
Abstract
Ecotropic viral integration site 1 (Evi1) was discovered in 1988 as a common site of ecotropic viral integration resulting in myeloid malignancies in mice. EVI1 is an oncogenic zinc-finger transcription factor whose overexpression contributes to disease progression and an aggressive phenotype, correlating with poor clinical outcome in myeloid malignancies. Despite progress in understanding the biology of EVI1 dysregulation, significant improvements in therapeutic outcome remain elusive. Here, we highlight advances in understanding EVI1 biology and discuss how this new knowledge informs development of novel therapeutic interventions. EVI1 is overexpression is correlated with poor outcome in some epithelial cancers. However, the focus of this review is the genetic lesions, biology, and current therapeutics of myeloid malignancies overexpressing EVI1.
Similar content being viewed by others


MECOM locus discovery
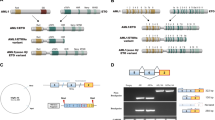
Evi1 was discovered by Mucenski et al. as a common site of ecotropic viral integration in mice that caused virally induced myeloid malignancies1. Through its rearrangements in human acute myeloid leukemia (AML), the human EVI1 gene was mapped to the long arm of chromosome 3 at q 26.2 (3q26.2)2. The EVI1 gene in humans is ~92% homologous to the mouse Evi12. EVI1 is encoded from the MDS1 and ecotropic viral integration site 1 (EVI1) complex locus (MECOM), which includes several alternative transcripts3. EVI1 exists either as a shorter single gene or as spliced to the short myelodysplastic syndrome 1 (MDS1) gene, present more than 350 kb upstream to EVI1, creating the longer MDS1-EVI1 gene3. The shorter isoform of EVI1 is abundant and oncogenic4,5. A truncated variant of the EVI1 transcript conserved in both mice and humans, EVI1∆324, lacks part of the first zinc finger domain and the ability to transform (Fig. 1)4.

C2H2-zinc finger motifs are shown in blue. Zinc finger motifs 1–7 and motifs 8–10 form the N-terminal and C-terminal zinc finger domains respectively. The repressive domain and acidic domain are depicted in tan and red, respectively. The numbers beneath the schematic indicate the amino acid positions of the zinc finger domains, the repressive domain and the acidic domain of EVI1.
EVI1: domain-structure and function
Human EVI1 is a 145 kilo Dalton (kDa) protein that contains 1051 amino acids. EVI1 localizes to the nucleus and binds DNA through its zinc finger (ZF) domains5. EVI1 contains ten zinc fingers that are arranged in two separate sets, one N-terminal containing seven zinc fingers, another C-terminal containing three zinc fingers5 (Fig. 1). Through electrophoretic mobility shift assays and chromatin immunoprecipitation (ChIP) assays, the N-terminal ZF domain was determined to bind TAGA/TCTA or CAGAGA/TCTCTG GATA-like simple sequence repeats (SSRs)5,6. The C-terminal ZF domain recognizes a CCATATAA ETS-like motif5,6. In the region between the ZF domains, is the repressor region that contains the interaction sites for the co-repressor CtBP (C-terminal binding protein 1)7,8. EVI1 also contains an acidic domain at its C terminus5 (Fig. 1).
Disruption of the full-length Evi1 transcript by mutagenesis in mice led to severe developmental defects in the heart and central nervous system, and homozygous mutants died at approximately embryonic day 10.59. Additionally, adult mice with conditional knockout of Evi1 had a marked reduction in their long-term hematopoietic stem cells (LT-HSCs), and upon transfer into irradiated mice were unable to engraft and repopulate efficiently10. The self-renewal ability of LT-HSCs is linked to EVI1 expression, and many LT-HSC-associated genes have EVI1 binding sites in their regulatory regions11. Furthermore, increased EVI1 expression is a common immortalizing factor of murine primary bone marrow after retroviral infection12. For example, MSCV integration promoted increased expression of EVI1 causing immortalization of immature myeloid cells, but they were unable to induce leukemia in transplanted hosts12. Thus EVI1 supports HSC self-renewal, but EVI1 expression alone is not enough to drive leukemogenesis12,13.
In addition to LT-HSC self-renewal, expression of EVI1 blocks hematopoietic differentiation of the granulocyte, erythroid, dendritic, and monocytic lineages14,15. EVI1 expression in primary mouse myeloid progenitor cells upregulated HSC-associated genes and decreased DNA replication and repair genes14. EVI1 transcripts are decreased in human CD34+ cells after stimulation of differentiation induced by cytokine administration, suggesting that downregulation of EVI1 is an important step in terminal differentiation of many hematopoietic lineages15. Forced expression of Evi1 in the mouse bone marrow cell line 32Dcl3 inhibits differentiation response to granulocytes and erythrocytes due to granulocyte colony-stimulating factor (G-CSF) and erythropoietin, respectively16,17. Extrinsic EVI1 expression blocked G-CSF-induced differentiation through transcriptional repression of the lineage-specific gene myeloperoxidase and the myeloid transcription factors C/EBPα (CCAAT enhancer binding protein alpha) and RUNX1 (runt-related transcription factor 1, also known as AML1)16,18. Erythroid differentiation was blocked by EVI1 through binding and subsequent inhibition of transcriptional activity of the myeloid transcription factors GATA1 (GATA binding protein 1) and PU.1 (transcription factor PU.1)17,19. In the megakaryocyte lineage, EVI1 is expressed in early precursor cells20. In a transgenic mouse model recapitulating human inv3(q21q26) AML that overexpresses EVI1 and also has GATA2 haploinsufficiency, EVI1 and GATA2 dysregulation together skewed hematopoiesis toward the megakaryocyte lineage more so than EVI1 overexpression alone21. This suggests that EVI1 may work in concert with other factors to promote the megakaryocyte lineage21. In general, for most myeloid lineages, EVI1 functions to promote a stem or early progenitor transcriptional program11,14. Forced EVI1 expression maintains the stem-like program while simultaneously suppressing myeloid transcription factors involved in myeloid differentiation16,18,19,21. Notably, endogenous EVI1 is generally downregulated under normal differentiation14,15. However, the degree to which endogenous EVI1 blocks differentiation and what factors normally downregulate EVI1 during differentiation largely remain an unknown.
MDS1-EVI1 and EVI1Δ324
In 1994, Nucifora et al. identified a transcript of unknown function that they termed MDS1, which formed a fusion protein with RUNX1 and/or EVI1 in several myelodysplastic syndrome (MDS) patients22. Currently, the function of the MDS1 protein itself is still unknown. In MDS1-EVI1, exon 2 of MDS1 is fused in-frame to EVI1 exon 2, which adds 188 amino acids upstream of the normal start codon of EVI1 in exon 322. A part of these extra N-terminal amino acids contains the PR domain, which shares homology with the B cell factor positive regulatory domain 1-binding factor (PRD1-BF1) and retinoblastoma binding protein RIZ13,12. The PR domain is related to a subset of the methyltransferase SET domains3,23 (Fig. 1). The combination of a PR domain and zinc finger domains in MDS1-EVI1 makes it a part of the PRDM (PR/SET domain) family and thus is also called PRDM3, which was characterized as a mono-methyl H3K9 methyltransferase23. Although the specific role of MDS1-EVI1 is not always separated from the role of EVI1, loss of MDS1-EVI1 is also associated with embryonic lethality, developmental defects, and dysregulation of hematopoiesis10,15.
