
Abstract
DREAMM-2 (NCT03525678) is an ongoing global, open-label, phase 2 study of single-agent belantamab mafodotin (belamaf; GSK2857916), a B-cell maturation antigen-targeting antibody-drug conjugate, in a frozen-liquid presentation in patients with relapsed/refractory multiple myeloma (RRMM). Alongside the main study, following identical inclusion/exclusion criteria, a separate patient cohort was enrolled to receive belamaf in a lyophilised presentation (3.4 mg/kg, every 3 weeks) until disease progression/unacceptable toxicity. Primary outcome was independent review committee-assessed overall response rate (ORR). Twenty-five patients were enrolled; 24 received ≥1 dose of belamaf. As of 31 January 2020, ORR was 52% (95% CI: 31.3–72.2); 24% of patients achieved very good partial response. Median duration of response was 9.0 months (2.8–not reached [NR]); median progression-free survival was 5.7 months (2.2–9.7); median overall survival was not reached (8.7 months–NR). Most common grade 3/4 adverse events were keratopathy (microcyst-like corneal epithelial changes, a pathological finding seen on eye examination [75%]), thrombocytopenia (21%), anaemia (17%), hypercalcaemia and hypophosphatemia (both 13%), neutropenia and blurred vision (both 8%). Pharmacokinetics supported comparability of frozen-liquid and lyophilised presentations. Single-agent belamaf in a lyophilised presentation (intended for future use) showed a deep and durable clinical response and acceptable safety profile in patients with heavily pre-treated RRMM.
Similar content being viewed by others


Introduction
Despite improved outcomes with currently available therapies, including proteasome inhibitors (PIs), immunomodulatory agents and anti-CD38 monoclonal antibodies (mAbs), multiple myeloma (MM) remains a challenging disease that is incurable for most patients1,2,3,4. The typical MM clinical course includes frequent relapses and development of refractory disease5. With each successive line of treatment, the duration of response (DoR) and progression-free survival (PFS) get shorter5,6. Patients refractory to anti-CD38 mAbs have a poor prognosis and limited treatment options, with newer agents used in combination (such as selinexor plus dexamethasone) resulting in an overall response rate (ORR) of 26% in patients refractory to at least one PI, one immunomodulatory agent and daratumumab7. Thus, there remains a need for novel targets and therapies in relapsed/refractory MM (RRMM).
B-cell maturation antigen (BCMA), a member of the tumour necrosis factor receptor family, is expressed on the surface of all normal plasma cells and late-stage B cells, as well as on all malignant cells in all patients with MM8,9. BCMA promotes the maturation and long-term survival of normal plasma cells and is also essential for proliferation and survival of malignant plasma cells in MM9. Belantamab mafodotin (belamaf; GSK2857916) is a first-in-class, BCMA-targeted antibody-drug conjugate (ADC) consisting of a humanised, afucosylated anti-BCMA mAb fused to the cytotoxic payload monomethyl auristatin F (MMAF) by a protease-resistant maleimidocaproyl linker10. Belamaf specifically binds to BCMA and eliminates myeloma cells by a multimodal mechanism, including delivering mafodotin to BCMA-expressing malignant cells thereby inhibiting microtubule polymerisation, and inducing immune-independent ADC-mediated apoptosis; immune-dependent enhancement of antibody-dependent cellular cytotoxicity and phagocytosis; and release of markers characteristic of immunogenic cell death—a form of regulated cell death involving the release of a series of damage-associated molecular patterns (such as calreticulin and high-mobility group box 1) leading to an adaptive immune response10,11,12,13.
In the first-in-human, phase 1 DREAMM-1 study (NCT02064387), single-agent belamaf administered as a frozen-liquid presentation induced clinically meaningful (ORR: 60%; 95% confidence interval [CI]: 42.1–76.1), deep (54% of patients with a very good partial response [VGPR] or better) and durable responses (PFS: 12 months, 95% CI: 3.1–not estimable; DoR: 14.3 months, 95% CI: 10.6–not estimable) with median duration of follow-up of 12.5 months in patients previously treated with alkylators, PIs and immunomodulatory agents and refractory to the last line of therapy14,15. In a sub-group of patients previously treated with anti-CD38 mAbs, and refractory to both a PI and an immunomodulatory agent, an ORR of 38.5% was reported in patients receiving 3.4 mg/kg single-agent belamaf every 3 weeks (Q3W)15.
