
Abstract
The inflammatory responses involving infiltration and activation of liver macrophages play a vital role in acute liver failure (ALF). In the liver of ALF mice, cannabinoid receptor 2 (CB2R) is significantly upregulated on macrophages, while CB2R agonist GW405833 (GW) could protect against cell death in acute liver damage. In this study, we investigated the molecular mechanisms underlying the protective effects of GW against ALF in vivo and in vitro from a perspective of macrophage glycometabolism. Mice were pretreated with GW (10 mg/kg, i.p.), then were injected with D-GalN (750 mg/kg, i.p.) and LPS (10 mg/kg, i.p.) to induce ALF. We verified the protective effects of GW pretreatment in ALF mice. Furthermore, GW pretreatment significantly reduced liver macrophage infiltration and M1 polarization, and inhibited the release of inflammatory factors TNF-α and IL-1β in ALF mice. These protective effects were eliminated by CB2R antagonist SR144528 or in CB2R−/− ALF mice. We used LPS-stimulated RAW264.7 cells as an in vitro M1 macrophage-centered model of inflammatory response, and demonstrated that pretreatment with GW (10 μM) significantly reduced glucose metabolism by inhibiting glycolysis, which inhibited LPS-induced macrophage proliferation and inflammatory cytokines release. We verified these results in a stable CB2R−/− RAW264.7 cell line. Moreover, we found that GW significantly inhibited the expression of hypoxia inducible factor 1α (HIF-1α). Using a stable HIF-1α−/− RAW264.7 cell line, we confirmed that GW reduced the release of inflammatory cytokines from macrophages and inhibited glycolysis by downregulating HIF-1α expression. In conclusion, activation of CB2Rs inhibits the proliferation of hepatic macrophages and release of inflammatory factors in ALF mice through downregulating HIF-1α to inhibit glycolysis.
Similar content being viewed by others

Introduction
Acute liver failure (ALF), a serious liver damage caused by infection, metabolism, immune reactions, drugs and other factors, has a mortality rate of more than 50% and can lead to multiple organ dysfunction [1,2,3]. Liver transplantation is an effective treatment, but its performance is limited by a shortage of donor livers and expensive surgical treatment [4]. The vast majority of treatments for ALF are symptomatic and supportive treatments. Efforts have been made to explore the pathogenesis and to screen effective treatments [1].
Macrophages are key cellular components of the liver, with 20 to 40 macrophages for every 100 hepatocytes in the mouse liver [5]. At the earliest stage of liver injury, macrophages can mediate the inflammatory cascade effect to trigger liver cell apoptosis, magnify liver injury and promote the sustainable development of ALF, so macrophages are important inflammatory effector cells for ALF [6, 7]. Macrophages can adjust their phenotype and function in response to environmental changes. Activated macrophages are generally divided into two forms of polarization (M1 and M2) [8]. Both M1 macrophages and M2 macrophages are closely associated with inflammatory responses. M1 macrophages express CD80, CD86 or CD16/32 and secrete proinflammatory cytokines such as TNF-α and IL-1β, which are mainly involved in proinflammatory responses. M2 macrophages express CD206, ARG-1, IL-10, CCL17 and CCL22, which are mainly involved in anti-inflammatory responses [8]. An average of 51.5% of Kupffer cells are activated [9] and polarized to the M1 phenotype to recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) from injured hepatocytes, release inflammatory cytokines and regulate inflammatory responses, subsequently leading to ALF through aseptic inflammation and hepatocyte apoptosis [5, 10, 11]. Therefore, targeting mediators of the macrophage inflammatory response is useful for exploring a possible treatment for ALF.
The activation, polarization, and involvement of macrophages in inflammation are based on metabolic adaptations including glycolytic reprogramming, which not only provides energy to support immune activities in a specific environment but also directly affects the functions of immune cells by regulating transcriptional and posttranscriptional events [12,13,14]. Glycolytic reprogramming is an important metabolic pathway and is characteristic of M1 macrophages, and downregulation of glycolysis can inhibit the proinflammatory functions of M1 macrophages; thus this reprogramming is a therapeutic target for multiple sclerosis, inflammatory bowel disease and other inflammatory diseases [15]. We hypothesized that inhibition of glycolytic reprogramming can reduce the proinflammatory response of macrophages and reduce hepatocyte apoptosis caused by their recognition of DAMPs, thereby improving ALF.
Our previous study showed that CB2R activation protected against acute liver injury in mice induced by concanavalin (Con A) [16]. The endocannabinoid system (ECS) consists of CBRs, including CB1R and CB2R, as well as endogenous ligand synthesis and degradation enzymes, and has a variety of physiological and pharmacological functions. CB2R is a G protein-coupled receptor that is mainly expressed in cells associated with innate immunity, such as B cells, T cells and macrophages [17]. Recent studies have shown that CB2R plays an important anti-inflammatory role in many inflammatory diseases, such as autoimmune myocarditis and inflammatory bowel disease [4]. The CB2R agonist JWH-133 against alcoholic liver injury in mice by regulating macrophage polarization and autophagy [1, 18]. JWH-133 also ameliorates D-galactosamine (D-GalN) /lipopolysaccharide (LPS)-induced ALF by inducing macrophage M2 polarization and regulating unique miR-expression of key molecules involved in targeting Toll-like receptor 4 (TLR4) pathway [19]. Many studies have shown that activation of CB2R can reduce the polarization of macrophages to the M1 type and promote M2 type polarization [20,21,22], regulate the release of inflammatory cytokines [23, 24] and reduce the occurrence of inflammation. Although the protective effect of activated cannabinoid receptor 2 of macrophages on acute liver injury has been verified by various experiments, the specific mechanism needs to be further explored. It has been reported that activation of CB1R and CB2R inhibits pyruvate kinase isoform M2 (PKM2), mediating glycolysis in pancreatic cancer cells [25]. Therefore, we hypothesized that inhibition of macrophage-associated inflammation by activation of CB2R based on glycolytic reprogramming would reduce hepatocyte apoptosis secondary to DAMP recognition and thus ameliorate ALF.