EVI1Δ324 is a variant transcript of EVI1 with an internal 972 nucleotide deletion that removes the 6th and 7th zinc finger from the N-terminal ZF domain4 (Fig. 1). ChIP assays with FLAG-tagged EVI1 or EVI1Δ324 in an ovarian carcinoma cell line (SKOV3) showed an ~71% overlap in binding peaks between the two24. Additionally, the transcriptional profile of HeLa cells overexpressing EVI1-FLAG or EVI1Δ324-FLAG was almost identical24. However, EVI1Δ324 does not replicate the transformative effects of EVI1 in rat fibroblasts, and is not known to have oncogenic activity nor is it linked currently with any myeloid malignancy24.
EVI1 regulation
Epigenetic regulation of EVI1
The region 5′ of EVI1 contains two CpG islands, one close to the transcription start site of EVI1 and a second located near MDS125. In an AML cell line that has low EVI1 expression, the CpG islands related to EVI1 and MDS1 had a marked increase in methylation, suggesting that EVI1 expression can be regulated by methylation in AML cells25. Furthermore, AML cell lines with high EVI1 expression displayed active chromatin marks, with histone acetylation and enrichment of H3K4me3 (histone 3 lysine 4 tri-methylation) at the EVI1 promoter. In contrast cell lines with low EVI1 expression have enrichment of the repressive histone mark H3K27me3 (histone 3 lysine 27 tri-methylation)25.
EVI1 promoter
The minimal promoter of EVI1 was localized to a 318 nucleotide region 5′ of the EVI1 transcription start site that does not contain a traditional TATA or CAAT box26 (Fig. 2). In the EVI1 minimal promoter, analysis of binding motifs and site directed mutagenesis identified active binding motifs for RUNX1, ELK1 (ETS transcription factor ELK1), RELA (RELA proto-oncogene, NF-κB Subunit), GATA1, and MYB (MYB proto-oncogene, transcription factor). Knockdown of RUNX1 and/or ELK1 in HEL cells decreased EVI1 mRNA and protein levels26. Furthermore, interactions between RUNX1 and EVI1 at the minimal promoter appear to positively regulate EVI1 activity26. MDS1-EVI1 and EVI1Δ324 bind further downstream of the minimal promoter of EVI1 and reduce its transcription26,27. MDS1-EVI1 and EVI1∆324 are reported to be co-expressed with EVI115,24. Although not further studied, present upstream of the minimal EVI1 promoter are the consensus binding motifs for GATA1, GATA2, and C/EBPα, suggesting that MDS1-EVI1 and EVI1∆324 may work in concert with other transcription factors to repress EVI127. CML (chronic myeloid leukemia) blast crisis patient-derived cells express high EVI1 and β-catenin levels28. Knockdown of β-catenin or its related co-transcription factor LEF1 (lymphoid enhancer factor 1) decreased EVI1 levels28. Bioinformatic analysis indicated two potential tandem LEF1/β-catenin-binding sites present 1.44 kb upstream of EVI1, which are bound by LEF1, as determined by ChIP assays28. Additional studies are needed to further clarify regulation of EVI1 by LEF1/β-catenin, RUNX1, GATA1, and/or ELK1.

Within the upstream EVI1 promoter, the positions and nucleotide sequences/binding sites of specific transcription factors are shown.
Post-translational modifications on EVI1
EVI1 has been reported to be phosphorylated at serine 196 (S196), S538, S858, and S86029,30. Stable isotope labeling of amino acids followed by mass spectrometry (SILAC-MS) identified EVI1-associated proteins. CK2 (casein kinase 2) was confirmed to phosphorylate EVI1 residues S538 and S858. Loss of phosphorylation was mediated by PP1α (protein phosphatase 1 alpha), and it decreased DNA-binding by the C-terminal ZF domain29. In contrast, phosphorylation of S196 on the 6th zinc finger in the N-terminal ZF domain decreases DNA binding and repression by EVI1 of promoters containing GATA-like motifs30. Although phosphorylation of Ser858 and Ser860 did not affect EVI1 DNA binding, loss of these phosphorylations blunted EVI1 transcriptional repression after cellular stress through reduced interaction of EVI1 with co-repressor CtBP131. EVI1 is also acetylated by CBP (CREB binding protein or KAT3A)/p300 (EP300, or KAT3B) and PCAF (P300/CBP-associated factor or KAT2B)32. CBP-induced acetylation increased EVI1 transcriptional activity in luciferase assays32. In contrast, PCAF-mediated acetylation of EVI1 has been reported to exhibit opposing effects on EVI1 activity. Co-expression of EVI1 with PCAF abrogated EVI1-mediated Bcl-xL expression, suggesting that EVI1 acetylation blocked EVI1 transactivation activity at the Bcl-xL promoter33. In contrast, PCAF-mediated acetylation of K564 on EVI1 increased its ability to transactivate GATA2, and this ability was lost in a K564A mutant that cannot be acetylated34. Overall, it is unclear whether these post-translational modifications can occur simultaneously, or whether one modification can hinder the acquisition of another.
Transcriptional regulation by EVI1
Transcriptional repression by EVI1
EVI1 co-immunoprecipitates with the H3K9me3 methyltransferase SUV39H1 (suppressor of variegation 3–9 homolog 1) and the related H3K9me1/2 methyltransferase G9a (euchromatic histone lysine methyltransferase 2)35,36 (Table 1A). EVI1 and SUV39H1 interaction required the N-terminal ZF domain of EVI1 and the C-terminal domain of SUV39H1. Histone methyltransferase assays showed SUV39H1 had methyltransferase activity alone or in a complex with EVI1. Furthermore, it was observed by the Nucifora and Delwel groups that EVI1-mediated repression of a GAL4 luciferase construct was enhanced by SUV39H1 co-expression35,36.
EVI1 represses PTEN (phosphatase and tensin homolog) through its N-terminal ZF domain and via recruitment of the polycomb repressor complex 2 (PRC2), including EZH2 (enhancer of zeste 2), by binding upstream of the PTEN transcription start site37. This increased accumulation of the repressive H3K27me3 mark and reduced histone acetylation at the PTEN locus has been observed in human AML patient samples37.
EVI1 interacts through its N-terminal ZF domain with the de novo DNA methyltransferases DNMT3A and 3B38,39. EVI1 expression correlated with differential hypermethylation of over 200 genes, as compared to normal CD34+ cells, or to a previously reported DNA methylation profile in a separate cohort of 344 AML patients39. Unbiased motif analysis of differentially methylated gene promoters showed an enrichment of the motif recognized by the N-terminal ZF domain of EVI139. DNMT3A was also found to be highly expressed in EVI1-high AML samples compared to other AML subtypes. EVI1 expression levels correlated positively with a stronger hypermethylation signature in AML patient samples39.
Interaction with co-repressor CtBP
A region just left to the C-terminal ZF domain of EVI1 was associated with transcriptional repression activity of EVI1 and shown to be required for EVI1 transformation of rat fibroblasts40. This region was also critical for EVI1 repression of TGF-β (transforming growth factor beta) signaling and was thus termed the repressive domain (Rp) (Fig. 1)41. Two consensus binding motifs for the transcriptional co-repressor CtBP were identified in the EVI1 Rp region. The PLDLS sequence at the residue 584 of EVI1 is the major site of CtBP interaction7,8. Mutation of the CtBP binding site at residue 584 abolished the ability of EVI1 to repress TGF-β-mediated growth arrest and transformation of rat fibroblasts7,8.
