
Abstract
Pure erythroid leukemia (PEL), also known as acute erythroid leukemia (AEL), is recognized as a distinct morphologic entity by both the 2016 and 2022 World Health Organization (WHO) classification system. By contrast, the 2022 International Consensus Classification (ICC) includes PEL under a broader category of “acute myeloid leukemia with mutated TP53”. We identified 41 Mayo Clinic cases of PEL (mean age 66 years, range 27–86; 71% males) and provide a comprehensive account of bone marrow morphology, immunophenotype, cytogenetic and mutation profiles. PEL was primary in 14 cases, therapy-related in 14, secondary in 12, and undetermined in one. All cases expressed biallelic TP53 alterations, including TP53 deletion/single TP53 mutation (68%), two TP53 mutations (29%) or two TP53 deletions (3%); additional mutations were infrequent. Karyotype was complex in all cases and monosomal in 90%. Treatment details were available in 29 patients: hypomethylating agent (HMA) alone (n = 5), HMA + venetoclax (n = 12), intensive chemotherapy (n = 4), supportive care/other (n = 8); no responses or allogeneic stem cell transplants were documented, and all patients died at a median 1.8 months (range 0.2–9.3). The current study highlights a consistent and reproducible set of morphologic and genetic characteristics that identify PEL as a distinct AML variant whose dismal prognosis requires urgent attention.
Similar content being viewed by others


Introduction
Pure erythroid leukemia (PEL) is a rare and aggressive subtype of acute myeloid leukemia (AML) and accounts for approximately 1% of all AML diagnoses [1, 2]. While this diagnosis may seem relatively straightforward from the literature description, in clinical practice, in fact, PEL is often a diagnostic struggle even for the most seasoned of hematopathologists. In addition, pathologists may experience trepidation in rendering such a diagnosis given the gravity of a reported approximately <6 month overall median survival. Originally described by DiGuglielmo, numerous investigators, over the last several decades, have subsequently studied PEL and further refined its distinctive clinicopathologic features. PEL previously was a diagnostic subcategory in the revised fourth edition of the WHO classification [1, 2]. Currently PEL is classified as acute myeloid leukemia with mutated TP53 in the International Consensus Classification [3] and as acute erythroid leukemia in the 5th edition of the WHO [4].
PEL is typically a disease of adults with median age at presentation of 66–68 years and its characteristics include complex/monosomal karyotype, TP53 protein overexpression, and biallelic TP53 mutations. PEL might present de novo or arise as progression from antecedent myelodysplastic syndrome, myeloproliferative neoplasm, or as a therapy-related neoplasm [1, 2., 5,6,7,8] While there are reports of PEL/erythroblastic sarcoma in the pediatric population, there is evidence that unique genetic features exist in pediatric PELs, and likely represent a distinctly different disease process from that of adults [9]. Given that PEL is a rare and often difficult to diagnose disease with dismal prognosis, we looked into our institutional experience with the disease, with the objective to provide a comprehensive account of its pathology and clinical outcome; to the best of our knowledge, the current work constitutes the largest series of PEL in adults, to date.
Methods
Selection of cases
We retrospectively identified bone marrow cases seen and diagnosed at Mayo Clinic Rochester with a diagnosis of pure erythroid leukemia (PEL) based on the revised fourth edition World Health Organization criteria from the period of June 2017 to May 2022 [1, 2]. Case inclusion criteria required morphology slides available for review to confirm the diagnosis of PEL, confirmatory immunohistochemical and/or flow cytometric studies, cytogenetics and next-generation sequencing targeting genes commonly mutated in myeloid disease.
Immunophenotyping studies
-
(i)
Immunohistochemistry. Immunohistochemical analysis was performed on 4 μm, formalin-fixed, paraffin-embedded sections in all cases. A broad immunohistochemical panel was utilized in the evaluation of the cases. Primary antibodies included CD34 (QBEnd/10, Leica (Novocastra)), CD117 (YR145, Cell Marque), CD71 (MRQ-48, Cell Marque), E-Cadherin (4A2C7, Life Technologies), Myeloperoxidase (Dako), Hemoglobin (Cell Marque), Glycophorin A (GA-R2, Ventana), CD61 and TP53 (DO-7, Ventana).