In this study, we used a D-GalN/LPS-induced ALF mouse model to verify the protective effect of a CB2R agonist and explore its possible mechanism from the perspective of macrophage glycometabolism.
Materials and methods
Animals and drug treatments
Six- to eight-week-old (20–25 g weight) male specific pathogen free (SPF) wild type (WT) mice (BALB/c, C57BL/6) were used in this study. The animals were housed in the Department of Laboratory Animals of Central South University (Changsha, China) in a standard controlled environment (20–25 °C, 50% ± 5% humidity, 12 h dark/light cycle) and fed a standard laboratory animal diet and water. Before the experiment, the mice were acclimatized to the new surroundings for 1 week. BALB/c mice were divided into a control group (phosphate buffered saline, PBS), model group (D-GalN + LPS), intervention group (GW405833 + D-GalN + LPS) and antagonistic group (SR144528 + GW405833 + D-GalN + LPS). The model group was treated with D-GalN (750 mg/kg, i.p.) and LPS (10 mg/kg, i.p.) for modeling of ALF. Thirty minutes before D-GalN + LPS injection, the intervention group was pretreated with GW405833 (GW, 10 mg/kg, i.p.), and the antagonist group was pretreated with GW (10 mg/kg, i.p.) and SR144528 (SR, 1 mg/kg, i.p.) 20 min later. The control group was injected with the same volume of PBS. Eighe hours later, serum was collected by eyeball operation after anesthesia, and liver samples were harvested after perfusion.
CB2R−/− mice from Jackson Laboratories, USA (B6.129P2-Cnr2tmlDgen.005786), were housed and fed in the same manner as WT mice. Experiments in CB2R−/− mice were performed with WT C57BL/6 mice as controls.
The research work followed the principles of humane treatment to ensure the welfare of the laboratory animals and strictly followed the rules and regulations of the Department of Laboratory Animals of Central South University, under the supervision of the Laboratory Animal Welfare Ethics Committee of Central South University and the laboratory animal manager.
Histological and immunohistochemical analysis
Liver samples were fixed with 4% paraformaldehyde, embedded in a paraffin sectioned to a thickness of 5 μm. The liver sample sections were stained with hematoxylin and eosin (H&E) for histopathological analysis, and with TUNEL (KeyGene, KGA704), an F4/80+ primary antibody (1:100; CST, 70076T), a CD86 primary antibody (1:100; CST, 19589) and a CD206 primary antibody (1:100; CST, 24595) for immunohistochemistry (IHC). The stained sections were observed by light microscope (Nikon, Tokyo, Japan) and analyzed with Image-Pro Plus. The red cytoplasm of hepatocytes and the bleeding area were identified as the positive area, and the measured area (mean) was used as the evaluation parameter for histopathological analysis. In TUNEL staining, the cells with brown nuclei were positive; in F4/80+, CD86 and CD206 immunohistochemical staining, the cells with brown membranes were positive. The measured area (mean) was used as the evaluation parameter for TUNEL and F4/80+ staining, and the measured IDO (mean) was used for CD86 and CD206 staining analysis with Image-Pro Plus.
Cell culture and treatment
RAW264.7 cells (ATCC Cat# TIB-71) were cultured with Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS), and 1% penicillin‒streptomycin. After RAW264.7 cells were seeded in 96-well plates (8 × 103/well) and 6-well plates (5 × 105/well) overnight, the control group was pretreated with PBS, and the experimental group were pretreated or not with the CB2R antagonist SR (1 μmol/L) for 20 min and then treated with the CB2R agonist GW (10 μmol/L) for 1 h. Each group was then treated with LPS 100 ng/mL for 24 h. The culture medium and cells of 6-well plates were collected separately for subsequent experiments. In the in vitro experiments, two consistent results of three independent experiments prevailed.
Cell Counting Kit-8(CCK-8) assay
The proliferation of cells in 96-well plates was investigated via a CCK8 kit (Beyotime, China) after treatment for 6 h, 12 h, 24 h, 48 h, and 72 h. The absorbance value was measured at 450 nm with the blank each time as the control.
Enzyme-linked immunosorbent assay (ELISA)
RAW264.7 cells were stimulated with LPS and cultured in low-serum culture medium with 1% FBS, and the supernatants were collected at the 24 h time point. The concentrations of TNF-α and IL-1β in the collected culture medium and serum of animals were measured following the instructions of the manufacturer (Dakewei, CN). The standard curve was plotted to calculate the concentration with blank as a control.
Mitochondrial stress test and glycolysis stress test
RAW264.7 cells (8 × 103/well) were seeded in Seahorse XF96 cell culture microplates (Angilent Technologies, Cat# 101085-004) overnight and were grouped for pretreatment as previously mentioned. Oxidative phosphorylation and glycolysis flux analysis were performed according to the manufacturer’s recommendations (Angilent Technologies, USA). XF assay medium (Angilent Technologies, Cat# 102365-100) was used for the mitochondrial stress test with 4 mM glutamine, 4.5 g/L glucose, and 1 mM pyruvate, and for the glycolysis stress test with 2 mM pyruvate. After the culture media were changed, the plates were incubated in a CO2-free incubator at 37 °C for 1 h. For the mitochondrial stress test, cells were treated first with oligomycin (1 μM), then with FCCP (0.5 μM), and finally with a combination of rotenone and antimycin A (0.5 μM). For the glycolysis stress test, cells were treated first with glucose (10 mM), then with oligomycin (1 μM), and finally with 2-deoxy-glucose (50 mM). The oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR) at each time point were measured following the Seahorse Biosciences protocol.
Lactate, pyruvate, and enzyme activity
According to Bioassay Systems’ instructions, ATP production, lactate levels, pyruvate levels, and lactate dehydrogenase and pyruvate kinase enzyme activity levels were detected. The kits used were an ATP assay kit (EATP-100), an L-Lactate assay kit (EFLLC-100), a pyruvate assay kit (EPYR-100), a lactate dehydrogenase assay kit (D2DH-100) and a pyruvate kinase assay kit (EPRK-100).
