
Abstract
Systemic light-chain (AL) amyloidosis is a rare and debilitating disease. Advances have been made in new treatments in recent years, yet real-world data on the management of the disease are scarce. EMN23 is a retrospective, observational study of patients who initiated first-line treatment in 2004–2018 in Europe, presenting the demographics, clinical characteristics, treatment patterns, and outcomes, from 4480 patients. Regimens based on bortezomib were the most frequently used as first-line therapy; only 6.2% of the patients received autologous stem cell transplant. Hematologic responses improved post-2010 (67.1% vs 55.6% pre-2010). The median overall survival (OS) was 48.8 (45.2–51.7) months; 51.4 (47.3–57.7) months pre-2010 and 46.7 (41.3–52.2) months post-2010. Early mortality was 13.4% and did not improve (11.4% vs 14.4% pre- and post-2010); furthermore, it remained high in patients with advanced cardiac disease (over 39% for stage IIIb). There was a significant improvement for stage IIIa (14.2 vs 30.7 months, p = 0.0170) but no improvement for stage IIIb patients (5.0 vs 4.5 months). This European real-world study of AL-amyloidosis emphasizes the unmet needs of early diagnosis, and the lack of improvement in survival outcomes of the frail stage IIIb population, despite the introduction of new therapies in recent years.
Similar content being viewed by others
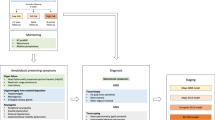


Introduction
Systemic light chain (AL) amyloidosis is a rare disease caused by misfolded clonal immunoglobulins produced by a plasma cell or, less often, a B cell clone [1, 2]. The immunoglobulin oligomers are toxic to tissues and organs [3], and amyloid deposition disrupts their normal function [4], leading to debilitating and commonly lethal complications [1, 2]. AL amyloidosis is associated with non-specific symptoms and substantial heterogeneity in clinical presentation [1, 2]. Correct diagnosis requires a high degree of suspicion and specialized techniques [5, 6], while the multisystemic nature of this disease requires multidisciplinary management and significant healthcare resources use [1, 2]. As a result, diagnosis and treatment of AL amyloidosis is often delayed [7] until after irreversible organ damage has occurred.
Current treatments for AL amyloidosis target the plasma/B cell clone aiming to reduce the production of toxic light chains. Until recently there were no approved therapies for AL amyloidosis and this disease was managed with adapted off-label therapies indicated for multiple myeloma or Waldenström’s Macroglobulinemia [8] following advances in these areas. New treatments for the underlying clonal disease in the past 10–15 years (such as bortezomib) [9,10,11,12,13,14,15,16,17] changed the therapeutic management of AL amyloidosis, but treatment patterns remained heterogeneous.
Because of the above, the approach to the diagnosis and management of AL amyloidosis has been based mostly on single referral center studies and much less on prospective or randomized studies [15, 17,18,19]. Data from tertiary referral centers from US indicate improvements over the past decades [20, 21] but a comprehensive, real-world overview is lacking. This is especially relevant for Europe, which is characterized by variations in the organization of the health care systems and allocation of healthcare resources in the different countries, including availability of diagnostic methods and approved therapies.
Methods
Study design
EMN23 is a retrospective, observational study aiming to collect real-world evidence for the management of systemic AL amyloidosis in patients who initiated first-line treatment from January 2004 to December 2018 in Europe. The study was sponsored by the European Myeloma Network (EMN). The duration from initiation until the database lock was approximately 24 months (Q2-2019 to Q2-2021). Collection of data and analysis of the main outcomes of interest consisted of patient demographic and clinical characteristics at diagnosis, treatments (at first and second line), and survival outcomes (overall survival (OS); time on treatment (ToT)) analyzed by treatment initiation period, cardiac stage, and hematologic responses. Landmark analyses on time to event endpoints for hematologic responses at 3 and 6 months, were performed for patients with available data.
One of the study aims was to investigate potential differences between the cohorts of patients who initiated first-line treatment in 2004–2010 (pre-2010) and 2011–2018 (post-2010), in order to depict changes in disease management secondary to the wider introduction of bortezomib-based regimens after 2010, especially of bortezomib, cyclophosphamide, and dexamethasone (VCD), in the everyday clinical practice [11, 12, 14, 22].