The pivotal, randomised, phase 2, DREAMM-2 study (NCT03525678) was designed to further assess the efficacy and safety of single-agent belamaf in patients who are refractory to an immunomodulatory agent and a PI and refractory and/or intolerant to a CD38-targeted mAb. Results from the DREAMM-2 primary analysis, in which patients received either belamaf 2.5 or 3.4 mg/kg in a frozen-liquid presentation intravenously Q3W, have been previously reported16,17. After approximately 13 months of follow-up, an ORR of 32% (97.5% CI: 21.7–43.6) and 35% (97.5% CI: 24.8–47.0) for the 2.5 and 3.4-mg/kg cohorts, respectively, was demonstrated in this heavily pre-treated patient population. Responses were deep (VGPR or better) in 58% and 66% of responders in each cohort, respectively. At the time of data cut-off, median PFS was 2.8 (95% CI: 1.6–3.6) and 3.9 (95% CI: 2.0–5.8) months in the 2.5-mg/kg and 3.4-mg/kg cohorts, respectively. The median DoR estimate was 11.0 (95% CI: 4.2–NR) and 6.2 (95% CI: 4.8–NR) months; median OS estimate was 13.7 (95% CI: 9.9–NR) and 13.8 (95% CI: 10.0–NR) months in the 2.5 and 3.4-mg/kg groups, respectively.
A refrigerated lyophilised powder presentation of belamaf was developed to improve supply chain robustness by eliminating frozen shipments and storage, and is the presentation intended for future clinical use. In order to gain clinical experience with the lyophilised presentation of belamaf, an independent, exploratory cohort of patients was included in the DREAMM-2 study to receive this alternative presentation. Herein, we report the analysis for this cohort.
Methods
Study design and treatment
The DREAMM-2 full study design has been reported previously16. In brief, this phase 2, open-label, two-arm, global, multicentre study consisted of a screening/baseline period after which patients in the main study were randomised to receive intravenous belamaf in a frozen-liquid presentation (2.5 or 3.4 mg/kg Q3W). An independent cohort of patients was enrolled to receive belamaf in a lyophilised presentation (3.4 mg/kg Q3W, selected on the basis of the results from the DREAMM-1 study15). As per International Conference on Harmonisation Q5E (ICHQ5E) guidance18, the liquid and lyophilised drug products have been deemed comparable for the purpose of safety and efficacy as both are administered intravenously, have the same formulation, are essentially identical upon dilution for administration, and have been demonstrated to be analytically comparable through extensive biochemical and functional characterisation studies (including primary and higher-order structures, bioassay and binding assays), and stability testing. This new presentation was supplied as a refrigerated lyophilised powder to be reconstituted with water for injection prior to dilution in normal saline. It was administered intravenously over ≥30 min on Day 1 of each 3-week cycle, until disease progression or unacceptable toxicity. No systemic pre-medication was given unless deemed necessary by the investigator. Corticosteroid eye drops and preservative-free lubricant eye drops were used in both eyes to mitigate corneal events, a known toxic effect of MMAF19 and commonly reported in DREAMM-1. At the discretion of patient and investigator, cooling eye masks could be applied from the start of belamaf infusion for approximately 1 h, and up to 4 h, as tolerated. Dose modifications (delays or reductions) were permitted to manage adverse events (AEs), or for medical or surgical and logistical reasons unrelated to treatment. Criteria for dose modifications and patient withdrawal from the study are shown in the study protocol (Supplementary Material). Patients in the lyophilised cohort followed the same assessments and procedures as in the main DREAMM-2 study16.
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines following approval by ethics committees and institutional review boards at each study site. All patients provided written informed consent.
Patient population
Inclusion/exclusion criteria were the same for patients in the lyophilised study cohort and the main DREAMM-2 study16.