-
(ii)
Flow cytometry. After isotonic erythrocyte lysis, flow cytometric immunophenotyping was performed on anticoagulated bone marrow aspirate specimens using previously described methods [10]. Samples were examined with flow cytometric immunophenotyping using two eight-color tubes containing antibodies from BD Biosciences (Tube1: CD13 PE, CD15 V450, CD16 APC-H7, CD33 PE-Cy7, CD34 PerCP-Cy5.5, CD45 V500, CD117 APC, HLA-DR FITC and Tube2: CD2 FITC, CD7 PE, CD34 PerCP-Cy5.5, CD36 APC, CD38 V450, CD45 V500, CD56 PE-Cy7, CD64 APC-H7). A total of 100, 000 events were collected per case. The data were analyzed using Kaluza software (Beckman-Coulter, Brea, CA) and/or Diva software (BD Biosciences).
Cytogenetics
Giemsa-banded (G-banded) chromosome analysis was performed on bone marrow samples according to conventional methods. When available, at least 20 metaphases were analyzed. Karyotypes of G-banded chromosomes were described according to the 2020 International System of Human Cytogenetic Nomenclature [11]. Abnormal clones were defined as two or more cells with the same structural abnormality, the same extra chromosomes, or the presence of three or more cells with loss of the same chromosome. Although technically clonal, cases with –Y, –X, + 15, and +Y were not considered a pathologic clonal finding but regarded as an age-related phenomenon. A complex karyotype is defined as ≥ 3 structural and/or numerical abnormalities. Monosomal karyotype is defined by the presence of one single autosomal monosomy (AM) in association with at least one additional AM or one structural chromosomal abnormality (excluding core-binding factor acute myeloid leukemia and acute promyelocytic leukemia).
Fluorescence in situ hybridization
Interphase fluorescence in situ hybridization (FISH) for TP53 locus-specific probe was performed on bone marrow aspirate interphase nuclei using standard FISH pretreatment, hybridization, and fluorescence microscopy protocols. A total of 200 interphase nuclei were evaluated in each case, with 100 nuclei evaluated independently by two qualified clinical cytogenetic technologists and interpreted by a board-certified (American Board of Medical Genetics and Genomics) clinical cytogeneticist. The expected typical abnormal FISH signal pattern for TP53 deletion is 1R2G.
Next-generation sequencing
Targeted NGS was performed to detect gene mutations commonly found in myeloid hematologic malignancies. DNA is extracted from the peripheral blood or bone marrow sample and following library preparation by hybrid capture, subjected to next-generation sequencing (NGS) with post-sequencing analysis of tumor-associated mutations. Performance characteristics of NGS panel: Single base substitutions: accuracy >99%; reproducibility 100% (intra- and interassay); sensitivity 5–10% variant allele fraction with a minimum depth coverage of 250X; Insertion/deletion events: accuracy >99%; reproducibility 100% (intra- and interassay); sensitivity 5–10% variant allele fraction with a minimum depth coverage of 250X. NGS testing performed at Mayo Clinic Rochester includes 42 commonly mutated genes in the panel, including TP53 exons 4–9. (ANKRD26 (NM_014915.2) 5’UTR, exons 1–4, intron c.172, ASXL1 (NM_015338.5) exons 10–13, BCOR (NM_001123385.1) exons 4–15, CALR (NM_004343.3) exon 9, CBL (NM_005188.3) intron 7 last 100 bps before start of exon 8, exon 8, intron 8, and exon 9, CEBPA (NM_004364.4) exon 1, CSF3R (NM_000760.3) exons 14 and 17, DDX41 (NM_016222.2) exons 1–17, DNMT3A (NM_022552.4) exons 8–23, ELANE (NM_001972.2) exons 1–5, ETNK1 (NM_018638.4) exons 2–5, ETV6 (NM_001987.4) exons 3–8, EZH2 (NM_004456.4) exons 2–20, FLT3 (NM_004119.2) exons 14–20, GATA1 (NM_002049.3) exons 2 and 4, GATA2 (NM_001145661.1) exons 3–7, intron 5, c.1017 + 1–1017 + 730, IDH1 (NM_005896.3) exon 4, IDH2 (NM_002168.3) exon 4, JAK2 (NM_004972.3) exons 12–16, KDM6A (UTX) (NM_021140.3) exons 1–29, KIT (NM_000222.2) exons 8–11 and 17, KRAS (NM_033360.3) exons 2–3, MPL (NM_005373.2) exons 10–12, NPM1 (NM_002520.6) exons 9–11, to −30 before exon 11, NRAS (NM_002524.4) exons 2 and 3, PHF6 (NM_001015877.1) exons 2–10, PTPN11 (NM_002834.3) exons 3–4 and 12–13, RAD21 (NM_006265.2) exons 1, 2, 4–7, 9–11, 13, 14, exon 10 flank 15 bp, RUNX1 (NM_001001890.2) exons 1–6, intron 4 c.725–13 T > A and intron 5 c.886 + 1–4del, SETBP1 (NM_015559.2) partial exon 4; amino acids 400–950, SH2B3 (LNK) (NM_005475.2) exon 2–8, SF3B1 (NM_012433.2) exons 13–16, SRP72 (NM_006947.3) exons 6, 10, SMC3 (NM_005445.3) exons 7, 8, 13, 17, 19, 21, 29, SRSF2 (NM_003016.4) exons 1 and 2, STAG2 (NM_001042750.1) exons 4–34, 12, 17 and 22 flank 15 bp, TERT (NM_198253.2) exons 2–16, TET2 (NM_001127208.2) exons 3–11, TP53 (NM_000546.4) exons 4–9, U2AF1 (NM_001025203.1) exons 2, 6, and 8, WT1 (NM_024426.2) exons 1–10, and ZRSR2 (NM_005089.3) exons 1–11).
Results
Clinical characteristics
We identified 41 cases of PEL that met our inclusion criteria (Fig. 1). There were 12 females and 29 males with a mean age of 66 years (range 27–86). As seen in Table 1, these cases included both secondary and de novo forms of PEL: 14 cases primary/de novo, 14 therapy-related, 12 secondary to progression of a myeloid disorder [MDS (5), MPN (5), MDS/MPN (2)], and one with no available clinical history. 100% of patients are dead of disease (41/41) with an overall mean survival of 3.3 months (median 2 months; range 0.1–23).

Infographic depicting the key elements that are highly reproducible in pure erythroid leukemia.
Hematologic indices and peripheral blood smear findings
All 40 patients with an available complete blood cell count presented with anemia (100%), essentially all with thrombocytopenia (98%) and 63% with neutropenia (Table 1). The average hemoglobin level was 8.1 g/dL with 48% of patients having severe anemia defined as <8.0 grams/deciliter. The average platelet count was 32 × 10(9)/L (range for-146) with 78% of patients demonstrating marked thrombocytopenia (defined as less than 50 × 10(9)/L). The average absolute neutrophil count was 1.9 × 10(9)/L (range 0–11.5). 25% of patients had marked neutropenia (<0.5 × 10(9)/L) 20% moderate neutropenia (0.5–1.0 × 10(9)/L); 18% mild neutropenia (1.01–1.5 × 10(9)/L). In those cases with sufficient numbers of neutrophils for cytologic evaluation, no cases showed significant dysplastic features (N = 21). There was an insufficient number of cases with sufficient platelets to accurately assess for platelet dysplasia. Circulating blasts were seen in 68% of cases with an average of 11.5% and a median of 4%.
Bone marrow findings
Table 1 provides a list of the salient bone marrow findings. In 56% of cases (23/41), the bone marrow aspirate smears/touch preparations were of suboptimal/inadequate quality for cytologic review due to dry tap, hemodilution and/or numerous broken cells/bare nuclei. Therefore, an accurate differential count could not be performed in these cases and the assessment of bone marrow involvement by the neoplastic erythroid population was informed by the bone marrow core biopsy (Fig. 1). In all cases, greater than 80% of bone marrow nucleated elements consisted of erythroid precursors. For cases with an aspirate available, the percentage of proerythroblasts was ≥30% of all erythroblasts. 85% of all cases (35/41), including all cases with a dry tap aspirate, showed tumor cells predominantly at the pronormoblast stage of maturation. The remaining 15% of cases showed some minimal maturation past the pronormoblastic stage but no case showed a significant population of terminally differentiated nucleated red blood cell precursors. 80% of cases had dysplastic megakaryocytes characterized by small, monolobated forms and small forms with hyperchromatic nuclei. In all cases, there was insufficient granulopoiesis to assess for dysplasia and non-erythroid blasts were not increased. In 20% of the cases the pattern of infiltration by the pronormoblasts was cohesive/focal/nodular mimicking a metastatic tumor. 3 cases (7%) showed a distinctive sinusoidal pattern. Ring sideroblasts were detected in 71% of cases with an average percent of 26 (range; rare – 80).