Patient data were collected from ten European countries: Austria, Czech Republic, France, Germany, Greece, Italy, the Netherlands, Portugal, Spain, and the United Kingdom (UK). According to local regulations, data were retrieved from patients’ medical records (paper/electronic and electronic databases) and were transferred directly to the study database (Germany, Greece, Italy, Spain, and the UK) and/or via an electronic case report form (all countries except Italy, Spain, and the UK).
The hematologic responses were assessed by investigators according to the protocol as per consensus criteria. In best hematologic responses, and in 3- and 6-month landmark analyses, patients who died less than 3 months after first-line treatment initiation and did not have a post-baseline assessment, and who were to be included in the landmark analysis were categorized under the “no-response” group. Patients who participated in clinical trials or patients who were under the “no-response” group were not included in the estimation of the hematologic response rates. Organ involvement was assessed according to criteria defined in each chronological period [23, 24].
The study protocol and amendments were reviewed by an Independent Ethics Committee or Institutional Review Board as appropriate per local laws and the study was conducted in accordance with Good Pharmacoepidemiology Practice (GPP) guidelines. Data collection and source data verification were always performed in accordance with local requirements and legislations.
Statistical analysis
Descriptive statistics for continuous variables included mean, standard deviation, median, Q1, Q3 quartile, minimum value, maximum value and 95% CI. Categorical variables were described using absolute frequencies and proportions. Significant differences between subgroups were examined using Mann-Whitney non-parametric test continuous variables and chi-square tests for categorical variables. Descriptive statistics for time-to-event variables (OS, ToT) were estimated using the Kaplan–Meier method, while to compare the survival distributions between subgroups, log rank test was used, based on the nature of the data.
Overall Response Rate (ORR) was defined as the proportion of patients with a best hematologic response of partial response (PR) or better. Patients without available response data were excluded from the denominator.
OS was defined as the time from treatment initiation (first treatment dose; the date of ASCT was used for the patients who underwent upfront ASCT) to death due to any cause or to the last follow-up (censored).
ToT was defined as the time from treatment initiation to discontinuation of first line treatment for any reason (e.g., scheduled treatment completion, disease progression (PD), toxicity, death, lost to follow-up). Patients who did not discontinue treatment were censored at the date of the last follow-up or on 31 December 2018 (whichever occurred first).
Although no formal hypothesis testing was performed, comparisons between groups were statistically assessed to provide a clearer evaluation of the results. All reported p values were two-sided, and p < 0.05 was considered statistically significant. All analyses were performed using SAS (v9.4 SAS Institute, Cary, NC). This study is registered with ClinicalTrials.gov, NCT04937777.
Results
Overall, 4480 patients were enrolled; Table S1 lists the participating centers. Patients who participated in clinical trials (N = 177 for first-line treatment and N = 38 for second line) were only included in the analyses for patient characteristics. Table 1 presents the demographic and clinical characteristics at diagnosis for patients who started first-line treatment in 2004–2018 (N = 4480), pre-2010 (N = 1415), and post-2010 (N = 3065).
In 967 (21.6%) patients with available data, the median time from symptoms to diagnosis was 5.1 months (5.4 and 5.0 months pre- and post-2010, respectively) and the median time from diagnosis to initiation of first-line treatment was 0.7 months.
Clonal disease was immunoglobulin light chain only in 41.0% while 4.4% of patients had IgM-related AL amyloidosis (Table S2). Heart (67.9%) and kidneys (66.1%) were the most frequently involved organs. Most patients (74.1%) had ≤2 organs involved and 34.3% had only one organ involved.
Cardiac stage (Mayo 2004 with European modification) [25, 26] at the time of diagnosis was available for 3775 (84.3%) patients, of whom 696 (18.4%) were stage I, 1413 (37.4%) stage II, 1003 (26.6%) stage IIIa, and 663 (17.6%) stage IIIb. Per Mayo 2012 staging [27] (available for 3065 patients), 19.8%, 24.2%, 29.2%, and 26.7% were rated as stage I, II, III, and IV, respectively. Cardiac stage distribution at diagnosis was similar for the two chronological periods (Table 1).