Generation of stable cell lines
To establish stable CB2R- and HIF-1α- knockdown RAW264.7 cell lines, cells were transduced with lentiviruses and shRNA specific for CB2R and HIF-1α to generate RAW264.7-shCB2R and RAW264.7-shHIF-1α cells, respectively. Positive cells were screened using puromycin (Invitrogen, #A1113803, USA). The knockdown efficiency was analyzed by Western blot analysis.
Western blotting analysis
Total protein was extracted from cells that were homogenized in RIPA lysis buffer (Beyotime, China) containing proteinase and phosphatase inhibitors (Roche) and centrifuged. The harvested proteins (30 μg/lane) were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (Millipore, IPVH00010). The membranes were incubated with primary antibodies after being blocked with fat-free milk. The primary antibodies utilized were against LDHA/LDHC (1:1000; CST, Cat# 3558), PKM2 (1:1000; CST, Cat# 4053), HK1 (1:1000; CST, Cat# 2024), PI3K (1:1000; CST, Cat# 42280), Akt (1:1000; CST, Cat# 4691), HIF-1α (1:1000; CST, Cat# 36169), CB2R (1:500; abcam, Cat# ab3561), phospho-Akt (1:1000; Immunoway, YP0006), β-actin (1:1000; Santa Cruz Biotechnology, Cat# sc-69879), and Tubulin (1:1000; Santa Cruz Biotechnology, Cat# sc-166729). The binding of antibodies was detected with a secondary antibody conjugated with horseradish peroxidase (HRP) and displayed with enhanced chemiluminescence reagents. Image Lab software was used for density analysis to measure the expression level of the target protein relative to the control (β-actin or tublin).
Immunofluorescence
Immunofluorescence for mouse liver tissue sections was performed as described previously [26]. The sections were successively incubated with primary antibodies against F4/80+ (1:500; Proteintech, 28463-1-AP) and HIF-1α (1:200; Bioss, bs-0737R) and secondary antibodies conjugated with Alexa Fluor 488 and counterstained with DAPI (Solarbio, C0060). Fluorescence signals were observed under a fluorescence microscope (Nikon Eclipse TI-SR, Japan).
Drug dosage
GW405833 is a selective agonist of CB2R [27] with high affinity for human and rodent CB2R [28]. It has been reported that high-dose GW (30~100 mg/kg) intraperitoneal injection induced off-target effects beyond those on CB2R in mice, while 10 mg/kg intraperitoneal injection does not [28, 29]. The TRPV1 pathway is activated when the GW concentration is above 10 μM [30]. Therefore, according to the literature, the intraperitoneal injection dose of the mice in this study was chosen to be 10 mg/kg, and the cell culture concentration in vitro was 10 μM.
SR144528 is a selective antagonist of CB2R. In mice, SR was used to antagonize the action of the CB2R agonist at one-tenth of the dose: SR at 1 μM can antagonize the binding of DAMGO (an analog of endocannabinoid 2-AG) to CB2R in vitro [31]. Therefore, we selected a dose of 1 mg/kg for intraperitoneal injection in mice and a concentration of 1 μM for use in vitro.
Statistical analysis
IBM SPSS Statistics 25 and GraphPad Prism 9.3 (GraphPad Software, Inc.) were used for the statistical analysis. The results are presented as the mean ± standard error of the mean (SEM). Comparisons between groups were performed using Student’s t-test. Comparisons of repeated measurement variables between groups were analyzed using two-way ANOVA. P < 0.05 was considered to indicate statistical significance.
Blinding
The experimental animals were randomized into the groups. In the experiments for detection of serum ALT, AST, TBIL, TNF-α, and IL–1β and the pathological and immunohistochemical investigations, the researchers were blinded to the groups.
Results
GW ameliorated D-GalN/LPS-induced acute liver injury
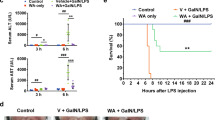
According to methods previous reported in the literature, we used D-GalN/LPS to induce acute liver failure in mice [19]. The experimental animals were classified into 4 groups: the control group, model group, intervention group, and antagonist group. Survival analysis showed that the death of liver cells occurred within 24 h. Within 24 h, the survival rates in the control, model, intervention and antagonist groups were 100%, 20.513% ± 11.956%, 90% ± 9.487%, and 60% ± 15.492%, respectively (Fig. 1). Statistical analysis showed that compared to that of the model group, the survival rates of the control and intervention groups were significantly higher (P < 0.001, 0.0014, respectively). There was no significant difference in the survival rate between the model group and the antagonist group.

Kaplan-Meier survival curves of BALB/C mice (n = 8, 13,10, 10 for group with PBS, D-GalN +LPS, GW + D-GalN +LPS, and GW + SR + D-GalN +LPS treatment respectively). (ns, P > 0.05; **P < 0.01; ***P < 0.001)
As shown in Fig. 2a–c, ALT, AST and TBIL levels in the model group were significantly higher than those in the PBS control group (P < 0.0001, <0.0001 and P < 0.05, respectively), and ALT and AST levels in the intervention group were lower than those in the model group (both P < 0.01). In addition, the model group exhibited massive hepatocyte necrosis, hemorrhaging with inflammatory cell infiltration, hepatic sinus enlargement or structural damage with small thrombus formation, and these pathological changes were alleviated in the group pretreated with GW (Fig. 2d). The pathological damage of the antagonist group (GW + SR + D-GalN +LPS) was more severe than that of the GW intervention group, but there were no significant differences in ALT, AST or TBIL levels (Fig. 2e).