Flow cytometric findings
Twenty-eight of 41 cases (68%) had sufficient bone marrow aspirate material to identify the population of interest (pronormoblasts) (Fig. 1). Interestingly, we found that none of the tested antigens (except myeloperoxidase) showed a uniform presence or absence pattern of expression (see Table 1). The vast majority (96%) of pronormoblasts expressed bright CD36 and were negative for HLA-DR (79%). 75% of cases showed aberrant positivity for CD7 (36% uniform: 39% partial). CD45 was weakly expressed in exactly half of the cases. In terms of “blast” markers, CD117 was expressed in 82% of cases but only 61% showed uniform positive expression. CD34 was expressed in 25% of cases which is critical in considering the incorrect interpretation of a myeloid blast population since CD13 is expressed in 68% of cases (uniform positive 47%; partial positive 21%) and CD33 in 64% of cases (25% uniform positive; 39% partial positive). Myeloperoxidase, as would be expected, was uniformly negative.
Immunohistochemical findings
Immunohistochemical studies were performed in a subset of cases, typically depending on the confidence of confirming erythroid lineage with an initial handful of markers (Table 1 and Fig. 1). The pronormoblasts in pure erythroid leukemia were essentially uniformly positive for CD71 (96%) with the majority being uniformly positive for CD117 (68%) and E-cadherin (88%). A subset of cases only showed partial positivity for these markers (CD117 19%; CD71 4%; E-cadherin 6%). Although uncommon, the pronormoblasts may be negative for CD117 (10%) and E-cadherin (6%). CD34 was uniformly positive in 16% of cases. Greater than 1/2 of the cases exhibited hemoglobin positivity in a subtle but convincing subset of blasts (58%) indicating that this marker is useful to confirm erythroid lineage when present. Similarly, glycophorin was convincingly positive in a small subset of pronormoblasts (33%). The majority of pronormoblasts /blasts were negative for CD61; however, 3 cases (9%) did show a distinctive small positive subset indicating that rare cases may show both erythroid and megakaryocytic differentiation.
Cytogenetic characteristics
100% (41/41) demonstrated an abnormal karyotype (Table 2 and Fig. 1). Of the 41 cases, 73% met criteria for a highly abnormal karyotype (≥10 numerical and/or structural abnormalities) and 90% satisfied criteria for a monosomal karyotype. 78% of cases demonstrated a deletion abnormality involving chromosome 5/5q and 66% demonstrated a deletion abnormality involving chromosome 7/7q. Of the 41 cases, 29 (71%) demonstrated a presumed deletion of the TP53 locus (17p13) of which 14 (34%) were confirmed by FISH.