Key inclusion criteria
To be eligible for inclusion, patients had to be 18 years or older with an Eastern Cooperative Oncology Group performance status of 0–2 and a histologically or cytologically confirmed diagnosis of MM according to the International Myeloma Working Group (IMWG) criteria20. They must have undergone stem cell transplant (>100 days before enrolment) or been considered transplant-ineligible; had disease progression after ≥3 prior lines of anti-myeloma treatment; were refractory to both an immunomodulatory agent and a PI, and refractory and/or intolerant to an anti-CD38 mAb; and meet the criteria for adequate organ system function. Patients with mild or moderate renal impairment and history of cytopenias (without active conditions) were eligible.
Key exclusion criteria
Patients were excluded if they had received prior allogeneic stem cell transplant, BCMA-targeted therapy, had corneal epithelial disease at screening (except mild punctate keratopathy) or any serious and/or unstable medical, psychiatric disorder or other condition that could interfere with the patient’s safety, ability to provide informed consent or compliance to the study procedures. Full inclusion and exclusion criteria are included in the Supplementary Material.
Endpoints and assessments
Analysis of the lyophilised cohort was an exploratory objective of the main DREAMM-2 study. Key endpoints were ORR (defined as the percentage of patients with a partial response or better, according to IMWG criteria)20 assessed by independent review committee (IRC), clinical benefit rate (minimal response or better), time to response (TTR), time to best response, DoR, time to progression, PFS, OS and safety. Investigator-assessed ORR was also recorded and will be reported elsewhere. The safety profile of lyophilised belamaf was monitored with clinical and laboratory assessments, the reporting of AEs graded (with the exception of keratopathy) according to the Common Terminology Criteria for Adverse Events (2010, version 4.03; see Supplementary Material)21 and the rate of discontinuations and dose adjustments. Keratopathy (defined as microcyst-like epithelial changes [MECs] to the corneal epithelium observed by eye examination, with or without symptoms), thrombocytopenia and infusion-related reactions (IRRs) were monitored as AEs of special interest (AESI). Baseline and subsequent eye examinations were performed pre-dose Q3W by an ophthalmologist or optometrist (full details are provided in the Supplementary Material). Corneal examinations and best-corrected visual acuity assessments (BCVA) were combined and graded on the basis of a keratopathy and visual acuity scale.
Pharmacokinetic analysis
The pharmacokinetic (PK) profile of belamaf was assessed by measurement of belamaf, total mAb (with and without the cytotoxic payload MMAF) and cysteine-maleimidocaproyl MMAF (cys-mcMMAF; the cytotoxic moiety released from belamaf) in plasma collected at Cycles 1 and 3 from all patients. The bioanalytical methods used to quantify concentrations of these analytes were selective, accurate and reproducible (data not shown). The assay methods for belamaf and total mAb measure both free and soluble BCMA-complexed molecules. Individual PK parameters were calculated using standard non-compartmental methods.
Statistical methods
The full analysis population comprised all patients enrolled in the lyophilised cohort of DREAMM-2, regardless of treatment administration. All patients who received ≥1 dose of lyophilised belamaf were included in the safety population. The sample size for this cohort was chosen based on feasibility in order to gain clinical experience with the lyophilised presentation. The probability of observing a ≥20% ORR was retrospectively calculated, with the assumption made that the true ORR was 33%, there would be a 95% probability of observing ≥20% ORR with 25 patients. For the ORR, two-sided exact 95% CI were reported; 95% CI are reported for other data. PFS, DoR and TTR were analysed using the Kaplan–Meier method. Descriptive statistics were used for efficacy endpoints, pre-treatment characteristics, AEs and PK parameters. All efficacy endpoints were assessed by the IRC. This study was overseen by an independent data monitoring committee. Direct comparisons to the main study were not intended or made, due to the non-randomised nature of enrolment into the lyophilised cohort and the relatively small numbers of patients enrolled. Analyses were carried out using Statistical Analysis System software (version 9.4).