Repression of other transcription factors by EVI1
EVI1 can also directly bind several transcription factors and inhibit their activity (Table 1B). EVI1 was able to repress GATA1-mediated activation of a synthetic promoter. However, EVI1 does not bind to the canonical GATA1 motif42. Instead, EVI1 zinc fingers one and six directly interact with the C-terminal zinc finger of GATA1 in GST-fusion pull-down assays. Also, EVI1 interaction with GATA1 decreased GATA1 DNA-binding ability. Mutation of EVI1 zinc fingers one and six abolished GATA1 interaction and restored differentiation potential to 32Dcl3 cells in response to erythropoietin42.
The 6th and 7th zinc finger of EVI1 was shown to directly interact with the C-terminal ETS-domain of PU.1 through co-immunoprecipitation and GST-fusion pull-down assays. Binding of EVI1 to PU.1 did not prevent DNA-binding ability of PU.1; instead it blocked association of PU.1 with c-Jun (Jun Proto-Oncogene), a subunit of the transcription factor AP-1. Mutation of the 6th and 7th EVI1 zinc fingers mitigated EVI1 interaction with PU.1 and restored differentiation potential to 32Dcl3 cells in response to G-CSF19. The 8th zinc finger in the C-terminal ZF domain of EVI1 was shown to interact with RUNX143. Binding of EVI1 repressed transcriptional activity of RUNX1 by decreasing its DNA-binding43. However, RUNX1 interaction with EVI1 had no effect on EVI1 DNA-binding. EVI1 interacts with the transcription factor SMAD3 through its N-terminal ZF domain41. EVI1 interaction repressed SMAD3 activity leading to blocked TGF-β mediated growth inhibition41.
Transcriptional activation by EVI1
A number of gene targets are upregulated by EVI1 (Table 2). EVI1 interaction with histone acetyltransferases has been reported to promote EVI1-mediated transcriptional activation32,34. EVI1 interaction with AP-1 subunits c-Fos and c-Jun was noticed as early as 1994 by Tanaka et al.44. EVI1-expressing cells exhibited increased c-Fos and c-Jun levels, and the C-terminal ZF domain of EVI1 was critical for activation of the c-Fos promoter44. Loss of EVI1 decreased c-Fos occupancy on the DNA, suggesting that EVI1 and AP-1 may act cooperatively at some loci6. A SILAC-MS screen also confirmed c-Fos and c-Jun interaction with EVI129. This screen also identified several additional transcription factors and co-factors that interact with EVI1, and 65% of EVI1-regulated genes were upregulated6,29. This highlighted the role of EVI1 as a transcriptional activator.
EVI1 dysregulation in myeloid leukemia
Chromosome 3 lesions leading to EVI1 overexpression
In the World Health Organization (WHO) classification of AML and related neoplasms, inversion or translocation of chromosome 3 at the MECOM locus [inv3(3;3)(q21q26)/inv,(3) t(3;3)(q21;q26.2)/t(3;3)] have been recognized as recurrent genetic abnormalities45 (Fig. 3). Inv(3)/t(3;3) is observed in ~1–2.5% of MDS and in a similar percentage of AML patients46,47. Inv(3)/t(3;3) rearrangements can also be observed in up to 25–40% of CML patients in blast crisis48,49. Despite their existence as distinct clinical entities, MDS, AML, and CML with inv(3)/t(3;3) rearrangements have similar cytogenetic abnormalities, molecular alterations, pathological features, and poor prognosis46,50,51,52. In inv,(3) breaks most frequently occur in a region between RPN1 (Ribophorin 1) and C3orf27, downstream of GATA2, that contains a distal GATA2 hematopoietic enhancer (−77 kb, G2DHE) and the region between C3orf50 and the first exon of the MECOM locus that encodes for the MDS1-EVI1 transcript (Fig. 3)53. The EVI1 and EVI1Δ324 transcripts remain intact, but the MDS1-EVI1 transcript is frequently not expressed53. In t(3;3), the breakpoint frequently occurs in between the MDS1 promoter and the first EVI1 exon, and MDS1-EVI1 transcript is frequently lost (Fig. 3)46. In 2014, the Delwel group and the Yamamoto group identified that a new super enhancer of ~40 kb is formed from repositioning of the GATA2 distal hematopoietic enhancer that drives increased EVI1 expression in inv(3)/t(3;3) AML53,54. The new enhancer region generated by the chromosomal rearrangement was noted to contain a 9-kb region with a p300 binding site that interacts with the EVI1 promoter, and removal of this binding site attenuates EVI1 expression53. Transgenic mice in which the human inv(3) chromosomal abnormality was recapitulated through a bacterial artificial chromosome developed leukemia, but if the relocated GATA2 enhancer region was deleted, EVI1 expression declined and leukemia did not develop54. These seminal studies confirmed that EVI1 dysregulation in response to chromosomal rearrangements in myeloid disease occurred not from the typical generation of a fusion transcript, but rather from “enhancer hijacking”53,54. This highlighted a two-fold impact, one leading to overexpression of EVI1 and the second causing haploinsufficiency of GATA2, as it is no longer expressed in the rearranged chromosome53.

In both inv(3)(q21q26) or t(3;3)(q21q26), the breakpoints lead to juxtaposition of a region surrounding the distal GATA2 enhancer and the RPN1 gene in 3q21 with the EVI1 gene in 3q26. Breakpoints occur 3′ of the EVI1 gene in the inv(3)(q21q26) setting, whereas they occur 5′ of the EVI1 gene in the case of t(3;3)(q21q26). In both types of 3q21q26 rearrangement, the GATA2 enhancer induces EVI1 gene transcription instead of GATA2 expression and thus promotes leukemogenesis.
Atypical 3q26 rearrangements
In ~0.5–1% of AML and MDS patients, atypical chromosome 3 rearrangements occur involving the MECOM locus55. Most atypical 3q26 rearrangements have levels of EVI1 overexpression comparable to inv(3)/t(3;3) cases, similar phenotypic changes, and share a poor prognosis46,55. Atypical 3q26 rearrangements include, but are not limited to t(2;3)(q21;q26), t(3;7)(q26;q24), t(3;8)(q26;q24), and t(3;6)(q26;q25), which involve THADA (THADA armadillo repeat containing), CDK6 (cyclin dependent kinase 6), MYC (V-Myc avian myelocytomatosis viral oncogene homolog), and ARID1B (AT-rich interaction domain 1B), respectively55. Similar to the “enhancer hijacking” of the GATA2 distal hematopoietic enhancer in inv(3)/t(3;3), atypical 3q26 rearrangements also seem to overexpress EVI1 through repurposing of enhancer elements from the translocation partners. In ten cases of atypical 3q26 rearrangements, EVI1 was overexpressed and the translocation partner whose enhancer was repurposed had decreased expression, with the exception of MYC in t(3;8)(q26;q24)55.