a ALT (n = 16, 8, 12, 11 respectively), b AST (n = 16, 11, 11, 8 respectively), c TBIL (n = 16, 15, 15, 13 respectively) in serum of mice was detected 8 h after treatment with PBS, D-GalN +LPS, GW + D-GalN +LPS, or GW + SR + D-GalN +LPS. d HE staining of liver sections of mice at time point 8 h after PBS, D-GalN +LPS, GW + D-GalN +LPS, or GW + SR + D-GalN +LPS treatment. e Statistical analysis of red area in liver section HE staining (n = 4). f TNF-α (n = 13, 7, 14, 14 respectively), g IL-1β (n = 11, 8, 13, 14 respectively) in mice serum was detected 8 h after treatment with PBS, D-GalN +LPS, GW + D-GalN +LPS, or GW + SR + D-GalN +LPS. (ns, P > 0.05; *P < 0.05; **P < 0.01; ****P < 0.0001)
The inflammatory cytokines TNF-α and IL-1β in serum were also detected. As shown in Fig. 2f, g, the levels of both TNF-α and IL-1β were significantly increased in the model group (P < 0.01 and P < 0.05, respectively), while they were reduced in the intervention group compared with the model group (both P < 0.05), and the level of IL-1β in the antagonist group was close to model group.
GW inhibited the elevations in the levels of F4/80+, a marker of liver Kupffer cell, in ALF
Compared with that in the model group, the increase in TUNEL staining in the GW pretreatment group was significantly decreased (both P < 0.05, Fig. 3a, b). Immunohistochemical labeling with CB2R in liver sections was performed in the control group and model group (Fig. 3c), which showed upregulation of CB2R expression in the model group with D-GalN +LPS treatment (Fig. 3d). Immunohistochemical staining for F4/80+ (Fig. 3e), CD86 (Fig. 3f), and CD206 (Fig. 3g) in liver sections was also performed in the control group, model group, intervention group, and antagonist group. Staining of F4/80+ and CD86 showed that the staining area or IDO value was significantly higher in the model group than in the control group, lower in the GW pretreatment group than in the model group, and higher in the antagonist group than in the intervention group (Fig. 3h, i). The CD206 staining IDO value was increased in the model group, intervention group, and antagonist group, but was highest in the intervention group (Fig. 3j).

a TUNEL staining of liver sections of mice at 6 h after PBS, D-GalN +LPS, GW + D-GalN +LPS, GW + SR + D-GalN +LPS treatment. b Statistical analysis of TUNEL staining in liver section (n = 8, 6, 4, 9 respectively). c Immunohistochemically labeled with CB2R of mice liver sections 8 h after PBS and D-GalN +LPS treatment. d Statistical analysis of CB2R immunohistochemical staining in liver section (n = 3, 6 respectively). Immunohistochemically labeled with e F4/80+, f CD86, g CD206 of mice liver sections 8 h after PBS, D-GalN +LPS, GW + D-GalN +LPS, or GW + SR + D-GalN +LPS treatment. Statistical analysis of h F4/80+ (n = 7, 6, 9, 6 respectively), i CD86 (n = 3), j CD206 (n = 4, 6, 6, 4, respectively) immunohistochemical staining in liver section. (ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001)
GW affected the proliferation and the production of TNF-α and IL–1β in macrophages stimulated by LPS
The RAW264.7 cell line has the functions of macrophage-mediated immunity, metabolism, and phagocytosis [32], so we used RAW264.7 cells stimulated by LPS to construct an M1 macrophage-centered model of the inflammatory response in vitro. RAW264.7 cells were divided into four groups: the CON (PBS), model (LPS), intervention (LPS + GW), and antagonistic (GW + SR + LPS) groups. The RAW264.7 cells were stimulated with LPS (100 ng/mL), and a cell proliferation test was conducted via CCK-8 assay at 6 h, 12 h, 24 h, 48 h, and 72 h. We found that LPS induced the proliferation of RAW264.7 cells, which was inhibited by GW, and GW’s effect was abolished by SR (Fig. 4a). Then, we detected the concentrations of TNF-α and IL-1β in the supernatant of each group. As shown in Fig. 4b, c, the concentrations of TNF-α and IL-1β in the model group were increased by LPS (both P < 0.05), while GW inhibited the secretions of both proteins (both P < 0.05). And with SR pretreatment, the IL-1β concentration was close to that in the model group. These results suggest that GW inhibits the inflammatory response by reducing the secretion of TNF-α and IL-1β in RAW264.7 cells induced by LPS.

a Different proliferation of RAW264.7 cells after grouped treatment (n = 6). b TNF-α (n = 5), c IL-1β (n = 6) concentrations in cell supernatant after grouping treatment. (ns, P > 0.05; *P < 0.05; **P < 0.01; ****P < 0.0001)
GW reprogrammed the LPS-induced changes in macrophage metabolism
Glycolysis and aerobic phosphorylation of RAW264.7 cells in each group were detected using a Seahorse energy metabolism detection system. We found that both glycolysis and glycolytic capacity were increased in the model group with LPS treatment (both P < 0.01), while both were reduced by GW pretreatment in the intervention group (P < 0.05, P < 0.01) (Fig. 5a, b). We then examined the concentrations of lactate, and pyruvate in the supernatant, as well as lactate dehydrogenase (LDH) and pyruvate kinase (PK) activity in the cells. The results showed that in the model group, the lactate level was increased (P < 0.0001) (Fig. 5c) and the pyruvate level was reduced (P < 0.0001) (Fig. 5d), while both lactate dehydrogenase activity and cell pyruvate kinase activity were increased (P < 0.05, P < 0.01, respectively) (Fig. 5e, f). Compared to those in the model group, the lactate concentrations, LDH activity, and PK activity were all decreased in the intervention group, and the concentration of pyruvate was increased (P < 0.01, P < 0.0001, P < 0.01, and P < 0.001, respectively). The effects of GW on LDH and PK were partially offset by the addition of SR144528 (P < 0.05) (Fig. 5e, f). As shown in Fig. 5g, there was no difference in ATP production between the model group and the CON group. ATP production was lower in the intervention group than that in the model group and the antagonist group (P < 0.05, P < 0.01, respectively).