Results
Patient disposition and baseline characteristics
Between 5 December 2018 and 10 January 2019, 31 patients were screened for the lyophilised presentation cohort at 9 sites in the USA and Australia. Twenty-five patients were allocated to treatment with the lyophilised presentation of belamaf (full analysis population) and 24 received the allocated treatment (safety population; 1 patient instead received frozen-liquid presentation; Fig. 1). At the data cut-off date (31 January 2020), patients had received a median of 3.5 treatment cycles (range: 1–17); median time on study treatment was 16.6 weeks (range: 3–60). Median duration of follow-up was 11.2 months (range: 1.8–14.5). At data cut-off, 17% (4/24) of patients were still receiving study treatment, and 83% (20/24) of patients had discontinued treatment (primary reason: progressive disease [67%]). Ten deaths were reported in this cohort: nine due to the disease under study, one had another cause.

a Between June 2018 and January 2019, 293 patients were screened for inclusion in the entire DREAMM-2 study. Between 5 December 2018 and 10 January 2019, 31 patients were screened for inclusion in the lyophilised presentation cohort. b Patients could have more than one reason for exclusion. c Five patients were excluded due to pre-existing corneal disease, as specified in the study protocol. d The remainder of enrolled patients were included in the main DREAMM-2 study previously reported16. Two patients in the main study were re-randomised and counted twice (once per each randomisation). e One patient was randomised to the belamaf 3.4 mg/kg lyophilised presentation, but actually received 3.4 mg/kg frozen-liquid presentation as first dose, and never received lyophilised presentation during the study.
Baseline characteristics are presented in Table 1. At screening, patients had received a median of 5 prior lines of therapy (range: 3–11). As per the inclusion criteria, all patients had received prior treatment with, and upon analysis were refractory to, a PI, an immunomodulatory agent, and an anti-CD38 mAb (daratumumab). Patients with high-risk cytogenetics (per IMWG criteria)20, international staging system stage III disease and extramedullary disease were well represented.
Efficacy
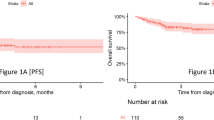
The IRC-assessed ORR was 52% (95% CI: 31.3–72.2). A VGPR was seen in 24% (6/25) of patients (46% [6/13] of responders) (Fig. 2 and Table 2). The IRC-assessed clinical benefit rate (minimal response or better) was 56% (95% CI: 34.9–75.6). The median (95% CI) TTR was 0.9 months (0.8–2.3) and the median time to best response was 1.4 months (0.8–2.9). At the time of data analysis, the median DoR was 9.0 months (95% CI: 2.8–NR) (Fig. 3A). Based on the Kaplan–Meier curve, the probability of maintaining a response ≥6 months was estimated to be 54% (95% CI: 25–76). The median PFS was 5.7 months (95% CI: 2.2–9.7 [Fig. 3B]). At data cut-off, median OS was not reached (95% CI: 8.7 months–NR).

Abbreviations: PR partial response, VGPR very good partial response. Responses were assessed by an independent review committee according to International Myeloma Working Group criteria20. Orange triangles represent patients with study treatment ongoing. Asterisks represent patients with follow-up ongoing. Responses are indicated at the time of the first report of ≥PR, followed by best response, unless the two occurred concurrently.

Responses were assessed by an independent review committee according to International Myeloma Working Group criteria20.
Safety
Overall, 100% (24/24) of patients experienced ≥1 AE. The most common AEs (any grade) were keratopathy (MECs, changes to the corneal epithelium observed by eye examination with or without symptoms), thrombocytopenia, fatigue, blurred vision, dry eye, anaemia and back pain (Table 3). The most common grade 3/4 AEs were keratopathy (MECs), thrombocytopenia, anaemia, hypercalcaemia, hypophosphatemia, neutropenia and blurred vision (Table 3). Serious AEs (SAEs) were reported in 63% of patients (Supplementary Table 1) and were considered treatment related in 17% of patients. There was one death due to an SAE (due to cardiac failure; unrelated to study treatment).