First-line treatment
Table 2 depicts regimen categories and the most common individual regimens that were used as first-line therapies. Bortezomib-based regimens (54.8%, N = 2454) were the most common first-line therapy, followed by chemotherapy (19.4%, N = 869) [mostly melphalan with dexamethasone (MDex)], and immunomodulatory drug (IMiD)-based regimens (N = 488, 10.9%). High-dose melphalan (HDM) with ASCT was used at first line in 6.2% (N = 278); 74.1% (N = 206) received induction prior to HDM-ASCT (bortezomib-based in 68.0% and IMiDs in 18.0%). First-line treatments among participating countries are shown in Table S3.
There was a significant change of first-line treatment type in the two chronological periods. Pre-2010, most patients received chemotherapy (in 42.6%, MDex in 32.4%), followed by IMiD-based regimens (30.3%), and bortezomib-based therapy (in 11.5%) while, after 2010, bortezomib-based regimens were used in the majority of patients (74.7%); VCD was the most frequently used bortezomib-based regimen (20.6% of all regimens post-2010), followed by bortezomib and dexamethasone (VD, 10.5%) and bortezomib with MDex (VMD, 5.4%). ASCT was performed in 7.6% of patients pre-2010 and in 5.5% post-2010.
Pre-2010, chemotherapy (mostly MDex) was the most frequently used regimen across all cardiac stages (range: 39.7% for stage I to 50.7% for stage IIIa). Post-2010, bortezomib-based regimens, and VCD in particular, were used in the majority of patients across all cardiac stages (range: 64.8% for stage I to 82.3% for stage IIIb) (Table S4). In patients with kidney involvement bortezomib-based therapy was used in 54.9%, chemotherapy in 19.2%, and IMiDs in 10.4%. Among patients with nerve involvement, 44.0% received bortezomib-based therapy, whereas 31.9% and 7.4% were given chemotherapy and IMiDs, respectively (Table S5). Most patients with IgM-related amyloidosis received rituximab-based regimens at first line, 33.3% (N = 23) and 39.8% (N = 51), pre- and post-2010, respectively (Table S6).
Response to first-line treatment
After first-line therapy, 63.4% of patients with available information achieved at least hematologic PR (including CR: 23.2% and VGPR: 21.2%, Table S7). The hematologic response rate was 68.4% with bortezomib-based regimens (CR: 26.1%, VGPR: 25.7%); 49.0% (CR: 14.3%, VGPR: 13.4%) with chemotherapy, and 53.7% (CR: 17.5%, VGPR: 9.3%) with IMiD-based regimens (Table S8). Hematologic response rates were similar among bortezomib-based regimens: 63.7% (CR: 19.2%, VGPR: 27.7%) for VCD, 62.1% (CR: 12.7%, VGPR: 27.4%) for VD, and 57.3% (CR: 20.5%, VGPR: 25.1%) for VMD. In ASCT-treated patients, 88.0% achieved ≥PR (CR: 43.4%, VGPR: 27.0%) (Table S9). The proportion of patients who failed to achieve a response or died within 3 months after starting therapy without any response assessment, pre- and post-2010, was 35.6% and 31.3% with bortezomib-based, 50.5% and 52.6% with chemotherapy, 43.9% and 65.4% with IMiD-based regimens, and 24.8% and 3.7% with ASCT (Table S10).
Hematologic response rates significantly improved post-2010 (67.1% vs 55.6% pre-2010, p < 0.001), with deeper responses (25.0% vs 19.5% CRs and 25 4% vs 12.4% VGPRs). As a consequence of the improvement in hematologic responses non-responding patients significantly decreased from 44.4% (SD: 16.7%, PD: 19.6%) pre-2010 to 32.9% (SD: 14.8%, PD: 9.4%) post-2010 (Table S11).