a ECAR at each point, b Glycolysis parameters of glycolysis stress test (n = 6 for each group). c Lactate, d Pyruvate concentration was detected in grouped treatment cell supernatant (n = 5 for each group). e LDH (n = 4), and f PK (n = 5) activity of grouped treatment cells were detected. g ATP production of grouped treatment cells (n = 3). h OCR at each point, i Aerobic respiration parameters of mitochondrial stress test of grouped treatment RAW264.7 cell (n = 6 for each group). (ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001)
Finally, we found that the maximal respiration and spare respiratory capacity in the model group were decreased (both P < 0.05) (Fig. 5h, i), but that these measurements were increased with GW pretreatment in the intervention group (P < 0.01, P < 0.0001, respectively). The OCR of ATP production was reduced in the intervention group (P < 0.05) and the antagonist group (P < 0.001).
CB2R knockdown magnified the inflammatory response of macrophages treated with LPS
To further evaluate the roles of CB2R in the macrophage inflammatory response, we transfected RAW264.7 cells with lentiviruses to express CB2R-specific shRNA, which effectively knocked down CB2R expression, as validated by Western blotting (Fig. 6a, b). As shown in Fig. 6c, CB2R knockdown resulted in an increased proliferation of cells stimulated by LPS (P < 0.05). Consistently, CB2R knockdown led to increased release of the inflammatory cytokines TNF-α, and IL–1β in LPS-treated cells (P < 0.05, Fig. 6d, e). More importantly, basal respiration, maximal respiration and spare respiratory capacity were reduced significantly (Fig. 6h, i), while the changes in glycolysis in cells stimulated with LPS were increased (Fig. 6f, g). These data suggest that the activation of CB2Rs in macrophages with LPS, can inhibit both cell proliferation and inflammatory factor release, which underlies the inhibition of inflammatory responses.

a CB2R knockdown efficiency of RAW264.7 cells were detected by Western blotting. (b) Semi-quantity statistical analysis of CB2R knockdown efficiency Western blotting (n = 3). c Time point 24 h cell proliferation test (n = 6 for each group). d TNF-α, e IL-1β concentration in cell supernatant after the indicated treatment (n = 5 for each group). f ECAR at each point, g Glycolysis parameters of glycolysis stress test of cells after the indicated treatment (n = 6 for each group). h OCR at each point, i Aerobic respiration parameter of mitochondrial stress test (n = 6 for each group). (ns, P > 0.05; *P < 0.05; **P < 0.01; ****P < 0.0001)
GW regulated glucose metabolism in macrophages through HIF-1α
Then we examined whether GW pretreatment could regulate the expression of proteins related to glycolysis by Western blotting (Fig. 7a). The expression level of LDHA/LDHC (Fig. 7b) was increased in the model group and the antagonist group and decreased in the intervention group, but the differences of expression levels of LDHA/LDHC between the groups were not statistically significant. The expression of PKM2(Fig. 7c) in the model group tended to be increased (P = 0.0720), and was decreased in the intervention group (P < 0.05). There were no differences in the expression of HK1 (Fig. 7d), PI3K (Fig. 7e), or Akt (Fig. 7f) among the groups. Both HIF-1α (Fig. 7g) and CB2R (Fig. 7h) were upregulated in the model group, and HIF-1α was downregulated in the intervention group. To further reveal the relationship between CB2R and HIF-1α, we infected RAW264.7 cells with lentiviruses to express HIF-1α specific shRNA and effectively knock down its expression (Fig. 7i, j). We found that with HIF-1α knockdown, GW pretreatment had no inhibitory effect on LPS-induced TNF-α and IL-1β release (Fig. 7k, l) or glycolysis alteration (Fig. 7m, n). These results indicate that GW inhibits macrophage glycolysis, cell proliferation, and inflammatory factors release through HIF-1α, inhibiting inflammation. In addition, we further detected the expression of phospho-Akt (p-Akt) (Figure S1a) by Western blotting, and the results showed that there was no difference in expression between NC and shCB2R RAW264.7 cells induced by LPS (Figure S1b) according to two-way ANOVA.

a Glycolysis related enzymes and signal proteins detected by Western Blotting. Semi-quantity statistical analysis of b LDHA/LDHC, c PKM2, d HK1, e PI3K, f Akt, g HIF-1α, h CB2R expression detected by Western blotting (n = 3). i HIF-1α knockdown efficiency of RAW264.7 cells were detected by Western blotting. j Semi-quantity statistical analysis of HIF-1α knockdown efficiency detected by Western blotting (n = 3). k TNF-α, l IL-1β concentration in cell supernatant after the indicated treatment (n = 5 for each group). m ECAR at each point, n Glycolysis parameters of glycolysis stress test of cells after the the indicated treatment (n = 6 for each group). (ns, P > 0.05; *P < 0.05; **P < 0.01)
GW protected against D-GalN/LPS-induced ALF by reducing macrophage infiltration and HIF-1α expression
CB2R−/− mice (6–8 weeks) were divided into control group (PBS), a model group (D-GalN+LPS), and an intervention group (GW + D-GaIN +LPS). Since CB2R-knockout mice were constructed on the C57BL/6 background, WT C57BL/6 mice were used as references. The C57BL/6 mice were divided into four groups: the control group (PBS), model group (D-GalN+LPS), intervention group (GW + D-GalN+LPS) and antagonist group (SR + D-GalN+LPS). The mice in the model group were intraperitoneally injected with D-GalN (350 mg/kg) and LPS (20 μg/kg) for acute liver failure modeling. GW (10 mg/kg) was injected thirty minutes before modeling in the intervention group, SR (10 mg/kg) was injected thirty minutes before modeling in the antagonist group, and the same volume of PBS was injected in the control group. The batches of C57BL/6 mice constructed with the CB2R−/− mice were not the same as those of WT mice. To avoid large individual differences that would affect the results, statistical analysis of differences between WT and CB2R−/− mice was not performed.
We detected serum ALT, AST, and TBIL levels and performed liver biopsy pathology (Fig. 8a–d). The results showed that D-GalN + LPS induced increase in ALT concentration, AST concentration, and liver pathological damage in both WT mice and CB2R−/− mice, while GW pretreatment reduced the serum concentrations of ALT and AST and liver pathological damage in WT mice but showed no effect in CB2R−/− mice (Fig. 8d, e).