EVI1 fusion proteins
Several translocations involving the MECOM locus do result in the generation of fusion proteins. The two most common being t(3;12)(q26;p13) and t(3;21)(q26;q24) that result in ETS variant transcription factor 6 (also TEL)-EVI1 and RUNX1-MDS1-EVI1 fusion proteins, respectively22,56 (Fig. 4). Both translocations are rare, found in less than 1% of myeloid malignancies56,57. Fluorescent in situ hybridization demonstrated that the t(3;12) breakpoints are between the ETV6 exon 2 and 3 and on heterogeneous regions in 3q26, both 3′ and 5′ of MDS1 as well as in between MDS1 and EVI156. The resulting translocation fuses the first two exons of ETV6 with the entire MDS1-EVI1 or EVI1 transcript. Since no known functional domain of ETV6 is added to EVI1 in the fusion protein, it is thought that the oncogenic properties of the fusion protein come from the inappropriate expression and function of EVI1 driven by the ETV6 promoter56. In line with this, similar to other 3q26 rearrangements, myeloid malignancies expressing the ETV6-EVI1 fusion are associated with dysmegakaryopoiesis and poor prognosis58. The t(3;21) can generate RUNX1-MDS1-EVI1 fusion protein, where the DNA-binding RUNT domain of RUNX1 is fused to the whole MDS1-EVI1 protein (Fig. 4)59. Expression of the RUNX1-MDS1-EVI1 protein is associated with disruption of RUNX1 and EVI1 regulatory networks. This is thought to be partly achieved by transcriptional repression of RUNX1 targets through recruitment of co-repressors by EVI1 in the fusion protein57. In mouse models of conditional RUNX1-MDS1-EVI1 expression or transplant models, the fusion protein is associated with development of hematopoietic dysplasia and acute megakaryoblastic leukemia60.

Schematic diagram of the EVI1, MDS1, MDS1-EVI1, RUNX1, RUNX1-MDS1-EVI1, ETV6, ETV6-EVI1, and ETV6-MDS1-EVI1 proteins.
EVI1 overexpression without chromosome 3 aberrations
Aberrant EVI1 expression can also occur in the absence of chromosome 3 rearrangements. EVI1 overexpression is observed in ~8–10% of MDS, 8% of de novo AML, and 30% of advanced CML, but it is unclear here how EVI1 overexpression occurs61. Several ChIP studies have shown that mixed lineage leukemia (MLL) and MLL fusion proteins, including MLL-AF9 and MLL-ENL bind to the EVI1 regulatory region, resulting in increased EVI1 expression62,63. In a recent report in which MLL-AF9 fusion gene was expressed either in murine Sca−Kit+ (LSK) HSCs or in granulocyte monocyte precursors (GMPs), LSK-MLL-AF9 cells had significantly higher levels of Evi1 than GMP-MLL-AF9 cells64. Additionally, AMLs with high EVI1 expression have been shown to be associated with inferior relapse-free and overall survival65.
Biologic consequences of 3q lesions and EVI1 overexpression
Genomic instability
Utilizing SILAC-MS studies to determine EVI1 interaction partners, Bard-Chapeau et al. observed enrichment in protein domains associated with DNA repair, chromatin remodeling, and transcription29. Furthermore, the EVI1 N-terminal ZF domain binds to GATA-like SSRs, and EVI1 ChIP analysis revealed an increase in recombination rates near EVI1 bound SSR13,24. How EVI1 increases genomic instability is not well characterized beyond its protein interactions. However, a gene therapy study using a Maloney murine leukemia virus vector to express NADPH-oxidase conducted in two patients to treat chronic granulomatous disease unfortunately caused integration of the vector at the MECOM locus. The patients developed clonal expansion of myeloid cells bearing activating insertions in the MECOM locus and EVI1 overexpression. Both patients developed monosomy 7 in the dominate clone, suggesting that EVI1 could favor expansion of clones with monosomy 7 or that EVI1 could contribute to the genomic instability leading to monosomy 713.
Effect of EVI1 on hematopoietic stem cell proliferation/differentiation
EVI1 is known to directly interact with and repress the activity of a number of myeloid transcription factors including GATA1, PU.1, and RUNX117,19,42,43. Enforced Evi1 expression transcriptionally repressed C/EBP-α in murine hematopoietic cells18. EVI1-mediated repression of C/EBP-α was also observed in the murine hematopoietic progenitor cell line 32Dcl317. Further confirmation that EVI1 represses C/EBP family members is needed through in vivo leukemia models and in patient-derived samples. EVI1 also regulates hematopoietic differentiation and proliferation through transcriptional repression of several miRNAs. EVI1 repressed miR-9 levels through binding to its regulatory region, recruiting DNMT3B, and inducing DNA methylation66. Decreased miR-9 led to increased levels of its target genes FOXO1 and 3 (Forkhead Box O1 and 3)66. EVI1 expression was also found to decrease miR-449A levels, and ChIP analysis showed EVI1 bound miR-449A regulatory region67. Repression of miR-449A by EVI1 increased expression of the miR-449A-targets Notch1 and Bcl-2 in human AML cell lines67.
EVI1 transcriptionally activates the hematopoietic proto-oncogene PBX1 (PBX homeobox 1) through binding to its promoter region68. Knockdown of PBX1 decreased EVI1-mediated transformation of primary mouse bone marrow cells68. Comparing tissues from wild type to those from EVI1+/− and EVI1−/− mice, at embryonic day 9.5, GATA2 expression was decreased in EVI1 depleted tissues69. EVI1 expression also correlated with high expression of megakaryocytic markers, including the thrombopoietin receptor MPL70. Furthermore, in a mouse model of EVI1 leukemia, thrombopoietin expression correlated with EVI1 expression, and double positive EVI1-thrombopoietin cells had enhanced secondary leukemia formation ability in a serial bone marrow transplant assay70. Collectively, in myeloid malignancies expressing EVI1, transcriptional alterations of specific myeloid transcription factors, and of miRNAs, contribute to myeloid dysplasia.
Increased drug resistance
Several pathways have been implicated in EVI1-mediated resistance to apoptosis leading to drug-resistance. High EVI1 expression correlated with high expression of the anti-apoptotic Bcl-xL protein in CML patient samples33. Conversely, knockdown of EVI1 was shown to decrease Bcl-xL levels by approximately five fold33. EVI1 interactions with the microenvironment are also implicated in apoptosis-resistance. The adhesion molecules ITGA6 (integrin subunit alpha 6) and GPR56 (adhesion G protein-coupled receptor G1) are highly expressed in EVI1-positive AML, and their knockdown leads to increased apoptosis in response to Ara-C treatment and loss of RhoA (ras homolog family member A) signaling, respectively71,72. In AML, cells high EVI1 expression correlated with high MYC and BCL2 expression, with poorer clinical outcome73.
Clinical phenotypes and outcome of EVI1-positive myeloid malignancies
MDS with EVI1 overexpression is commonly associated with dyserythropoiesis and with the presence of micro megakaryocytes51. Categorized as high risk, more than half of inv(3)/t(3;3) MDS patients with EVI1 overexpression progress to AML within ~2 years of diagnosis46,47. Furthermore, EVI1 overexpression correlates with shorter overall survival and poorer response to treatment51. Overall survival of patients with EVI1-positive MDS ranges from 13 to 17 months after diagnosis46,47. Like EVI1-positive MDS, AML with EVI1 overexpression often presents with myeloid dysplasia, particularly of the erythrocyte and megakaryocytic lineages46,51. Studies have also reported EVI1 expression as an independent prognostic factor for poorer overall survival in AML, and high EVI1 expression is associated with poorer response to therapy46,74,75. Several clinical studies have reported that, in 3q26-rearranged AML, the median overall survival after diagnosis remains approximately less than 1 year, whereas long-term overall survival is less than 15% (Table 3)47,74,76.
EVI1-expressing CML may also be associated with megakaryocytic dysplasia51. EVI1 expression is rarely detected in the chronic phase of CML, but is readily detected in a significant proportion (25–40%) of blast crisis of CML, suggesting that acquisition of EVI1 expression can drive progression into blast crisis48,49. EVI1 expression in CML blast crisis is correlated with poor response to therapy, and has been linked with acquisition of resistance to tyrosine kinase inhibitors48.