Among patients with available information, hematologic response was achieved in 58.6% of patients at 3 months from the start of first-line therapy (1594/2720) and in 70.1% of patients at 6 months (1633/2329). Three-month ORR was 45.6% (CR + VGPR: 24.8%) pre-2010 and 63.6% (CR + VGPR: 38.9) post-2010, and 6-month ORR was 58.3% (CR + VGPR: 31.0%) and 75.1% (CR + VGPR: 54.5%), respectively (Table S12).
Hematologic response rates at 3 months were 64.1% for bortezomib-based regimens, 42.2% for chemotherapy and 42.8% for IMiD-based regimens. At the 6-month landmark, response rates for patients with available information were 74.5% for bortezomib-based regimens, 54.1% for chemotherapy, and 55.1% for IMiD-based regimens (Table S13).
Median ToT for first-line therapy was 4.7 (range: 4.4–5.0) months; similar in the pre- and post-2010 era (4.7 vs 4.8 months) and was shorter in patients with more advanced disease stages (Table S14).
Second-line therapy
Overall, 1759 patients received second-line treatment. After the second-line therapy, 58.7% of patients with available information achieved at least hematologic PR (including CR: 15.3% and VGPR: 20.1%, Table S7). Median time from start of first line to second-line therapy was 9.5 (0.6–165.7) months and was shorter post-2010 (8.6 vs 11.3 months). Pre-2010, bortezomib-based regimens were the most frequent second-line treatment (N = 349, 45.0%) while post-2010 IMiD-based regimens became the major choice (N = 410, 41.7%) (Table 3). Notably, 15.6% of patients who received bortezomib at first line and required a second-line treatment, received bortezomib-based therapy again (Table S15). Per cardiac stage, 50.8% of patients with stage I, 46.6% with stage II, 33.1% with stage IIIa, and only 18.6% with stage IIIb received second-line treatment. Among patients who received first-line ASCT and required second-line therapy (N = 122), 30.3% received bortezomib-based, and 33.6% IMiD-based regimens. Salvage therapy for patients who failed to respond to primary therapy was based on bortezomib and IMiDs in most cases, with bortezomib used in most patients after primary failure to IMIDs (55.7%), and IMiDs (51.8%) in most patients after primary failure to bortezomib.
Survival
The median follow-up for the whole cohort was 54.5 (51.4–57.8) months (Table S16). The median OS was 48.8 (45.2–51.7) months; 51.4 (47.3–57.7) months for the pre-2010 group and 46.7 (41.3–52.2) months for the post-2010 group (Table S17).
Early mortality (i.e., within 3 months from start of therapy) was 13.4%, did not improve (11.4% in the pre-2010 and 14.4% in the post-2010 era, Table S18) and was higher in patients with severe cardiac disease in both pre-2010 and post-2010 periods: 1.1% and 2.1% for stage I patients, 7.8% and 7.7% for stage II, 22.7% and 15.6% for stage IIIa, and 39.3% and 39.8% for stage IIIb, respectively (Table 4). The median OS for stage I patients was 116.5 months for the pre-2010 group and was not reached for post-2010 patients (log-rank p = 0.670); for stage II patients, median OS was 47.3 months pre-2010 and 67.0 months post-2010 (log-rank p = 0.0539); for stage IIIa patients was 14.2 and 30.7 months, respectively (log-rank p = 0.0170) and for stage IIIb patients, 5.0 and 4.5 months (log-rank p = 0.530), respectively (Fig. 1). The OS per revised Mayo 2012 was not reached in stage I patients, was 69.7 in stage II patients, 24.7 in stage III patients, and 10.6 months for stage IV patients (Fig. S1).

Stage I, Stage II, Stage IIIa and Stage IIIb represent the comparison of survival curves in the pre- and post-2010, separately for the respective patient subgroups defined by Mayo 2004/European stage at diagnosis.