Mutations
40 of 41 (98%) cases demonstrated at least one TP53 pathogenic mutation as detected by NGS (Table 2). In the single case with no TP53 (or other) mutation detected by NGS, there were 2 TP53 deletions detected by FISH. All 41 cases showed double TP53 alterations as exemplified by either two TP53 deletions by karyotype (3%; N = 1), double TP53 mutations by NGS (29%; N = 12) or a combination of a TP53 deletion and TP53 mutation (68%; N = 28). Twelve patients (29%) showed additional pathogenic mutations in genes associated with myeloid disorders, in addition to the above-described TP53 mutation(s). Therefore, an interesting overall finding is that aside from a single or double TP53 mutation, the NGS findings are relatively “non-complex/simple” on a next-generation sequencing panel (42 genes in this study). Five of the 12 patients (42%) comprised disease progression from an underlying myeloproliferative neoplasm (MPN) and showed NGS evidence of a JAK2 V617F mutation. The underlying MPN in these five patients included two cases of polycythemia vera, 2 cases of the essential thrombocythemia and 1 case favor primary myelofibrosis. Other pathogenic mutations that were detected in the non-MPN patients included abnormalities in several clonal hematopoiesis of indeterminate potential (CHIP) genes (e.g., DNMT3A, TET2 and SF3B1); however, all exhibited variant allele frequencies greater than 20%, and some even higher in the 50% and 60% range. Of these non-JAK2 mutation cases, the NRAS, CSF3R and TET2 c.4319_4329del; p.Arg1440Hisfs*34 were in the context of therapy-related disease; the remaining 4 other mutations (TET2, TET2, DNMT3A, and SF3B1) were in the context of disease progression from myelodysplastic syndrome.
Treatment details and survival
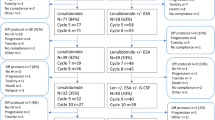
Treatment details were available in 29 patients seen and managed at the Mayo Clinic (Table 3). As was the case for the entire study population of 41 patients, all of these 29 patients expressed biallelicTP53 alterations and complex karyotype, with 90% showing monosomal karyotype. As listed in Table 3, treatment included hypomethylating agent (HMA) alone (n = 5), HMA + venetoclax (n = 12), intensive chemotherapy (n = 4), supportive care (n = 7), and quizartinib (n = 1); no responses or allogeneic stem cell transplants were documented, and all patients died at a median 1.8 months (range 0.2–9.3). Figure 2 illustrates survival data on these 29 patients, stratified by treatment type. PEL was universally fatal with no evidence of survival impact from typical AML therapies, including combination of hypomethylating agents and venetoclax; the 3- and 12-months survival rates were 24% and 0%, respectively. Univariate analysis of multiple host or disease factors failed to identify a statistically significant predictor of survival.

Overall survival data of 29 patients with pure erythroid leukemia (a), stratified by treatment received (b). HMA = hypomethylating agent; *One of 8 patients included in the supportive care category received quizartinib.
Discussion
Herein, we provide a detailed and comprehensive analysis of the pathologic characteristics of 41 cases of pure erythroid leukemia. To our knowledge, this represents the largest series of pure erythroid leukemia cases reported to date. Based on our composite findings, we would argue that pure erythroid leukemia is a unique pathologic entity and should be considered to remain a pathologic diagnosis in hematologic classification systems. While the overall natural disease course may be similar to other acute myeloid leukemias with TP53 mutation(s), it seems that, in this particular disease, the features are overall highly reproducible and provide a clear indication to the healthcare team as to disease expectations. Given that a diagnosis of pure erythroid leukemia may be challenging from a pathologic perspective, and that the prognostic implication from such a diagnosis is grave (median overall survival 2 months), a comprehensive and systematic approach with the utilization of a diverse set of ancillary tools may be necessary in the elucidation of these rare cases.
Pure erythroid leukemia, in adults, can be summarized with the following features (Fig. 1): presentation with anemia and thrombocytopenia, suboptimal bone marrow aspirate smears/touch preparations, distinctive cytology with predominantly pronormoblasts and minimal presence of maturing erythroid elements, may mimic metastatic disease in the core biopsy (25%), association with ring sideroblasts (70%), cytogenetic complexity (100%) with hypodiploidy and often doubling of that clone, deletion abnormalities of chromosomes 5 and 7 (78% and 66%, respectively), extraordinarily simple NGS mutational profile (71% show no myeloid disease associated gene abnormality aside from TP53 mutation) and double TP53 genetic alterations (100%). The immunophenotypic assessment of these cases typically requires a combination of both flow cytometry and immunohistochemistry to confidently determine that the blasts are truly of erythroid lineage.