Monosomy 7 and MLL translocations
Loss of one copy of chromosome 7 (monosomy 7, −7) or deletion of the long arm of chromosome 7 (−7q) is observed in 30–70% of MDS and AML with inv(3)/t(3;3)74. Retrospective studies have shown that inv(3)/t(3;3) MDS/AML with −7/−7q display worse prognosis than inv(3)/t(3;3) alone74,77. Which genetic alteration occurs first is unclear, and likely varies on a case-by-case basis, given the heterogeneity of the myeloid malignancies. As noted above, in two cases where gene therapy activated EVI1 expression through retroviral insertion, both cases developed monosomy 7 in the dominant leukemic clone, suggesting that EVI1 at least favors events leading to monosomy 713. The q arm of chromosome 7 contains several key genes whose haploinsufficiency is considered to be a loss of tumor-suppressor and thus contribute to leukemia transformation. These genes include EZH2 and MLL3, as well as the cytoplasmic cellular regulators SAMD9 (Sterile Alpha Motif Domain Containing 9) and SAMD9L78. Perhaps due to the ability of MLL and MLL-fusion proteins to upregulate EVI1 transcription, EVI1 overexpression can be observed in ~30% of cases with MLL translocation, and here EVI1 expression correlates with poor prognosis63,65.
Transcription factor mutations
Approximately 20% of MDS and AML patients with inv(3)/t(3;3) express mutations in RUNX179. Another transcription factor IKZF1 (IKAROS family zinc finger) is also mutated in up to 25% of cases of inv(3)/t(3;3) MDS or AML. Since IKZF1 is located on chromosome 7, IKZF1 mutations occur in clones without chromosome 7 deletions77. Although not a mutation, almost all MDS, AML, and CML with inv(3)/t(3;3) have GATA2 haploinsufficiency due to the re-location of the GATA2 distal hematopoietic enhancer53,54. This was shown to contribute to EVI1-driven leukemia transformation21. Of note, despite loss of expression from one allele of GATA2, 15% of inv(3)/t(3;3) can carry additional mutations in GATA2 on the non-rearranged allele52,77.
Activating mutations in signaling pathways
A significant proportion of inv(3)/t(3;3) MDS and AML cases have activating mutations in RAS GTPase family member (NRAS or KRAS), or in other RAS-signaling pathway proteins, including PTPN11 (protein tyrosine phosphatase non-receptor type 11), and NF1 (neurofibromin 1), which promote dysregulated RAS signaling and uncontrolled proliferation52,77,79. These mutations are observed in 66–98% of inv(3)/t(3;3) MDS/AML52,77,79. A greater percentage of AML cases with inv(3)/t(3;3) AML carried RAS family mutations, as compared to the MDS cases79.
Mutations in epigenetic machinery
Low frequency of mutations in DNMT3, TET2 (tet methylcytosine dioxygenase 2), and IDH1/2 (isocitrate dehydrogenase 1/2) were observed in AML or MDS with inv(3)/t(3;3)46,51,77. However, mutations in the polycomb group protein ASXL1 (ASXL transcriptional regulator 1) were reported in ~20% of AML cases with inv(3)/t(3;3)77. Mutations in splicing factors SF3B1 (splicing factor 3b subunit 1) and U2AF1 (U2 small nuclear RNA auxiliary factor 1) were also found in ~30–60% of inv(3)/t(3;3) MDS or AML cases52,77. The biologic impact of these ‘epimutations’ in myeloid malignancies on the transcriptional signature attributed to inv(3)/t(3;3) and EVI1 overexpression remains to be elucidated.
Mutations inversely correlated with EVI1
Mutations in NPM1 (nucleophosmin 1) and C/EBP-α inversely correlate with inv(3)/t(3;3) and EVI1 expression55,77,79. Why EVI1 overexpression is not seen with NPM1 or C/EBP-α mutations is unknown. One possibility could be that survival of the clones with high EVI1 expression in combination with a NPM1 or C/EBP-α mutation is impaired.
Clinical outcome with standard therapy of myeloid malignancies with inv(3)/t(3;3)
Standard front-line treatment of MDS includes DNA de-methylating agents like azacitidine and decitabine. In advanced, high-risk MDS carrying inv(3)/t(3;3) with an increased percent of bone marrow blasts between 5 and 20%, or in overt transformation of MDS to secondary AML (sAML), chemotherapy with cytarabine (Ara-C) and the anthracyclines, idarubicin and daunorubicin is commonly employed. In CML, treatment in the chronic phase generally begins with a tyrosine kinase inhibitor (TKI) such as imatinib, dasatinib or nilotinib. In transformation of CML into blast crisis (CML-BC) carrying inv(3)/t(3;3) or t(3;21), therapy with a second generation tyrosine kinase inhibitors and/or chemotherapy is utilized. EVI1-positive myeloid malignancies have been documented to be relatively refractory to current therapies. There is no statistical difference in the overall 5-year survival rates between MDS and AML with inv(3)/t(3;3), which averages at 3–5%46,47,51,74. In a study by the Delwel group, allogenic hematopoietic stem cell transplant following the first clinical remission yielded increased survival odds in AML patients with MLL translocation with EVI1 overexpression65. However, greater than 40% of the patients still succumbed to their disease65. Overall survival rate of patients with CML who initially respond but later progress on TKI therapy and acquire EVI1 overexpression is half compared to those without EVI1 expression48.
Potential targeted therapies for EVI1-positive myeloid malignancies
To date, following treatment of myeloid malignancies with inv(3)/t(3;3) or EVI1 overexpression with targeted therapies, including DNA hypomethylating drugs, venetoclax or glasdegib, or with FLT3 TKI or IDH1/2 inhibitors, clinical outcome data are unavailable80. A targeted agent has yet to be identified and developed that exhibits clinical efficacy specifically against EVI1-overexpressing myeloid malignancies.
Treatment with ‘epimodifiers’
One promising target is the chromatin reader protein BRD4 (bromodomain containing 4), which is involved in transcriptional activation, especially via sustaining the activity of super enhancers, such as those of MYC, CDK4/6 and BCL2/Bcl-xL81. By also inhibiting GATA2 super enhancer, treatment with BET (bromodomain and extra-terminal motif) inhibitor could repress EVI1, as well as reverse EVI1-dependent transcriptional programs through inactivating enhancers and super enhancers of the key oncogenes (Fig. 5). Preclinical use of BET inhibitor treatment was reported to inhibit growth and induce apoptosis of an EVI1-overexpressing AML cell line82. Since several BET inhibitors are already undergoing clinical evaluation, they represent an attractive therapy option for myeloid malignancies with inv(3)/t(3;3) and/or EVI1 overexpression53.

In AML cells with inv(3)(q21q26) or t(3;3)(q21q26), the hijacked GATA2 enhancer interacts with the EVI1 promoter resulting in high expression of EVI. This leads to aberrant regulation of multiple transcriptional programs including metabolism, stem cell phenotype, growth/survival, environmental interactions, and immune surveillance in AML cells. Treatment with BET inhibitors that evict BET proteins such as BRD4 from the chromatin of the GATA2 enhancer leads to downregulation of EVI1 and its activity in AML cells.