The OS was similar between regimen groups (45.3 vs 48.6 vs 41.1 months for bortezomib-, IMiD-, and chemotherapy-based therapy, respectively), while the 3-month OS rate of patients undergoing ASCT was 97.8%. Median OS for patients with stage I disease was not reached for bortezomib-based regimens and was 81.1 and 88.5 months for IMiD-based and chemotherapy regimens, respectively. For stage II, the median OS was 67.0 vs 39.4 vs 44.8 and for stage IIIa 32.8 vs 7.3 vs 14.3 for bortezomib-based, IMiD-based, and chemotherapy regimens, respectively. However, in patients with stage IIIb disease median OS was poor across all regimen groups (4.6 vs 5.3 vs 4.6 respectively) (Table S19). The median OS in patients with IgM-related AL amyloidosis was 39.7 months (51.7 months pre-2010 and 29.8 months post 2010 era, p = 0.2772; Table S20).
To evaluate the effect of early hematologic responses on OS we performed a landmark analysis at 3 and at 6 months after treatment initiation. In 2711 evaluable patients the median OS for patients achieving CR at 3 months was 109.4 (range: 97.3–149.7) months, 82.8 (range: 66.0–103.7) months with VGPR, 61.7 months with PR (range: 52.7–69.8), and 21.0 (range: 16.8–25.9) months for those who did not achieve a response (p < 0.0001 by log-rank; Fig. 2). At 6 months, the median OS for patients (N = 2322) with CR was 142.6 (range: 104.2−NR) months vs 78.2 months for VGPR (range: 64.5–100.8), 59.9 (range: 51.8–69.8) months for PR and 20.2 (range: 14.8–24.3) months for those who did not achieve a response (p < 0.0001 by log-rank; Fig. S2).

CR complete response, PD progressive disease, PFS progression-free survival, PR partial response, SD stable disease, VGPR very good partial response.
Analysis by hematologic response at 3 months per Mayo2004/European stage showed that patients with stage I or II disease achieving CR did not reach a median OS, while for patients with stages IIIa and IIIb, median OS was 56.2 and 47.0 months, respectively. The respective median OS for patients with stage I, II, IIIa, and IIIb disease achieving VGPR was 81.1, 111.6, 60.5 and 35.7 months, and 83.5, 70.2, 44.6, and 18.5 months for those with PR. Finally, patients failing to achieve a hematologic response by 3 months had a median OS of 96.4, 23.3, 8.9, and 3.3 months. The between-response difference in OS was statistically significant for all stages (p < 0.001 by log-rank; Fig. S3).
Discussion
To the best of our knowledge, EMN23 is the largest study assessing real-word characteristics and outcomes of patients with systemic AL amyloidosis. The analysis of 4480 patient records provides a mosaic that is representative of the heterogeneity of this disease and the challenges associated with its management.
For AL amyloidosis, due to the difficulties associated with the diagnosis and management of the patients, a substantial proportion of them are treated in referral centers, often when their disease has advanced. At the same time, large-scale real-world data can only be collected from Sites with the relevant expertise, as these centers are usually the ones which have the required resources and specialized knowledge to perform systematic and reliable data collection.
Delayed diagnosis remains a major challenge in AL amyloidosis. Despite advances in diagnosis (introduction of serum free light chain measurement and the use of cardio-biomarkers), time to diagnosis did not improve, with half of the patients being diagnosed more than 5 months after the first symptoms, often at an advanced cardiac stage, with more than one third presenting with stage III disease. These observations strongly support the need to further increase disease awareness and invest in the screening of high-risk populations.
Early mortality remained high and essentially unchanged within more than 15 years of therapeutic advances and the introduction of bortezomib, reflecting the delayed diagnosis and the advanced cardiac disease at treatment initiation. These real-world outcomes differ from those published from tertiary referral centers in the United States, where improvements in survival have been observed [20, 21]. The more extensive use of ASCT in these experienced centers also indicates a more selected population.
An unmet need is to improve the outcomes of stage IIIb patients, in whom early mortality remains at 40%. New treatment strategies, beyond clonal-targeted therapy, are needed; however, early and deep hematologic response remains the most effective strategy and can improve survival (OS of stage IIIa and IIIb complete responders at 3 months was 56.2 and 47.0 months, respectively). Unfortunately, the optimal regimen for this frail population is yet to be developed. In contrast, the prognosis of stage I patients is good; these patients may also benefit from intensive therapies. ASCT-related mortality was low (3-month OS rate 97.8%) indicating that in low-risk and carefully selected patients, ASCT is also safe [28]. Patients with stage II and stage IIIa disease seem to benefit the most from new therapies; OS in this patient population improved after 2010 (although it did not reach statistical significance for stage II), probably due to the introduction of bortezomib. These patients are fitter and tolerate bortezomib better than stage IIIb patients, while they may still have reversible organ damage.