The differential diagnosis of PEL includes florid reactive erythroid hyperplasias and other myeloid neoplasms with a predominance of erythroid precursors. For the reactive etiologies, we would not expect to identify dysplastic megakaryocytes, a cytogenetic abnormality or an aberrant phenotype (eg. CD7 positivity) on the pronormoblasts. Similarly, strong uniform nuclear expression of TP53 by immunohistochemistry would argue against a non-neoplastic possibility [8]. As for the myeloid neoplasms with an abundance of erythroid precursors, this can be trickier to sort out but if the criteria for PEL are systematically applied, a correct diagnosis is rendered. For example, in acute myeloid leukemia with myelodysplasia-related changes, there are ≥20% myeloid-lineage blasts. In myelodysplastic syndrome, particularly when high grade, one may see a complex abnormal karyotype or monosomal karyotype as well as erythroid predominance, but erythroid precursors should not comprise >80% of bone marrow nucleated elements.
A diagnosis of PEL may be established by assessing for the characteristic distinctive pronormoblastic cytology, variable expression of the erythroid-specific antigens hemoglobin and glycophorin A, and presence of biallelic TP53 alterations that are always associated with complex/monosomal karyotype [1, 2, 7, 12, 13]. Two new classification systems for hematopoietic tumors have recently been published; in the International Consensus Classification (ICC), PEL is no longer recognized as a separate entity and is instead included in a broader category of AML with mutated TP53 [3]; in the proposed 5th edition of the WHO document, The WHO alters the terminology from PEL to acute erythroid leukemia (AEL) [4]. The ICC system emphasizes in its category of acute myeloid leukemia that one intention was to “move to a more genetically-defined classification”. The newly defined AML with mutated TP53, includes cases with single (or multiple) somatic TP53 mutation with a variant allele frequency of ≥10% and ≥20% peripheral blood or bone marrow myeloid blasts/blast equivalents, or cases which meet criteria for pure erythroid leukemia. We agree with the findings reported in the literature that AML with mutated TP53 is typically associated with complex including monosomal karyotype and an overall dismal clinical outcome. That said, it should be remembered that there is underlying premise in the practice of medicine that there is an “art” to what we do as physicians and as members of the health care team as we serve our communities and our patients. We would suggest that in this particular entity, pure erythroid leukemia, there is an art to this diagnosis through a careful assessment of its unique morphology and laboratory, immunophenotypic, genetic and clinical findings. Perhaps this could be included as a qualifier to the diagnosis of AML with mutated TP53.
In PEL, the unique cytogenetic and molecular genetic characteristics might suggest therapeutic targets beyond TP53; and its invariable association with biallelic TP53 alterations, implies an even worse diagnosis, compared to single-hit TP53 abnormalities. The current study also underscores the limited value of AML-like treatment in PEL, including that of HMA + venetoclax combination. We have previously reported the value of HMA + venetoclax in both newly diagnosed and relapsed/refractory AML [14,15,16]. In one of these reports involving newly diagnosed AML, [14] unlike the case in the current study, responses to HMA + venetoclax were documented in 8 (32%) of 25 TP53-mutated AML cases, most of whom expressed monoallelic TP53 alteration. It is possible that the difference in response rates between the two studies might be related to their difference in TP53 allelic state. Regardless, the fact that not even one patient responded to any of the AML-like treatment instituted highlights the urgent need for new investigational agents. In a recent multi-center report of 291 patients with TP53 mutated AML, treated with various induction regimens, complete remission with or without count recovery (CR/CRi), was achieved in only 29%, and 46 patients were bridged to ASCT [17]; in the particular study, HMA + venetoclax combination therapy was the most successful in inducing CR/CRi and outcome was favorably influenced by ASCT and not TP53 variant allele frequency. These observations suggest the potential value of ASCT in PEL but the current lack of chemotherapy regimens capable of inducing CR/CRi remains to be a challenge.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Arber DA, Brunning RD, Le Beau MM, Falini B, Vardiman JW, Foucar K et al. Acute myeloid leukemia and precursor neoplasm. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon; 2017. pp.129–71.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. The International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphological, Clinical, and Genomic Data. In Press; 2022.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(Jul):1703–19. https://doi.org/10.1038/s41375-022-01613-1. Epub 2022 Jun 22PMID: 35732831; PMCID: PMC9252913.
Weinberg OK, Arber DA. Erythroleukemia: an Update. Curr Oncol Rep. 2021;23:69.
Arber DA. Revisiting erythroleukemia. Curr Opin Hematol. 2017;24:146–51.