a ALT, b AST, c TBIL (n = 6, 7, 6, 6, 4, 5, 4, respectively) in serum of mice, and d statistical analysis of red area in liver section HE staining (n = 3, 5, 6, 6, 3, 5, 3, respectively) detected 6 h after the indicated treatment with PBS, D-GalN +LPS, GW + D-GalN +LPS, or SR + D-GalN +LPS in WT mice, and PBS, D-GalN +LPS, or GW + D-GalN +LPS in CB2R−/− mice; e Pathological changes of mouse liver tissue after indicated treatment were examined by HE staining. (ns, P > 0.05; *P < 0.05; ***P < 0.001; ****P < 0.0001)
The liver sections were also costained for immunofluorescence of F4/80+ and HIF-1α (Fig. 9a, b). The expression levels of F4/80+ (Fig. 9c) and HIF-1α (Fig. 9d) were increased in the model group in both WT (P < 0.05, P < 0.01, respectively) and CB2R−/− mice (P < 0.05, P < 0.001, respectively) compared with PBS-treated controls. GW pretreatment showed a tendency to inhibit the overexpression of F4/80+ (P = 0.0539) and significantly inhibited the expression of HIF-1α (P < 0.05) in WT mice but not in CB2R−/− mice.

Liver sections of mice were treated with F4/80+ and HIF-1α immunofluorescence co-staining 6 h after indicated treatment (×400) in a WT mice and b CB2R−/− mice. c Quantitative analysis of F4/80+ expression by Image J. d Analysis of liver macrophage with HIF-1α staining positive. (n = 4, 6, 5, 5, 4, 5, 4, respectively). (ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001)
Discussion and conclusion
Accumulating lines of evidence have shown the protective effects of cannabinoid receptor 2 agonists on liver injury [1, 16, 18, 19] and even the ameliorative effect of JW133 on acute liver failure [19]. In this study, we performed both biochemical analyses (of ALT, AST, and TBIL as references) and pathological analysis as the gold standard for ALF analysis and found that GW significantly protected liver cells against D-GalN/LPS-induced ALF in both BALB/c mice and C57BL/6 mice. GW-induced protection did not occur in CB2R−/− mice and was reversed by the CB2R antagonist SR, which suggests that the protective effects of GW on D-GalN/LPS-induced ALF are mediated through the activation of CB2R.
The two vital parts of the pathophysiology of acute liver failure are direct liver injury and the inflammatory response, and innate immune cells called macrophages play an important role [33]. The forms of macrophages are heterogeneous, but these cells can generally be divided into two extreme forms, proinflammatory M1 macrophages (induced by LPS, TNF-α, or IFN-γ) and anti-inflammatory M2 macrophages (induced by IL-13 and IL-4), and the polarization is related to the microenvironment [34]. Activated macrophages are mostly of the M1 type in ALF mice and release IL-1β, TNF-α, and other inflammatory factors to mediate the subsequent apoptosis of liver cells [5, 10, 11]. In our study, liver macrophage infiltration, M1 polarization and M1 macrophage-related inflammatory factors levels were increased in D-GalN/LPS -induced ALF mice. The number of liver macrophages, M1 polarization, and the production of inflammatory factors were inhibited. Increased numbers of macrophages were polarized to the M2 type with GW activation of CB2R, which was reversed by pretreatment with the CB2R antagonist SR in ALF mice. In vitro, we also observed that CB2R activation inhibited the macrophage proliferation and proinflammatory cytokine (TNF-α and IL-1β) secretion upon stimulation with LPS. Therefore, we conclude that CB2R activation inhibits liver macrophages proliferation, M1 polarization, and proinflammatory cytokines production in ALF.
The state of macrophages and their response to inflammation depend on metabolic reprogramming, so the concept of “immune metabolism” has been introduced [12, 35]. Unlike M2 macrophages, which have a complete tricarboxylic acid (TCA) cycle, LPS-induced M1 macrophages utilize glycolysis [14, 36] and the pentose phosphate pathways (PPP) to meet ATP requirements, and their TCA cycle is broken at two points: at the step of the enzyme isocitrate dehydrogenase and then at the post-succinic acid step [13, 37]. The results of our study showed that PK activity was increased in LPS-treated macrophages, suggesting that glucose utilization is upregulated without a corresponding increase in pyruvate, which is likely associated with high pyruvate consumption. Pyruvate can be used to generate lactate by LDH through the glycolysis metabolism pathway to produce energy quickly or enter the mitochondrial TCA cycle to produce abundant ATP [38, 39]. The incomplete TCA cycle in LPS-induced macrophages results in limited consumption of pyruvate, and a large amount of pyruvate enters the glycolysis pathway, which was verified in our study by the results of increased LDH activity and lactate plus undifferentiated total ATP production in the model group. The ATP sources include glycolysis and oxidative phosphorylation pathways. The OCR of ATP production in LPS macrophages indicated a decrease utilization of oxidative phosphorylation pathways, indicating an increase in ATP production from the glycolysis pathway. Moreover, the results of glycolytic stress tests and mitochondrial stress tests also revealed the direct transformation of LPS-induced macrophage metabolism from oxidative phosphorylation (OXPHOS) to glycolysis. Interestingly, with GW pretreatment, these shifts were modified to show decreased PK activity, LDH activity, and lactate production. Moreover, the comparison between the antagonist group and the intervention group showed that the total ATP production increased while the OCR of ATP production decreased in the antagonist group, indicating that increased amounts of ATP came from the glycolysis pathway. These results suggest that CB2R activation can downregulate glycolysis in macrophages induced by LPS.
