myeloma - a meta-analysis of 2596 trial patients. Haematologica. 2021;106:2754–8.
Mohan M, Gong Z, Ashby TC, Al Hadidi S, Thanendrarajan S, Schinke C, et al. Concomitant deletion of the short arm (Del 1p13. 3) and amplification or gain (1q21) of chromosome 1 by fluorescence in situ hybridization are associated with a poor clinical outcome in multiple myeloma. Cancer. 2023;129:2491–8.
Mohan M, Weinhold N, Schinke C, Thanedrarajan S, Rasche L, Sawyer JR, et al. Daratumumab in high-risk relapsed/refractory multiple myeloma patients: adverse effect of chromosome 1q21 gain/amplification and GEP70 status on outcome. Br J Haematol. 2020;189:67–71.
Mohyuddin GR, Koehn K, Abdallah AO, D WS, Rajkumar SV, Kumar S, et al. Use of endpoints in multiple myeloma randomized controlled trials over the last 15 years: A systematic review. Am J Hematol. 2021;96:690–7.
Moreau P, Dimopoulos MA, Mikhael J, Yong K, Capra M, Facon T, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet. 2021;397:2361–71.
Avet-Loiseau H, Attal M, Campion L, Caillot D, Hulin C, Marit G, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol. 2012;30:1949–52.
Kubo K, Hori M, Ohta K, Handa H, Hatake K, Matsumoto M, et al. Elotuzumab plus lenalidomide and dexamethasone for newly diagnosed multiple myeloma: a randomized, open-label, phase 2 study in Japan. Int J Hematol. 2020;111:65–74.
Grosicki S, Simonova M, Spicka I, Pour L, Kriachok I, Gavriatopoulou M, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. Lancet. 2020;396:1563–73.
Kumar SK, Jacobus SJ, Cohen AD, Weiss M, Callander N, Singh AK, et al. Carfilzomib or bortezomib in combination with lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma without intention for immediate autologous stem-cell transplantation (ENDURANCE): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2020;21:1317–30.
Neben K, Lokhorst HM, Jauch A, Bertsch U, Hielscher T, van der Holt B, et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood. 2012;119:940–8.
Gay F, Musto P, Rota-Scalabrini D, Bertamini L, Belotti A, Galli M, et al. Carfilzomib with cyclophosphamide and dexamethasone or lenalidomide and dexamethasone plus autologous transplantation or carfilzomib plus lenalidomide and dexamethasone, followed by maintenance with carfilzomib plus lenalidomide or lenalidomide alone for patients with newly diagnosed multiple myeloma (FORTE): a randomised, open-label, phase 2 trial. Lancet Oncol. 2021;22:1705–20.
Richard S, Chari A, Delimpasi S, Simonova M, Spicka I, Pour L, et al. Selinexor, bortezomib, and dexamethasone versus bortezomib and dexamethasone in previously treated multiple myeloma: Outcomes by cytogenetic risk. Am J Hematol. 2021;96:1120–30.
Panopoulou A, Cairns DA, Holroyd A, Nichols I, Cray N, Pawlyn C, et al. Optimizing the value of lenalidomide maintenance by extended genetic profiling: an analysis of 556 patients in the Myeloma XI trial. Blood. 2023;141:1666–74.
Richardson PG, Perrot A, San-Miguel J, Beksac M, Spicka I, Leleu X, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): follow-up analysis of a randomised, phase 3 study. Lancet Oncol. 2022;23:416–27.
Jackson GH, Pawlyn C, Cairns DA, de Tute RM, Hockaday A, Collett C, et al. Carfilzomib, lenalidomide, dexamethasone, and cyclophosphamide (KRdc) as induction therapy for transplant-eligible, newly diagnosed multiple myeloma patients (Myeloma XI+): interim analysis of an open-label randomised controlled trial. PLoS Med. 2021;18:e1003454.
Dimopoulos MA, Dytfeld D, Grosicki S, Moreau P, Takezako N, Hori M, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N Engl J Med. 2018;379:1811–22.
Chari A, Kaufman JL, Laubach JP, Sborov DW, Reeves B, Rodriguez C, et al. Daratumumab plus lenalidomide, bortezomib, and dexamethasone (D-RVd) in transplant-eligible newly diagnosed multiple myeloma (NDMM) patients (Pts): final analysis of griffin among clinically relevant subgroups. Blood. 2022;140:7278–81.