Several transcriptional regulators including EVI1 are acetylated by CBP/p30032. For example, RUNX1 interacts with EVI1 and is positively regulated by acetylation32,83,84. Furthermore, the GATA2 distal enhancer that is relocated and transactivates EVI1 in inv(3) and t(3;3) chromosomal aberrations contains a p300 binding site that is critical in driving EVI1 expression54. Additionally, the GATA2 enhancer has increased read-through of enhancer RNAs (eRNAs) at the breakpoints that cause its repositioning, and the CBP/p300 inhibitor GNE-049 is reported to particularly inhibit eRNAs53,85. Therefore, targeted combination therapy with this HAT inhibitor would simultaneously cause deacetylation of the transcription factors, leading to decreased transcriptional activity, as well as cause the loss of super enhancer function, thereby effectively shutting down EVI1 transcriptional program.
EVI1 can repress transcription through recruitment of DNMT3A or B resulting in de novo methylation39,86. EVI1 expression is also associated with hypermethylation of over 200 genes in AML samples39,86. Consistent with this, DNA methyltransferase inhibitors have exhibited clinical activity in EVI1-overexpressing AML87. However, since monotherapy with DNA hypomethylating agent as a first-line treatment for MDS exhibits only a modest clinical efficacy, combination with other targeted therapies is likely to achieve superior efficacy against EVI1-expressing MDS. Use of EZH2 inhibitor to abrogate dependency of EVI1-expressing MDS or AML with monosomy 7 on the residual normal EZH2 function may exert added efficacy.
Targeted therapy with BH3-mimetic apoptosis inducers
Recently, co-treatment with venetoclax, a BH3-mimetic inhibitor of Bcl-2 protein, with azacitidine was approved for therapy of AML88. EVI1-mediated upregulation of the anti-apoptotic Bcl-xL suggests that BH3-mimetic inhibitor targeting this anti-apoptotic protein could also have therapeutic value, alone or in combinations against EVI1-expressing myeloid malignancies. Recently, the small molecule compound pyrrole-imidazole polyamide, which inhibits the DNA-binding activity of N-terminal ZF domain of EVI1, was shown to induce apoptosis of EVI1-expressing AML cells due to downregulation of the EVI1 target GRP56, which was linked to EVI1-mediated resistance to apoptosis71. Several therapies targeting anti-apoptotic proteins are in clinical trials, and these agents could potentially be employed in treatment of EVI1-expressing myeloid malignancies.
Targeting β-catenin-TCF7L2 activity
In CML with inv(3)/t(3;3), β-catenin/TCF1 (T cell factor 1) signaling was reported to be activated, which positively regulated EVI128. Therefore, along with treatment with BCR-ABL1 targeted TKI inhibition of β-catenin/TCF signaling may be a promising strategy. Additionally, several groups have demonstrated that resistance to BET inhibitors in AML is mediated by the activity of β-catenin/TCF7L2/c-Myc axis, resulting in re-expression of c-Myc despite treatment with BET inhibitor89. In this setting also, treatment with a β-catenin/TCF signaling inhibitor, e.g., BC2059 (tegavivint), may not only reverse BET inhibitor resistance but also exhibit synergy with BET inhibitor against AML with inv(3)/t(3;3) and/or EVI1 overexpression90.
Summary and future directions
While great strides have been made, there is still much more to be elucidated regarding EVI1 biology and its contributions to leukemogenesis. EVI1 is a transcriptional regulator that promotes a stem-like expression program in hematopoietic progenitors, crucial to their self-renewal, growth, and repopulating potential. The role of co-expression of alternative EVI1 transcripts in myeloid malignancies remains to be determined. EVI1 regulates transcription through recruitment of epigenetic modifiers. Recent studies have highlighted the dysregulated transcriptome and signaling pathways that underpin the aberrant biology, aggressive phenotype, and refractoriness to standard therapy of EVI1-overexpressing myeloid malignancies (Fig. 5). How commonly the co-occurring genetic alterations and mutations, e.g., monosomy 7 and RAS pathway mutations, and their order of acquisition, contribute to the aggressive phenotype and therapy-refractoriness of EVI1-overexpressing myeloid malignancies has yet to be fully characterized. New probes or alternative methodology need to be developed that will assist in probing the clonal architecture of the co-mutations that occur with EVI1 dysregulation at the single-cell level. Importantly, functional genomic studies need to be conducted to identify specific dependences that can be targeted to achieve superior efficacy against EVI1-overexpressing myeloid malignancies. Large scale screens with or without the presence of a promising therapeutic agent, e.g., by CRISPR technology, could also potentially yield new knowledge for designing effective combination therapies. Analysis of the 3D chromatin architecture of “hijacked enhancers” at EVI1 locus and their response to treatment may also provide insights into effective ways to decrease EVI1 expression, suggest promising treatments, and potential mechanisms of resistance. How dysregulated EVI1 expression in myeloid malignancies creates immune evasion and T cell exhaustion also remains to be fully elucidated. In this exciting era of novel immunotherapies new research avenues and potential strategies have already been illuminated. These are likely to involve harnessing of the innate or adaptive immune mechanisms to overcome immune tolerance or T cell exhaustion in eliminating myeloid malignancies, including those driven by EVI1 dysregulation.
References
Mucenski, M. L. et al. Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors. Mol. Cell. Biol. 8, 301–308 (1988).
Morishita, K. et al. The human Evi-1 gene is located on chromosome 3q24-q28 but is not rearranged in three cases of acute nonlymphocytic leukemias containing t(3;5)(q25;q34) translocations. Oncogene Res. 5, 221–231 (1990).
Fears, S. et al. Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family. Proc. Natl Acad. Sci. USA 93, 1642–1647 (1996).
Bordereaux, D., Fichelson, S., Tambourin, P. & Gisselbrecht, S. Alternative splicing of the Evi-1 zinc finger gene generates mRNAs which differ by the number of zinc finger motifs. Oncogene. 5, 925–927 (1990).
Perkins, A. S., Fishel, R., Jenkins, N. A. & Copeland, N. G. Evi-1, a murine zinc finger proto-oncogene, encodes a sequence-specific DNA-binding protein. Mol. Cell. Biol. 11, 2665–2674 (1991).
Bard-Chapeau, E. A. et al. Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc. Natl Acad. Sci. USA 109, 2168–2173 (2012).
Izutsu, K. et al. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood. 97, 2815–2822 (2001).
Palmer, S. et al. Evi-1 transforming and repressor activities are mediated by CtBP co-repressor proteins. J. Biol. Chem. 276, 25834–25840 (2001).
Hoyt, P. R. et al. The Evi1 proto-oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech. Dev. 65, 55–70 (1997).
Zhang, Y. et al. PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood. 118, 3853–3861 (2011).
Kataoka, K. et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J. Exp. Med. 208, 2403–2416 (2011).
Du, Y., Jenkins, N. A. & Copeland, N. G. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. 106, 3932–3939 (2005).
Stein, S. et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 16, 198–204 (2010).
Kustikova, O. S. et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia. 27, 1127–1138 (2013).
Steinleitner, K. et al. EVI1 and MDS1/EVI1 expression during primary human hematopoietic progenitor cell differentiation into various myeloid lineages. Anticancer Res. 32, 4883–4889 (2012).
Morishita, K., Parganas, E., Matsugi, T. & Ihle, J. N. Expression of the Evi-1 zinc finger gene in 32Dc13 myeloid cells blocks granulocytic differentiation in response to granulocyte colony-stimulating factor. Mol. Cell. Biol. 12, 183–189 (1992).
Kreider, B. L., Orkin, S. H. & Ihle, J. N. Loss of erythropoietin responsiveness in erythroid progenitors due to expression of the Evi-1 myeloid-transforming gene. Proc. Natl Acad. Sci. USA 90, 6454–6458 (1993).