Median dose intensity was 2.32 mg/kg Q3W (range: 1.0–3.4), which was lower than intended due to the incidence of dose reductions and delays. Dose reductions and delays occurred in 58% (14/24) and 71% (17/24) of patients, respectively. Of those with dose reductions, 71% (10/14) of patients had a single dose reduction to 2.5 mg/kg and 29% (4/14) had a second reduction to 1.92 mg/kg. In patients with dose delays, 59% (10/17) of patients had a single dose delay, 12% (2/17) had two dose delays and 29% (5/17) of patients had ≥3 dose delays. The median duration of dose delays was 21 days (range: 4–168). AEs leading to dose reductions (58%) and delays (79%) were common; 2 patients (8%) permanently discontinued treatment due to AEs (keratopathy [MECs] in 1 patient, cardiac failure in 1 patient). Permanent treatment discontinuation due to AEs was considered treatment related in 1 patient (4%). The most common AEs leading to dose reductions (occurring in ≥5% of patients) included keratopathy (MECs; in 46% of patients), thrombocytopenia (8%) and blurred vision (8%); patients could have more than one AE leading to dose reduction. Keratopathy (MECs; 75%) and blurred vision (25%) were the most common AEs leading to dose delays.
Thrombocytopenia (which included thrombocytopenia, haematoma and platelet count decreased) was reported in 46% of patients, with 21% of patients experiencing grade 3/4 events (Table 3). IRRs (including terms IRR, pyrexia, transfusion reaction and chills occurring ≤24 h of infusion) occurred in 17% (4/24) of patients, with no grade 3/4 events. In patients with IRRs, the first occurrence was typically with first infusion (in 75% [3/4] patients); 2/4 patients experienced a single IRR and 2/4 had two IRRs; IRRs resolved in all patients. Although not protocol mandated, 46% of patients received at least one prophylactic pre-medication for IRRs, with 29% of patients receiving prophylactic pre-medication at Cycle 1. In terms of drug class, 33% of patients in the safety population received an analgesic (paracetamol), 42% received an antihistamine and 25% received a steroid as prophylactic pre-medication for IRRs.
Keratopathy (MECs) was the most frequent AE (96%); grade 1/2 (mild/moderate) events were recorded in 21% (5/24) patients and grade 3 (severe) events in 75% (18/24) patients. No grade 4 events occurred. In patients with grade ≥2 events (n = 21), the median time to onset of the first occurrence of keratopathy (MECs) was 23 days (range: 18–283). The onset of keratopathy (MECs) was reported in 58% of patients in the safety population at Cycle 1, in 83% of patients by Cycle 2, in 92% of patients by Cycle 4 and reached the maximum reported incidence of 96% after Cycle 10. At data cut-off, 52% (11/21) of patients with ≥grade 2 keratopathy (MECs) recovered from the first occurrence, with a median duration of first occurrence of 127 days (range: 23–278). Among the 11 patients who recovered, 5 recovered after treatment discontinuation, 5 recovered with dose delay or dose reduction, and 1 recovered without dose modification. At data cut-off, the first occurrence of ≥grade 2 keratopathy (MECs) had not resolved in the remaining 48% (10/21) of patients. Of these, 20% (2/10) of patients were on treatment, 50% (5/10) were no longer in follow up due to death or loss to follow-up and 30% (3/10) were still in follow-up. Keratopathy (MECs) was the most common AE leading to dose delays (75%) and reductions (46%). Dose delays due to keratopathy (MECs) began at Week 4, while dose reductions began later, at Week 7.
BCVA declined to 20/50 or worse in the better seeing eye at least once during or after the treatment period in 33% (8/24) of patients. Median time to onset for the first occurrence was 57 days (range: 39–146). As of the last follow-up, 100% (8/8) patients had recovered (BCVA better than 20/50 in the better seeing eye) with median time to recovery of 21.5 days (range: 16–43). Two patients had a transient worsening of their vision (BCVA worse than or equal to 20/200) in one eye only; however, both patients saw an improvement in BCVA (i.e., returned to baseline during follow-up). In 1 patient, the event occurred 61 days after the last dose (treatment discontinued due to progressive disease) and had resolved 21 days later; in the other patient, the event occurred after the first dose but was resolved prior to administration of the second dose. This patient has remained on treatment without a further occurrence; follow-up is ongoing. No patients had a transient worsening of vision to 20/200 in their better seeing eye.