Bortezomib-based regimens were the most commonly used after 2010 and the variations (VCD, VD, and VMD) had similar efficacy. Responses to VCD and the other bortezomib-based regimens were lower than in clinical trials, probably due to the exclusion of frail, and of stage IIIb patients from prospective clinical trials [14, 16,17,18, 29]. These results highlight the importance of real-world data in the evaluation of treatments outside the strict context of clinical trials.
Importantly, first-line therapy, even with bortezomib-based regimens, was associated with primary failure in one in three patients and approximately half achieved deep hematologic responses. Management of patients, who do not respond to first-line therapy is challenging since available second-line options (such as IMiDs) show lower efficacy and are less well tolerated than in myeloma patients [13, 30]. Recent advances with the more extensive use of daratumumab or venetoclax in some selected patients have improved treatment options in the relapsed/refractory setting but, in our study, stage IIIb patients had lower probability to receive a salvage therapy: many died early or could not tolerate the available medication. Thus, for such patients it seems that there is only one chance to give an effective therapy and is therefore logical to use the most effective one at treatment initiation, without delay. The recently approved Daratumumab-VCD is associated with a significant improvement in CR and VGPR rates in patients with stage IIIa disease [18], but no data have been published in a journal so far for stage IIIb patients. The real-world efficacy of this new regimen remains also to be assessed.
Second-line therapy followed the evolution of myeloma therapies and the extensive use of bortezomib at first line. Class switch was a common strategy; re-use of successful therapies was also applied and of bortezomib in particular. To our knowledge such data have not been reported so far and are of clinical relevance, even though the treatment options continue to expand.
This real-world study highlighted the prognostic value of the currently used staging systems (modified Mayo 2004 and revised Mayo 2012), of current hematologic response criteria and of early evaluation of the hematologic responses [24]. To the best of our knowledge, this is the first real-world validation of the importance of rapid hematologic responses. Furthermore, the importance of rapid hematologic responses for patients with more advanced disease is emphasized, supporting the primary use of the most effective therapies and early regimen switch in non-responding patients.
Even though deeper hematologic responses are associated with improved survival, and despite the fact that these were more frequent and rapid in the post-2010 period, the overall survival did not seem to improve significantly. However, the benefit in survival post-2010 may be underestimated as potential improvements in the OS of stage I patients may be masked by the shorter follow-up time in the more recent cohort.
Limitations of the study include the heterogeneity of the cohort, lack of complete datasets for several patients, non-centrally evaluated hematologic response, change of response criteria over time (in 2005 and after 2012 [23, 24]), and difficulties in the assessment of organ responses. All these limitations are inherent to registry studies which depict the practice at the time of data recording, as well as the dynamic changes that occurred within 15 years. Nonetheless, real-world data provide a valuable guide for the design of clinical studies that evaluate therapies, highlight the deficiencies in specific aspects of disease management and can guide clinical research and the allocation of valuable health care resources.
This large real-world study captures the evolution of management of AL amyloidosis in Europe and provides valuable data on the outcomes of this rare disease. The results point to the unmet needs of early diagnosis, management of high-risk disease and of primary treatment failure. Based on the real-world evidence obtained from EMN23, efforts are required to improve early detection of AL amyloidosis; this can be realized through the intensification of education of the medical community to increase disease awareness, new biomarkers to identify high-risk groups, the design and evaluation of screening programs for high-risk populations, and the development of new diagnostic tools. Efforts are also required to develop safe but highly and rapidly effective anti-clonal therapies, while awaiting the results of studies that currently evaluate amyloid-targeting therapies.
Data availability
All data mentioned in the main text is accompanied by the relevant tables or figures in the “Results” section or in the Supplementary material. Individual patient data will not be shared.
References
Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387:2641–54.