Wilson, M. et al. EVI1 Interferes with Myeloid Maturation via Transcriptional Repression of Cebpa, via Binding to Two Far Downstream Regulatory Elements. J. Biol. Chem. 291, 13591–13607 (2016).
Laricchia-Robbio, L., Premanand, K., Rinaldi, C. R. & Nucifora, G. EVI1 Impairs myelopoiesis by deregulation of PU.1 function. Cancer Res. 69, 1633–1642 (2009).
Shimizu, S. et al. EVI1 is expressed in megakaryocyte cell lineage and enforced expression of EVI1 in UT-7/GM cells induces megakaryocyte differentiation. Biochem. Biophys. Res. Commun. 292, 609–616 (2002).
Yamaoka, A. et al. EVI1 and GATA2 misexpression induced by inv(3)(q21q26) contribute to megakaryocyte-lineage skewing and leukemogenesis. Blood Adv. 4, 1722–1736 (2020).
Nucifora, G. et al. Consistent intergenic splicing and production of multiple transcripts between AML1 at 21q22 and unrelated genes at 3q26 in (3;21)(q26;q22) translocations. Proc. Natl Acad. Sci. USA 91, 4004–4008 (1994).
Pinheiro, I. et al. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell. 150, 948–960 (2012).
Sayadi, A. et al. Functional features of EVI1 and EVI1Delta324 isoforms of MECOM gene in genome-wide transcription regulation and oncogenicity. Oncogene. 35, 2311–2321 (2016).
Vazquez, I. et al. Down-regulation of EVI1 is associated with epigenetic alterations and good prognosis in patients with acute myeloid leukemia. Haematologica. 96, 1448–1456 (2011).
Maicas, M. et al. Functional characterization of the promoter region of the human EVI1 gene in acute myeloid leukemia: RUNX1 and ELK1 directly regulate its transcription. Oncogene. 32, 2069–2078 (2013).
Maicas, M. et al. The MDS and EVI1 complex locus (MECOM) isoforms regulate their own transcription and have different roles in the transformation of hematopoietic stem and progenitor cells. Biochim. Biophys. Acta. Gene Regul. Mech. 1860, 721–729 (2017).
Manachai, N. et al. Activation of EVI1 transcription by the LEF1/beta-catenin complex with p53-alteration in myeloid blast crisis of chronic myeloid leukemia. Biochem. Biophys. Res. Commun. 482, 994–1000 (2017).
Bard-Chapeau, E. A. et al. EVI1 oncoprotein interacts with a large and complex network of proteins and integrates signals through protein phosphorylation. Proc. Natl Acad. Sci. USA 110, E2885–E2894 (2013).
White, D. J. et al. Phosphorylation of the leukemic oncoprotein EVI1 on serine 196 modulates DNA binding, transcriptional repression and transforming ability. PLoS ONE 8, e66510 (2013).
Paredes, R. et al. EVI1 carboxy-terminal phosphorylation is ATM-mediated and sustains transcriptional modulation and self-renewal via enhanced CtBP1 association. Nucleic Acids Res. 46, 7662–7674 (2018).
Chakraborty, S. et al. Interaction of EVI1 with cAMP-responsive element-binding protein-binding protein (CBP) and p300/CBP-associated factor (P/CAF) results in reversible acetylation of EVI1 and in co-localization in nuclear speckles. J. Biol. Chem. 276, 44936–44943 (2001).
Pradhan, A. K., Mohapatra, A. D., Nayak, K. B. & Chakraborty, S. Acetylation of the proto-oncogene EVI1 abrogates Bcl-xL promoter binding and induces apoptosis. PLoS ONE 6, e25370 (2011).
Shimahara, A., Yamakawa, N., Nishikata, I. & Morishita, K. Acetylation of lysine 564 adjacent to the C-terminal binding protein-binding motif in EVI1 is crucial for transcriptional activation of GATA2. J. Biol. Chem. 285, 16967–16977 (2010).
Cattaneo, F. & Nucifora, G. EVI1 recruits the histone methyltransferase SUV39H1 for transcription repression. J. Cell. Biochem. 105, 344–352 (2008).
Spensberger, D. & Delwel, R. A novel interaction between the proto-oncogene Evi1 and histone methyltransferases, SUV39H1 and G9a. FEBS Lett 582, 2761–2767 (2008).
Yoshimi, A. et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood. 117, 3617–3628 (2011).
Senyuk, V. et al. The oncoprotein EVI1 and the DNA methyltransferase Dnmt3 co-operate in binding and de novo methylation of target DNA. PLoS ONE 6, e20793 (2011).
Lugthart, S. et al. Aberrant DNA hypermethylation signature in acute myeloid leukemia directed by EVI1. Blood. 117, 234–241 (2011).
Bartholomew, C., Kilbey, A., Clark, A. M. & Walker, M. The Evi-1 proto-oncogene encodes a transcriptional repressor activity associated with transformation. Oncogene. 14, 569–577 (1997).
Kurokawa, M. et al. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature. 394, 92–96 (1998).
Laricchia-Robbio, L. et al. Point mutations in two EVI1 Zn fingers abolish EVI1-GATA1 interaction and allow erythroid differentiation of murine bone marrow cells. Mol. Cell. Biol. 26, 7658–7666 (2006).
Senyuk, V. et al. Repression of RUNX1 activity by EVI1: a new role of EVI1 in leukemogenesis. Cancer Res. 67, 5658–5666 (2007).
Tanaka, T. et al. Evi-1 raises AP-1 activity and stimulates c-fos promoter transactivation with dependence on the second zinc finger domain. J. Biol. Chem. 269, 24020–24026 (1994).
Sun, J. et al. De novo acute myeloid leukemia with inv(3)(q21q26.2) or t(3;3)(q21;q26.2): a clinicopathologic and cytogenetic study of an entity recently added to the WHO classification. Mod. Pathol. 24, 384–389 (2011).
Lugthart, S. et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J. Clin. Oncol. 28, 3890–3898 (2010).
Cui, W. et al. Myelodysplastic syndrome with inv(3)(q21q26.2) or t(3;3)(q21;q26.2) has a high risk for progression to acute myeloid leukemia. Am. J. Clin. Pathol. 136, 282–288 (2011).
Paquette, R. L. et al. Frequent EVI1 translocations in myeloid blast crisis CML that evolves through tyrosine kinase inhibitors. Cancer Genet 204, 392–397 (2011).
Wang, W. et al. Clinical and prognostic significance of 3q26.2 and other chromosome 3 abnormalities in CML in the era of tyrosine kinase inhibitors. Blood. 126, 1699–1706 (2015).
Summerer, I. et al. Prognosis of MECOM (EVI1)-rearranged MDS and AML patients rather depends on accompanying molecular mutations than on blast count. Leuk. Lymphoma. 61, 1–4 (2020).
Rogers, H. J. et al. Complex or monosomal karyotype and not blast percentage is associated with poor survival in acute myeloid leukemia and myelodysplastic syndrome patients with inv(3)(q21q26.2)/t(3;3)(q21;q26.2): a Bone Marrow Pathology Group study. Haematologica. 99, 821–829 (2014).
Groschel, S. et al. Mutational spectrum of myeloid malignancies with inv(3)/t(3;3) reveals a predominant involvement of RAS/RTK signaling pathways. Blood. 125, 133–139 (2015).
Groschel, S. et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 157, 369–381 (2014).
Yamazaki, H. et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 25, 415–427 (2014).
Ottema, S. et al. Atypical 3q26/MECOM rearrangements genocopy inv(3)/t(3;3) in acute myeloid leukemia. Blood. 136, 224–234 (2020).