Cannabinoids can act not only on cannabinoid receptors, but also on other G protein receptors, such as GPCR55, peroxisome proliferator-activated receptor- α (PPARα) and transient receptor potential cation channel subfamily V member 1 (TRPV1), exerting a bias effect [40]. In our study, SR did not show perfect antagonistic performance against GW in tests of LDH activity, lactate concertrations, mitochondrial stress, and HIF-1α expression in liver macrophages, so we verified this with CB2R-knockdown (KD) macrophages and CB2R-knockout mice (KO). As expected, these results confirmed that glycolysis, which is an important metabolic transformation pathway in M1 macrophages induced by LPS, was aggravated in CB2R-KD RAW264.7 cells; and GW has no effect on the HIF-1α expression in liver macrophages in CB2R-KO mice. Unfortunately, in mitochondrial stress tests, the maximum respiratory OCR was increased in NC macrophages induced by LPS, suggesting that lentivirus transfection may affect mitochondrial respiration. In contrast, we still found that CB2R KD downregulated the OCR of basal respiration, maximal respiration, ATP production, and spare respiratory capacity. Nevertheless, CB2R activation reduced glycolysis in macrophages induced by LPS. Inhibition of glycolysis can affect many typical functions of many inflammatory phenotypes, such as aggression, mobility, ROS production and secretion of proinflammatory cytokines [14, 41]. In our study, with the metabolic reprogramming caused by CB2R activation, the proliferation ability of LPS-activated macrophages and the release of proinflammatory cytokines (TNF-α and IL-1β) were also decreased.
Reprogramming toward glycolysis leads to elevated succinic acid levels and mitochondrial ROS production in M1 macrophages, which are associated with HIF-1α activation and subsequent IL-1β formation [42]. Activation and stabilization of HIF-1α are necessary for the expression of multiple glycolysis-related enzymes, including glucose transporter 1(GLUT1), hexokinase (HK), phosphofructokinase, PK, LDHA, and GAPDH [43,44,45], supporting the conversion of pyruvate into lactate. HIF-1α also inactivates pyruvate dehydrogenase, thereby limiting pyruvate entry into the TCA cycle [46, 47]. PKM2 expression is upregulated in M1 macrophages and can activate HIF-1α [47,48,49], which is important in glycolysis and polarization [50]. In our study, it was also observed that the expression levels of HIF-1α, and the activities of LDH and PK in LPS-activated macrophages were significantly increased, but these effects were eliminated by GW. When we knocked down HIF-1α in macrophages, the effects of GW were restrained (including those related to glycolysis and the release of inflammatory factors), which is consistent with previous reports that CB2R may regulate microglial inflammation through the HIF-1α/Tim-3 signaling pathway [51]. PKM2 has been shown not to be regulated by LPS stimulation [47, 52] and to exist as a monomer or dimer without enzyme activity, or as a tetramer with enzyme activity [48, 49, 53]. The dimer of PKM2, when transferred to the nucleus, interacts with HIF-1α and then promotes downstream target gene expression [47, 54]. Thus, PKM2 enzyme activity is increased, glycolysis positive feedback, and HIF-1α expression is stabilized. However, once the expression of HIF-1α is inhibited by activated CB2R, this cycle is broken, PKM2 activity is decreased, glycolysis is downregulated, and macrophages polarization and inflammatory factor expression are affected. HIF-1α not only mediates glycolysis in macrophages induced by LPS but also plays an important role in the regulation of inflammatory diseases. For example, HIF-1α is involved in metabolic reprogramming to glycolysis and in the production of proinflammatory cytokines and chemokines during sepsis [55, 56]. HIF-1α contributes to hemostasis, sterile inflammation, and early hepatocellular necrosis in acetaminophen-induced ALF [57]. In our study, the expression of HIF-1α was increased in liver macrophages of ALF liver tissue, and the increase in expression accompanied by increased infiltration of macrophages, which exacerbated liver pathological damage.
Aerobic glycolysis can be regulated by not only the transcription factor HIF-1α but also the PI3K/Akt signaling pathway [58]. The PI3K/Akt signaling pathway can regulate HIF-1α expression and transcriptional activity during hypoxia, and regulate glycolysis through a HIF-independent mechanism [59]. Inhibition of the PI3K/Akt signaling pathway has been reported to protect against ALF by promoting chaperone-mediated autophagy [60], or by suppressing oxidative stress-related inflammation [61]. By Western blotting, we detected the expression of PI3K and Akt (including p-Akt), but we did not obtain enough evidence that CB2R activation could regulate glycolysis and the inflammatory response through the PI3K/Akt signalling pathway.
In conclusion, the CB2R activation of liver macrophages can alleviate liver injury, by modulating the macrophage-centered inflammatory response. For example, it decreases macrophage infiltration, inhibits M1 polarization, and reduces inflammatory factors secretion by downregulating HIF-1α expression and macrophage glycolysis and then reducing the apoptosis of hepatocytes in ALF. Our study provides information on “immune metabolism” for further study of the complex pathophysiology of ALF, and provides a novel pathway to aid in the search for treatment targets.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Louvet A, Teixeira-Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A, et al. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology. 2011;54:1217–26.
Chen J, Huang ZB, Li H, Zheng X, Chen JJ, Wang XB, et al. Early diagnostic biomarkers of sepsis for patients with acute-on-chronic liver failure: a multicenter study. Infect Dis Ther. 2021;10:281–90.
Chen J, Huang ZB, Fan XG, Hu XW, Qi M, Liao CJ, et al. Potential predictors for prognosis and postpartum recovery time of acute fatty liver of pregnancy. BMC Pregnancy Childbirth. 2020;20:601.
Olah A, Szekanecz Z, Biro T. Targeting cannabinoid signaling in the immune system: “High”-ly exciting questions, possibilities, and challenges. Front Immunol. 2017;8:1487.
Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–21.
Triantafyllou E, Woollard KJ, McPhail MJW, Antoniades CG, Possamai LA. The role of monocytes and macrophages in acute and acute-on-chronic liver failure. Front Immunol. 2018;9:2948.
Mihm S, Danger-Associated Molecular Patterns (DAMPs): Molecular triggers for sterile inflammation in the liver. Int J Mol Sci. 2018;19:3104.
Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090.
Kolodziejczyk AA, Federici S, Zmora N, Mohapatra G, Dori-Bachash M, Hornstein S, et al. Acute liver failure is regulated by MYC- and microbiome-dependent programs. Nat Med. 2020;26:1899–911.