Merlini G, Dispenzieri A, Sanchorawala V, Schönland SO, Palladini G, Hawkins PN, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Prim. 2018;4:38.
Shi J, Guan J, Jiang B, Brenner DA, Monte FD, Ward JE, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci USA. 2010;107:4188–93.
Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29:1924–33.
Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR 3rd, Dogan A. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–9.
Brambilla F, Lavatelli F, Di Silvestre D, Valentini V, Rossi R, Palladini G, et al. Reliable typing of systemic amyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood. 2012;119:1844–7.
Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light chain amyloidosis: patient experience survey from the Amyloidosis Research Consortium. Adv Ther. 2015;32:920–8.
Palladini G, Merlini G. How I treat AL amyloidosis. Blood. 2022;139:2918–30.
Kastritis E, Anagnostopoulos A, Roussou M, Toumanidis S, Pamboukas C, Migkou M, et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007;92:1351–8.
Kastritis E, Wechalekar AD, Dimopoulos MA, Merlini G, Hawkins PN, Perfetti V, et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. J Clin Oncol. 2010;28:1031–7.
Venner CP, Lane T, Foard D, Rannigan L, Gibbs SDJ, Pinney JH, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119:4387–90.
Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, Spong J, Reeder CB, et al. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood. 2012;119:4391–4.
Kastritis E, Terpos E, Roussou M, Gavriatopoulou M, Pamboukas C, Boletis I, et al. A phase 1/2 study of lenalidomide with low-dose oral cyclophosphamide and low-dose dexamethasone (RdC) in AL amyloidosis. Blood. 2012;119:5384–90.
Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126:612–5.
Sanchorawala V, Palladini G, Kukreti V, Zonder JA, Cohen AD, Seldin DC, et al. A phase 1/2 study of the oral proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis. Blood. 2017;130:597–605.
Kastritis E, Dialoupi I, Gavriatopoulou M, Roussou M, Kanellias N, Fotiou D, et al. Primary treatment of light-chain amyloidosis with bortezomib, lenalidomide, and dexamethasone. Blood Adv. 2019;3:3002–9.
Kastritis E, Leleu X, Arnulf B, Zamagni E, Cibeira MT, Kwok F, et al. Bortezomib, melphalan, and dexamethasone for light-chain amyloidosis. J Clin Oncol. 2020;38:3252–60.
Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385:46–58.
Dispenzieri A, Kastritis E, Wechalekar AD, Schönland SO, Kim K, Sanchorawala V, et al. A randomized phase 3 study of ixazomib-dexamethasone versus physician’s choice in relapsed or refractory AL amyloidosis. Leukemia. 2022;36:225–35.
Muchtar E, Gertz MA, Kumar SK, Lacy MQ, Dingli D, Buadi FK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017;129:2111–9.
Staron A, Zheng L, Doros G, Connors LH, Mendelson LM, Joshi T, et al. Marked progress in AL amyloidosis survival: a 40-year longitudinal natural history study. Blood Cancer J. 2021;11:139.
Jaccard A, Comenzo RL, Hari P, Hawkins PN, Roussel M, Morel P, et al. Efficacy of bortezomib, cyclophosphamide and dexamethasone in treatment-naïve patients with high-risk cardiac AL amyloidosis (Mayo Clinic stage III). Haematologica. 2014;99:1479–85.
Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol. 2005;79:319–28.
Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30:4541–9.
Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22:3751–7.
Wechalekar AD, Schonland SO, Kastritis E, Gillmore JD, Dimopoulos MA, Lane T, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013;121:3420–7.
Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30:989–95.
Sanchorawala V, Boccadoro M, Gertz M, Hegenbart U, Kastritis E, Landau H, et al. Guidelines for high dose chemotherapy and stem cell transplantation for systemic AL amyloidosis: EHA-ISA working group guidelines. Amyloid. 2022;29:1–7.
Manwani R, Cohen O, Sharpley F, Mahmood S, Sachchithanantham S, Foard D, et al. A prospective observational study of 915 patients with systemic AL amyloidosis treated with upfront bortezomib. Blood. 2019;134:2271–80.