Reinig EF, Greipp PT, Chiu A, Howard MT, Reichard KK. De novo pure erythroid leukemia: refining the clinicopathologic and cytogenetic characteristics of a rare entity. Mod Pathol. 2018;31:705–17.
Alexandres C, Basha B, King RL, Howard MT, Reichard KK. p53 immunohistochemistry discriminates between pure erythroid leukemia and reactive erythroid hyperplasia. J Hematopathology. 2021;14:15–22.
King RL, Siaghani PJ, Wong K, Edlefsen K, Shane L, Howard MT, et al. Novel t(1;8)(p31.3;q21.3) NFIA-RUNX1T1 translocation in an infant erythroblastic sarcoma. Am J Clin Pathol. 2020;156:129–38.
Morice WG, Kurtin PJ, Hodnefield JM, Shanafelt TD, Hoyer JD, Remstein ED, et al. Predictive value of blood and bone marrow flow cytometry in B-cell lymphoma classification: comparative analysis of flow cytometry and tissue biopsy in 252 patients. Mayo Clin Proc. 2008;83:776–85.
McGowan-Jordan J, Hastings RJ, Moore S, editors. ISCN 2020: An International System for Human Cytogenomic Nomenclature. Basel: Karger; 2020.
Montalban-Bravo G, Benton CB, Wang SA, Ravandi F, Kadia T, Cortes J, et al. More than 1 TP53 abnormality is a dominant characteristic of pure erythroid leukemia. Blood. 2017;129:2584–258.
Fang H, Wang SA, Khoury JD, El Hussein S, Kim DH, Tashakori M, et al. Pure erythroid leukemia is characterized by biallelic TP53 inactivation and abnormal p53 expression patterns in de novo and secondary cases. Haematologica. 2022;107:2232–2237.
Gangat N, Johnson I, McCullough K, Farrukh F, Al-Kali A, Alkhateeb H, et al. Molecular predictors of response to venetoclax plus hypomethylating agent in treatment-naïve acute myeloid leukemia. Haematologica 2022;107:2501–5.
Gangat N, Tefferi A. Venetoclax-based chemotherapy in acute and chronic myeloid neoplasms: literature survey and practice points. Blood Cancer J. 2020;10:122.
Morsia E, McCullough K, Joshi M, Cook J, Alkhateeb HB, Al-Kali A, et al. Venetoclax and hypomethylating agents in acute myeloid leukemia: Mayo Clinic series on 86 patients. Am J Hematol. 2020;95:1511–21. https://doi.org/10.1002/ajh.25978. Epub 2020 Sep 16.
Badar T, Atallah E, Shallis RM, Goldberg AD, Patel A, Abaza Y, et al. Outcomes of TP53-mutated AML with evolving frontline therapies: Impact of allogeneic stem cell transplantation on survival. Am J Hematol. 2022;97:E232–E235.
Author information
Authors and Affiliations
Contributions
All authors conceived and/or designed the work that led to the submission, acquired data, and/or played an important role in interpreting the results. KKR designed the study. KKR, CA, and PTG coordinated the study. KKR reviewed and collected the laboratory data, bone marrow slides, immunophenotypic data. MA and AT reviewed, collected and analyzed the clinical information. CA and PTG collected the genetic data and PTG and KKR analyzed the data. All authors reviewed and validated the data. All authors contributed to various aspects of the figures and tables preparation. KKR drafted the paper and all authors reviewed, revised and approved the final manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Informed consent
We obtained a waiver for informed consent for the study. This study was approved by the Mayo Clinic Institutional Review Board.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reichard, K.K., Tefferi, A., Abdelmagid, M. et al. Pure (acute) erythroid leukemia: morphology, immunophenotype, cytogenetics, mutations, treatment details, and survival data among 41 Mayo Clinic cases. Blood Cancer J. 12, 147 (2022). https://doi.org/10.1038/s41408-022-00746-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00746-x
This article is cited by
-
A lightweight deep learning model for acute myeloid leukemia-related blast cell identification
The Journal of Supercomputing (2024)
-
Acute erythroid leukemia with TP53 mutation and BCR/ABL1: challenges in classification and management
Annals of Hematology (2024)