Among patients with keratopathy (MECs), 83% reported symptoms (including blurred vision or subjective dry eye) and/or had a decrease in BCVA (2 or more lines decline in the better seeing eye). Overall, blurred vision and dry eye were the most common patient-reported corneal symptoms (38 and 25%, respectively), and were generally <grade 3 (Table 3). Median time to first occurrence of blurred vision and dry eye was 26 days (range: 19–247) and 45 days (range: 2–66), respectively. Median duration of first occurrence was 43 days (range: 26–178) and 115 days (range: 4–173), respectively. As of last follow-up, blurred vision had resolved in 56% (5/9) of affected patients, and dry eye resolved in 33% (2/6) of patients.
Pharmacokinetics
Exposure measures (AUC and Cmax) were generally similar for the three analytes (belamaf, total mAb and cys-mcMMAF) after administration of either the frozen-liquid or lyophilised presentations of belamaf (Table 4). In population PK analyses, presentation was not a significant factor for belamaf PK (data not shown). After accounting for key covariates, PK behaviour of the three analytes was similar after administration of the frozen-liquid and lyophilised presentations.
Discussion
In this exploratory cohort of patients with heavily pre-treated RRMM, single-agent belamaf (3.4 mg/kg Q3W) in a lyophilised presentation demonstrated deep and durable anti-myeloma activity, with an ORR of 52%. Responses were deep, with 46% (6/13) of responders achieving a VGPR. ORRs were similar to those in patients with RRMM who were refractory to a PI and an immunomodulatory agent and exposed to anti-CD38 mAbs receiving single-agent belamaf 3.4 mg/kg Q3W in both the first-in-human DREAMM-1 study (ORR: 38.5% in this sub-group of 13 patients) and the previously published main DREAMM-2 study (ORR: 35% at 13-month follow-up). The ORR reported in this study compares favourably with STORM, the only other clinical trial designed to prospectively evaluate an anti-myeloma treatment (combination selinexor plus dexamethasone) in patients refractory to at least one PI, one immunomodulatory agent, and daratumumab (as in DREAMM-2), in which an ORR of 26% was reported7. The STORM study recruited patients previously exposed to bortezomib, carfilzomib, lenalidomide, pomalidomide, daratumumab and an alkylating agent, a similar population to this DREAMM-2 cohort in which all patients were exposed to bortezomib, lenalidomide, pomalidomide and daratumumab, and 80% of patients were exposed to carfilzomib. The median DoR in this study was 9.0 months (95% CI: 2.8–NR) after median follow-up of approximately 11 months; a median DoR of 4.4 months was reported in STORM7, suggesting that clinical responses to belamaf are durable, as was the case in the DREAMM-1 study15. The median PFS in this patient cohort was 5.7 months, while median OS was not reached (95% CI: 8.7 months–NR) even at this later time point.
As in the main DREAMM-2 study, belamaf had an acceptable safety profile, with no new safety concerns identified with the lyophilised presentation16. Based on previous clinical experience with belamaf and literature reports of MMAF-containing ADCs, thrombocytopenia was an AESI19. In this study, while common, thrombocytopenia was considered self-limiting and did not lead to treatment discontinuation. IRRs, as expected for biological agents including belamaf, were common, but resolved in all patients.
As expected, keratopathy (MECs) on eye examination was common, but events were generally limited to the epithelium (the superficial layer of the cornea) and rarely led to treatment discontinuation. Dry eye and blurred vision events were also common, but as with keratopathy (MECs), were effectively managed with dose delays and/or reductions and concomitant use of preservative-free lubricant eye drops. Corneal events associated with belamaf may be adequately managed by close liaison with eye care professionals and dose modifications (both delays and reductions), as clinically warranted. For patients with grade 1 events, treatment should be continued at the current dose (on the basis of the 2.5-mg/kg results from the main study)17. For grade 2 events, dosing should be withheld until corneal exam findings and changes in BCVA improve to a grade 1 event or better, when dosing should resume at the current dose. For grade 3 events, treatment should be withheld until corneal exam findings and changes in BCVA improve to grade 1 or better, when dosing should resume at a reduced dose of 1.9 mg/kg. Treatment should be permanently discontinued for grade 4 events.