Zweegman S, van der Holt B, Mellqvist UH, Salomo M, Bos GM, Levin MD, et al. Melphalan, prednisone, and lenalidomide versus melphalan, prednisone, and thalidomide in untreated multiple myeloma. Blood. 2016;127:1109–16.
Morgan GJ, Davies FE, Gregory WM, Bell SE, Szubert AJ, Navarro Coy N, et al. Cyclophosphamide, thalidomide, and dexamethasone as induction therapy for newly diagnosed multiple myeloma patients destined for autologous stem-cell transplantation: MRC Myeloma IX randomized trial results. Haematologica. 2012;97:442–50.
Jenner MW, Pawlyn C, Davies FE, Menzies T, Hockaday A, Olivier C, et al. The addition of vorinostat to lenalidomide maintenance for patients with newly diagnosed multiple myeloma of all ages: results from ‘Myeloma XI’, a multicentre, open-label, randomised, phase III trial. Br J Haematol. 2023;201:267–79.
Kapoor P, Schmidt T, Jacobus S, Wei Z, Fonseca R, Callander NS, et al. OAB-052: Impact of chromosome 1 abnormalities on newly diagnosed multiple myeloma treated with proteasome inhibitor, immunomodulatory drug, and dexamethasone: analysis from the ENDURANCE ECOG-ACRIN E1A11 trial. Clin Lymphoma Myeloma Leukemia. 2021;21:S33–S4.
Richardson PG, Jacobus SJ, Weller EA, Hassoun H, Lonial S, Raje NS, et al. Triplet therapy, transplantation, and maintenance until progression in myeloma. N Engl J Med. 2022;387:132–47.
Attal M, Lauwers-Cances V, Hulin C, Leleu X, Caillot D, Escoffre M, et al. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med. 2017;376:1311–20.
Luo S, Su T, Zhou X, Hu WX, Hu J. Chromosome 1 instability in multiple myeloma: aberrant gene expression, pathogenesis, and potential therapeutic target. FASEB J. 2022;36:e22341.
Hanamura I. Gain/amplification of chromosome arm 1q21 in multiple myeloma. Cancers. 2021;256:13.
Martin T, Richardson PG, Facon T, Moreau P, Perrot A, Spicka I, et al. Primary outcomes by 1q21+ status for isatuximab-treated patients with relapsed/refractory multiple myeloma: subgroup analyses from ICARIA-MM and IKEMA. Haematologica. 2022;107:2485–91.
An G, Xu Y, Shi L, Shizhen Z, Deng S, Xie Z, et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica. 2014;99:353–9.
Shah V, Sherborne AL, Walker BA, Johnson DC, Boyle EM, Ellis S, et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular profiles of 1905 trial patients. Leukemia. 2018;32:102–10.
Fonseca R, Van Wier SA, Chng WJ, Ketterling R, Lacy MQ, Dispenzieri A, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20:2034–40.
Abdallah N, Greipp P, Kapoor P, Gertz MA, Dispenzieri A, Baughn LB, et al. Clinical characteristics and treatment outcomes of newly diagnosed multiple myeloma with chromosome 1q abnormalities. Blood Adv. 2020;4:3509–19.
Minguela A, Vasco-Mogorron MA, Campillo JA, Cabanas V, Remigia MJ, Berenguer M, et al. Predictive value of 1q21 gain in multiple myeloma is strongly dependent on concurrent cytogenetic abnormalities and first-line treatment. Am J Cancer Res. 2021;11:4438–54.
Gay F, Roeloffzen W, Dimopoulos MA, Rosiñol L, van der Klift M, Mina R, et al. Results of the Phase III Randomized Iskia Trial: Isatuximab-Carfilzomib-Lenalidomide-Dexamethasone Vs Carfilzomib-Lenalidomide-Dexamethasone As Pre-Transplant Induction and Post-Transplant Consolidation in Newly Diagnosed Multiple Myeloma Patients. Blood. 2023;142:4.
Costa LJ, Chhabra S, Medvedova E, Dholaria BR, Schmidt TM, Godby KN, et al. Minimal residual disease response-adapted therapy in newly diagnosed multiple myeloma (MASTER): final report of the multicentre, single-arm, phase 2 trial. Lancet Haematol. 2023;10:e890–e901.