Kumar SK, Hayman SR, Buadi FK, Roy V, Lacy MQ, Gertz MA, et al. Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long-term results from a phase 2 trial. Blood. 2012;119:4860–7.
Funding
This study was supported by Janssen and sponsored by the European Myeloma Network.
Author information
Authors and Affiliations
Contributions
EK, GP, SS, AJ, MCM, HA, and PS contributed to study design; GP, SS, GM, PM, AJ, FB, MAD, SR, UH, WR, MTC, HA, MCM, RB, RH, CJ, EK, and AW collected the data; AL and GC analyzed the data; all authors contributed to data interpretation and writing of the report, critically reviewed, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
GP reports participating in advisory boards for Alexion, Argobio, Janssen, and Protego; honoraria from Alexion, Argobio, Janssen, Protego, The Binding Site, Pfizer, Prothena, Sebia, and Siemens; research funding from Gate bioscience and The Binding Site. SS reports research support from Janssen, Prothena, and Sanofi; participating in advisory boards for Janssen, Telix, and Prothena; honoraria from Janssen, Takeda, Pfizer, and Prothena; travel and congress participation grants from Janssen, Prothena, Celgene, Binding Site, and Jazz. PM reports speaker honoraria from Janssen and Pfizer; participating in advisory board for Janssen. AJ reports honoraria from Servier, Amgen, Abbvie, and Pfizer; participating in advisory committees for Janssen; research funding from Sanofi and Janssen. FB reports lecture fees from Janssen, Astra Zeneca, GSK, and Novartis; consultancy for Janssen and Astra Zeneca. MAD reports honoraria from participation in advisory boards from Amgen, Takeda, Beigene, BMS, and Janssen. UH reports travel grants from Janssen, Prothena, and Pfizer; participating in advisory boards for Pfizer, Janssen, and Prothena; honoraria from Janssen, Pfizer, Alnylam, and Akcea; research funding from Janssen and Prothena. WR reports honoraria/advisory board from Janssen, MSD, Amgen, and Sanofi. MTC reports honoraria for lectures/advisory board for Janssen, Amgen, Akcea, and Sanofi. MCC reports honoraria from BMS; consultancy for Janssen-Cilag and Gilead; speaker’s bureau for Medscaoe. RH reports consultancy/advisory relationship with Janssen, Amgen, Celgene, Abbvie, BMS, Novartis, PharmaMar, and Takeda; honoraria from Janssen, Amgen, Celgene, BMS, PharmaMar, and Takeda; research funding from Janssen, Amgen, Celgene, BMS, Novartis, and Takeda. CJ reports scientific advisory for Takeda, BMS, Janssen, GSK, and Amgen; research funding from Takeda. AL reports current employment with Health Data Specialists. GC reports current employment with Health Data Specialists. PS reports research support from Janssen, BMS, Sanofi, and Amgen, all for EMN trials; participating in advisory boards for Janssen, BMS, Sanofi, Amgen, Cargen, and Pfizer. EK reports honoraria/advisory board from Genesis Pharma, Janssen, GSK, and Pfizer; research support from Amgen, Janssen, and Pfizer. AW reports honoraria/advisory board from Janssen, Alexion, Attralus, Celgene/BMS, Takeda, and GSK. All remaining authors have declared no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palladini, G., Schönland, S., Merlini, G. et al. The management of light chain (AL) amyloidosis in Europe: clinical characteristics, treatment patterns, and efficacy outcomes between 2004 and 2018. Blood Cancer J. 13, 19 (2023). https://doi.org/10.1038/s41408-023-00789-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-023-00789-8
This article is cited by
-
Timing and co-occurrence of symptoms prior to a diagnosis of light chain (AL) amyloidosis
Blood Cancer Journal (2024)
-
Enhancing prognostic guidance in renal light-chain amyloidosis: a new staging system incorporating pathological characters
International Urology and Nephrology (2024)
-
Advancing precision medicine in immunoglobulin light-chain amyloidosis: a novel prognostic model incorporating multi-organ indicators
Internal and Emergency Medicine (2024)
-
AmyKoS—a German real-world prospective cohort study of patients with suspected and proven amyloidosis
Journal of Rare Diseases (2024)