Raynaud, S. D. et al. Fluorescence in situ hybridization analysis of t(3; 12)(q26; p13): a recurring chromosomal abnormality involving the TEL gene (ETV6) in myelodysplastic syndromes. Blood. 88, 682–689 (1996).
Izutsu, K. et al. The t(3;21) fusion product, AML1/Evi-1 blocks AML1-induced transactivation by recruiting CtBP. Oncogene. 21, 2695–2703 (2002).
Shimada, K. et al. CML cells expressing the TEL/MDS1/EVI1 fusion are resistant to imatinib-induced apoptosis through inhibition of BAD, but are resensitized with ABT-737. Exp. Hematol. 40, 724–737 (2012).e2.
Takeshita, M. et al. AML1-Evi-1 specifically transforms hematopoietic stem cells through fusion of the entire Evi-1 sequence to AML1. Leukemia. 22, 1241–1249 (2008).
Maki, K. et al. Development of megakaryoblastic leukaemia in Runx1-Evi1 knock-in chimaeric mouse. Leukemia. 20, 1458–1460 (2006).
Russell, M. et al. Expression of EVI1 in myelodysplastic syndromes and other hematologic malignancies without 3q26 translocations. Blood. 84, 1243–1248 (1994).
Arai, S. et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 117, 6304–6314 (2011).
Bindels, E. M. et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood. 119, 5838–5849 (2012).
Cai, S. F. et al. Leukemia cell of origin influences apoptotic priming and sensitivity to LSD1 inhibition. Cancer Discov. 10, 1500–1513 (2020).
Groschel, S. et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J. Clin. Oncol. 31, 95–103 (2013).
Senyuk, V. et al. Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proc. Natl Acad. Sci. USA 110, 5594–5599 (2013).
De Weer, A. et al. EVI1-mediated down regulation of MIR449A is essential for the survival of EVI1 positive leukaemic cells. Br. J. Haematol. 154, 337–348 (2011).
Shimabe, M. et al. Pbx1 is a downstream target of Evi-1 in hematopoietic stem/progenitors and leukemic cells. Oncogene. 28, 4364–4374 (2009).
Yuasa, H. et al. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 24, 1976–1987 (2005).
Nishikawa, S. et al. Thrombopoietin/MPL signaling confers growth and survival capacity to CD41-positive cells in a mouse model of Evi1 leukemia. Blood. 124, 3587–3596 (2014).
Saha, H. R. et al. Suppression of GPR56 expression by pyrrole-imidazole polyamide represents a novel therapeutic drug for AML with high EVI1 expression. Sci. Rep. 8, 13741 (2018).
Yamakawa, N. et al. The increased expression of integrin alpha6 (ITGA6) enhances drug resistance in EVI1(high) leukemia. PLoS ONE 7, e30706 (2012).
Akhter, A. et al. Acute myeloid leukemia (AML): upregulation of BAALC/MN1/MLLT11/EVI1 gene cluster relate with poor overall survival and a possible linkage with coexpression of MYC/BCL2 Proteins. Appl. Immunohistochem. Mol. Morphol. 26, 483–488 (2018).
Groschel, S. et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J. Clin. Oncol. 28, 2101–2107 (2010).
Grimwade, D. et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 116, 354–365 (2010).
Sitges, M. et al. Acute myeloid leukemia with inv(3)(q21.3q26.2)/t(3;3)(q21.3;q26.2): Study of 61 patients treated with intensive protocols. Eur. J. Haematol. 105, 138–147 (2020).
Lavallee, V. P. et al. EVI1-rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood. 125, 140–143 (2015).
De Weer, A. et al. Identification of two critically deleted regions within chromosome segment 7q35-q36 in EVI1 deregulated myeloid leukemia cell lines. PLoS ONE 5, e8676 (2010).
Haferlach, C. et al. The inv(3)(q21q26)/t(3;3)(q21;q26) is frequently accompanied by alterations of the RUNX1, KRAS and NRAS and NF1 genes and mediates adverse prognosis both in MDS and in AML: a study in 39 cases of MDS or AML. Leukemia. 25, 874–877 (2011).
Short, N. J. et al. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 10, 506–525 (2020).
Bradner, J. E., Hnisz, D. & Young, R. A. Transcriptional Addiction in cancer. Cell. 168, 629–643 (2017).
Loven, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Bae, S. C. & Lee, Y. H. Phosphorylation, acetylation and ubiquitination: the molecular basis of RUNX regulation. Gene. 366, 58–66 (2006).
Filippakopoulos, P. & Knapp, S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356 (2014).
Raisner, R. et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 24, 1722–1729 (2018).
Dickstein, J. et al. Methylation and silencing of miRNA-124 by EVI1 and self-renewal exhaustion of hematopoietic stem cells in murine myelodysplastic syndrome. Proc. Natl Acad. Sci. USA 107, 9783–9788 (2010).
Wanquet, A. et al. Azacitidine treatment for patients with myelodysplastic syndrome and acute myeloid leukemia with chromosome 3q abnormalities. Am. J. Hematol. 90, 859–863 (2015).
Pollyea, D. A., et al. Venetoclax with azacitidine or decitabine in patients with newly diagnosed acute myeloid leukemia: long term follow-up from a Phase 1b study. Am. J. Hematol. 96, 208–217 (2020).
Saenz, D. T. et al. Mechanistic basis and efficacy of targeting the beta-catenin-TCF7L2-JMJD6-c-Myc axis to overcome resistance to BET inhibitors. Blood. 135, 1255–1269 (2020).
Saenz, D. T. et al. Targeting nuclear beta-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia. 33, 1373–1386 (2019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
T.M.K. receives research funding from BMS/Celgene, Amgen, AstraZaneca, Astellas, Pfizer, AbbVie, Genentech, JAZZ, Cellenkos, InCyte, and Ascentage; T.M.K. serves as a consultant/advisory board member for Agios, Pfizer, Abbvie, Genentech, JAZZ, Daiichi Sankyo and Novartis. C.D.D. receives research funding from Abbvie, Agios, Calithera, Cleave, BMS/Celgene, Daiichi-Sankyo, ImmuneOnc and Loxo. C.D.D. serves as a consultant to Abbvie, Agios, Celgene/BMS, Daiichi Sankyo, ImmuneOnc, Novartis, and Takeda. C.D.D. serves on an advisory board with stock options for Notable Labs. K.N.B. serves as a consultant to Iterion Therapeutics. All other authors have no conflict of interest to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Birdwell, C., Fiskus, W., Kadia, T.M. et al. EVI1 dysregulation: impact on biology and therapy of myeloid malignancies. Blood Cancer J. 11, 64 (2021). https://doi.org/10.1038/s41408-021-00457-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-021-00457-9
This article is cited by
-
Preclinical efficacy of targeting epigenetic mechanisms in AML with 3q26 lesions and EVI1 overexpression
Leukemia (2024)
-
Efficacy of novel agents against cellular models of familial platelet disorder with myeloid malignancy (FPD-MM)
Blood Cancer Journal (2024)
-
A distal super-enhancer activates oncogenic ETS2 via recruiting MECOM in inflammatory bowel disease and colorectal cancer
Cell Death & Disease (2023)
-
Identification of transcription factors dictating blood cell development using a bidirectional transcription network-based computational framework
Scientific Reports (2022)
-
AKT inhibition sensitizes EVI1 expressing colon cancer cells to irinotecan therapy by regulating the Akt/mTOR axis
Cellular Oncology (2022)