Callander N, Silbermann R, Kaufman JL, Godby KN, Laubach JP, Schmidt TM, et al. Analysis of transplant-eligible patients (Pts) who received frontline daratumumab (DARA)-Based quadruplet therapy for the treatment of newly diagnosed multiple myeloma (NDMM) with high-risk cytogenetic abnormalities (HRCA) in the Griffin and Master studies. Blood. 2022;140:10144–7.
Mohan M, Gong Z, Ashby TC, Al Hadidi S, Thanendrarajan S, Schinke C, et al. Concomitant deletion of the short arm (del(1p13.3)) and amplification or gain (1q21) of chromosome 1 by fluorescence in situ hybridization are associated with a poor clinical outcome in multiple myeloma. Cancer. 2023;129:2491–8.
Schavgoulidze A, Talbot A, Perrot A, Cazaubiel T, Leleu X, Manier S, et al. Biallelic deletion of 1p32 defines ultra-high-risk myeloma, but monoallelic del(1p32) remains a strong prognostic factor. Blood. 2023;141:1308–15.
Jackson GH, Davies FE, Pawlyn C, Cairns DA, Striha A, Collett C, et al. Response-adapted intensification with cyclophosphamide, bortezomib, and dexamethasone versus no intensification in patients with newly diagnosed multiple myeloma (Myeloma XI): a multicentre, open-label, randomised, phase 3 trial. Lancet Haematol. 2019;6:e616–e29.
Boyd KD, Ross FM, Chiecchio L, Dagrada GP, Konn ZJ, Tapper WJ, et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC Myeloma IX trial. Leukemia. 2012;26:349–55.
Acknowledgements
The authors thank Dr Lee Wade and Dr Muhammad Aziz from the University of Toledo, Ohio for their assistance in creating a search strategy. There was no funding for this study.
Author information
Authors and Affiliations
Contributions
GRM conceived the research idea. GRM and KAK performed an initial search and screening of studies. KN collected data, and GRM and KN performed data analysis. KN and GRM wrote the first draft of the manuscript. All authors provided critical input on the manuscript, the methodology of the study, reviewed the initial draft of manuscript and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
KN reports no conflict of interest. GRM reports receiving royalties for writing from MashupMD. GGF reports no conflict of interest. RD reports no conflict of interest. TS has served as a consultant for BiolineRx, Janssen, and Sanofi. RF serves the role of consulting: AbbVie, Adaptive Biotechnologies, AMGEN, AZeneca, Bayer, Binding Site, BMS (Celgene), Millenium Takeda, Jansen, Juno, Kite, Merck, Pfizer, Pharmacyclics, Regeneron, and Sanofi; Scientific Advisory Boards: Adaptive Biotechnologies, Caris Life Sciences, and Oncotracker; Board of Directors: Antegene (for profit), AZBio (not for Profit); and Patent for FISH in MM - ~$2000/year. RC serves the role of consulting for Sanofi, Janssen and Adaptive. KAK reports no conflicts of interest. MM is supported by AHW CTSI KL 2 Award. Institutional Research Funding: Sanofi S.A, GlaxoSmithKline plc, Takeda Pharmaceutical Company, Ionis Pharmaceuticals, Bristol-Myers Squibb Company, Celgene Corporation and Amgen Inc.Consultancy: Sanofi S.A. Honorarium: Sanofi S.A., Blood Cancer Today, MashupMD, and MJH life sciences. HM is supported by an early career award from Hamilton Health Sciences. Honoraria: Pfizer, Janssen, Takeda, GSK, BMS, Sanofi, FORUS, and Amgen. Research funding: Janssen. LJC has research support from Janssen, AbbVie, BMS, Genentech, Amgen, Gracel, and Caribou and consulting role with Amgen, Janssen, BMS, AbbVie, and Pfizer. DS reports consulting/Advisory Board for Janssen, Sanofi, Pfizer, Abbvie, BMS and GSK.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neupane, K., Fortuna, G.G., Dahal, R. et al. Alterations in chromosome 1q in multiple myeloma randomized clinical trials: a systematic review. Blood Cancer J. 14, 20 (2024). https://doi.org/10.1038/s41408-024-00985-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-024-00985-0