The belamaf frozen-liquid presentation was primarily used in DREAMM-1 and in the main cohort of the pivotal DREAMM-2 study to evaluate safety and efficacy14,15,16. The refrigerated lyophilised presentation is intended for future clinical use, has been demonstrated to be analytically comparable to the liquid presentation, and is a more robust presentation since it eliminates the frozen shipment and storage requirements. From the patient perspective, either presentation of the drug product is essentially identical upon dilution for intravenous administration. After correction for covariates, no significant difference in PK behaviour was observed for the two presentations, and presentation was not a significant factor in the population PK and exposure–response analyses for belamaf. Belamaf is the first anti-BCMA agent with a multimodal mechanism of action, convenient dosing schedule and no requirement for combination with dexamethasone, making it potentially attractive for use in the real-world setting22. The data presented here, in combination with previously published data from DREAMM-1 and DREAMM-2, support single-agent lyophilised belamaf as a practical and effective treatment option for patients with heavily pre-treated RRMM.
Data availability
Anonymised individual patient data and study documents can be requested from www.clinicalstudydatarequest.com.
References
Nooka, A. K. & Lonial, S. New targets and new agents in high-risk multiple myeloma. Am. Soc. Clin. Oncol. Educ. Book. 36, e431–e441 (2016).
Gandhi, U. H. et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 33, 2266–2275 (2019).
Pick, M. et al. Daratumumab resistance is frequent in advanced-stage multiple myeloma patients irrespective of CD38 expression and is related to dismal prognosis. Eur. J. Haematol. 100, 494–501 (2018).
Neri, P., Bahlis, N. J. & Lonial, S. New strategies in multiple myeloma: immunotherapy as a novel approach to treat patients with multiple myeloma. Clin. Cancer Res. 22, 5959–5965 (2016).
Yong, K. et al. Multiple myeloma: patient outcomes in real-world practice. Br. J. Haematol. 175, 252–264 (2016).
Verelst, S. G. R. et al. Long-term outcomes in patients with multiple myeloma: a retrospective analysis of the dutch Population-based HAematological Registry for Observational Studies (PHAROS). HemaSphere. 2, e45 (2018).
Chari, A. et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med. 381, 727–738 (2019).
Lee, L. et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br. J. Haematol. 174, 911–922 (2016).
O’Connor, B. P. et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 199, 91–98 (2004).
Tai, Y.-T. et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 123, 3128–3138 (2014).
Montes De Oca, R. et al. The anti-BCMA antibody-drug conjugate GSK2857916 drives immunogenic cell death and immune-mediated anti-tumor responses, and in combination with an OX40 agonist potentiates in vivo activity. in Proc 24th Congress of the European Hematology Association Amsterdam, The Netherlands. 2019: PF558.
Tai, Y. T. & Anderson, K. C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy. 7, 1187–1199 (2015).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Trudel, S. et al. Targeting B-cell maturation antigen with GSK2857916 antibody-drug conjugate in relapsed or refractory multiple myeloma (BMA117159): a dose escalation and expansion phase 1 trial. Lancet Oncol. 19, 1641–1653 (2018).
Trudel, S. et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 9, 37 (2019).
Lonial, S. et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 21, 207–221 (2020).
Lonial S. et al. Pivotal DREAMM-2 Study: single-agent belantamab mafodotin (GSK2857916) in patients with relapsed/refractory multiple myeloma (RRMM) refractory to proteasome inhibitors (PIs), immunomodulatory agents, and refractory and/or intolerant to anti-CD38 monoclonal antibodies (mAbs). in Proc 56th Annual Meeting of the American Society of Clinical Oncology Chicago, USA. 2020: EP436.
European Medicines Agency. ICH Q5E Biotechnological/biological products subject to changes in their manufacturing process: comparability of biotechnological/biological products. Reference number: CPMP/ICH/5721/03. 2005. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-5-e-comparability-biotechnological/biological-products-step-5_en.pdf.
Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs. 8, 659–671 (2016).
Rajkumar, S. V. et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 15, e538–e548 (2014).
Common Terminology Criteria for Adverse Events (CTCAE), Version 4.03. (National Cancer Institute, Rockwell, MD, 2009).
Richardson, P. G. et al. Interpreting clinical trial data in multiple myeloma: translating findings to the real-world setting. Blood Cancer J. 8, 109 (2018).
Acknowledgements
We thank the ophthalmology and optometry colleagues who provide ophthalmic examinations to patients enrolled in the study. Medical writing support was provided by Gemma Corr, DPhil, and Sarah Hauze, PhD, of Fishawack Indicia, Ltd., UK, and funded by GSK. Drug linker technology was licensed from Seattle Genetics (Bothell, WA, USA) and the monoclonal antibody was produced with POTELLIGENT Technology licensed from BioWa (Princeton, NJ, USA). Funding: This study was funded by GlaxoSmithKline (GSK). GSK contributed to the study design, implementation, data collection, interpretation and analysis.
Author information
Authors and Affiliations
Contributions
P.G.R.: Study design, data acquisition and interpretation; H.C.L.: Data acquisition; A.-O.A.: Data acquisition; A.D.C.: Data acquisition; P.K.: Data acquisition; P.M.V.: Data acquisition. A.H.: Study design, data analysis and interpretation; K.W.: Data analysis and interpretation; J.B.: Study design, data analysis and interpretation; T.P.: Study design, data analysis and interpretation; J.B.: Data analysis and interpretation; S.R.: Study design, data analysis and interpretation; R.C.J.: Study design, data analysis and interpretation; J.O.: Study design, data analysis and interpretation; I.G.: Study design, data analysis and interpretation; S.L.: Study design. All authors were involved in paper preparation and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
P.G.R. has received grant funding and personal fees from Celgene, Takeda, and Oncopeptides, and personal fees from Janssen, Karyopharm and Sanofi. H.C.L. has received grant funding and personal fees from Amgen, Celgene, Janssen, and Takeda; personal fees from GSK and Sanofi, and grant funding from Daiichi Sankyo. A.-O.A. declares no conflict of interest. A.D.C. has received grant funding from GSK, BMS, and Novartis; personal fees from Janssen, Takeda, Oncopeptides, Kite Pharma and Seattle Genetics, and personal fees and other association with GSK and Celgene. P.K. has received grant funding from GSK, Amgen, Sanofi, Takeda, Sorrento and Janssen (to the institution) and served on an advisory board for GSK (for which the institution received an honorarium). P.M.V. has received personal fees from Adaptive Biotechnologies, BMS/Celgene, Janssen, Novartis, Oncopeptides and TeneoBio. A.H., J. Baron, T.P. and J.O. are employees of and hold stocks and shares in GSK. K.W. and S.R. are employees of GSK. J. Byrne is an employee of GSK and holds stocks/shares in GSK, Adaptimmune, and Novartis. I.G. and R.C.J. are employees of GSK and hold stocks/shares in GSK and Novartis. S.L. has received grant funding and personal fees from Celgene and Takeda, and personal fees from Novartis, BMS, GSK, Amgen, Merck and Janssen.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Richardson, P.G., Lee, H.C., Abdallah, AO. et al. Single-agent belantamab mafodotin for relapsed/refractory multiple myeloma: analysis of the lyophilised presentation cohort from the pivotal DREAMM-2 study. Blood Cancer J. 10, 106 (2020). https://doi.org/10.1038/s41408-020-00369-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-020-00369-0
This article is cited by
-
Targeted immunotherapy: harnessing the immune system to battle multiple myeloma
Cell Death Discovery (2024)
-
BCMA-Targeted Biologic Therapies: The Next Standard of Care in Multiple Myeloma Therapy
Drugs (2022)
-
Myeloma research on the move
Blood Cancer Journal (2021)
-
Ocular Toxicity of Targeted Anticancer Agents
Drugs (2021)