Liang DY, Liu LM, Ye CG, Zhao L, Yu FP, Gao DY, et al. Inhibition of UII/UTR system relieves acute inflammation of liver through preventing activation of NF-kappaB pathway in ALF mice. PLoS One. 2014;8:e64895.
Zannetti C, Roblot G, Charrier E, Ainouze M, Tout I, Briat F, et al. Characterization of the inflammasome in human Kupffer cells in response to synthetic agonists and pathogens. J Immunol. 2016;197:356–67.
Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462.
Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–30.
Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289:7884–96.
Palsson-McDermott EM, O’Neill LAJ. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30:300–14.
Huang ZB, Zheng YX, Li N, Cai SL, Huang Y, Wang J, et al. Protective effects of specific cannabinoid receptor 2 agonist GW405833 on concanavalin A-induced acute liver injury in mice. Acta Pharmacol Sin. 2019;40:1404–11.
Huang Z, Wang H, Wang J, Zhao M, Sun N, Sun F, et al. Cannabinoid receptor subtype 2 (CB2R) agonist, GW405833 reduces agonist-induced Ca2+ oscillations in mouse pancreatic acinar cells. Sci Rep. 2016;6:29757.
Denaes T, Lodder J, Chobert MN, Ruiz I, Pawlotsky JM, Lotersztajn S, et al. The Cannabinoid Receptor 2 protects against alcoholic liver disease via a macrophage autophagy-dependent pathway. Sci Rep. 2016;6:28806.
Tomar S, Zumbrun EE, Nagarkatti M, Nagarkatti PS. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide-induced acute liver failure through regulation of macrophage polarization and microRNAs. J Pharmacol Exp Ther. 2015;353:369–79.
Du Y, Ren P, Wang Q, Jiang SK, Zhang M, Li JY, et al. Cannabinoid 2 receptor attenuates inflammation during skin wound healing by inhibiting M1 macrophages rather than activating M2 macrophages. J Inflamm. 2018;15:25.
Jiang P, Wang L, Zhang M, Zhang M, Wang C, Zhao R, et al. Cannabinoid type 2 receptor manipulates skeletal muscle regeneration partly by regulating macrophage M1/M2 polarization in IR injury in mice. Life Sci. 2020;256:117989.
Braun M, Khan ZT, Khan MB, Kumar M, Ward A, Achyut BR, et al. Selective activation of cannabinoid receptor-2 reduces neuroinflammation after traumatic brain injury via alternative macrophage polarization. Brain Behav Immun. 2018;68:224–37.
Zhou H, Du R, Li G, Bai Z, Ma J, Mao C, et al. Cannabinoid receptor 2 promotes the intracellular degradation of HMGB1 via the autophagy-lysosome pathway in macrophage. Int Immunopharmacol. 2020;78:106007.
Luo XQ, Li A, Yang X, Xiao X, Hu R, Wang TW, et al. Paeoniflorin exerts neuroprotective effects by modulating the M1/M2 subset polarization of microglia/macrophages in the hippocampal CA1 region of vascular dementia rats via cannabinoid receptor 2. Chin Med. 2018;13:14.
Dando I, Donadelli M, Costanzo C, Dalla Pozza E, D’Alessandro A, Zolla L, et al. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013;4:e664.
Bueno MJ, Jimenez-Renard V, Samino S, Capellades J, Junza A, Lopez-Rodriguez ML, et al. Essentiality of fatty acid synthase in the 2D to anchorage-independent growth transition in transforming cells. Nat Commun. 2019;10:5011.
Whiteside GT, Gottshall SL, Boulet JM, Chaffer SM, Harrison JE, Pearson MS, et al. A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur J Pharmacol. 2005;528:65–72.
Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia, and catalepsy. Neuropharmacology. 2005;48:658–72.
Li AL, Carey LM, Mackie K, Hohmann AG. Cannabinoid CB2 Agonist GW405833 suppresses inflammatory and neuropathic pain through a CB1 mechanism that is independent of CB2 receptors in mice. J Pharmacol Exp Ther. 2017;362:296–305.
Schuelert N, Zhang C, Mogg AJ, Broad LM, Hepburn DL, Nisenbaum ES, et al. Paradoxical effects of the cannabinoid CB2 receptor agonist GW405833 on rat osteoarthritic knee joint pain. Osteoarthr Cartil. 2010;18:1536–43.
Paldy E, Bereczki E, Santha M, Wenger T, Borsodi A, Zimmer A, et al. CB(2) cannabinoid receptor antagonist SR144528 decreases mu-opioid receptor expression and activation in mouse brainstem: role of CB(2) receptor in pain. Neurochem Int. 2008;53:309–16.
Hartley JW, Evans LH, Green KY, Naghashfar Z, Macias AR, Zerfas PM, et al. Expression of infectious murine leukemia viruses by RAW264.7 cells, a potential complication for studies with a widely used mouse macrophage cell line. Retrovirology. 2008;5:1.
Chung RT, Stravitz RT, Fontana RJ, Schiodt FV, Mehal WZ, Reddy KR, et al. Pathogenesis of liver injury in acute liver failure. Gastroenterology. 2012;143:e1–e7.
Motwani MP, Gilroy DW. Macrophage development and polarization in chronic inflammation. Semin Immunol. 2015;27:257–66.
Devanney NA, Stewart AN, Gensel JC. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol. 2020;329:113310.
Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–14.
Mills EL, O’Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol. 2016;46:13–21.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9.
O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–65.
Biringer RG. Endocannabinoid signaling pathways: beyond CB1R and CB2R. J Cell Commun Signal. 2021;15:335–60.
Pavlou S, Wang L, Xu H, Chen M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J Inflamm (Lond). 2017;14:4.
Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–70. e13
Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. 2007;178:7516–9.
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–42.
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–37.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–85.
Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65–80.
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3.
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2:ra73.
Fan XG, Pei SY, Zhou D, Zhou PC, Huang Y, Hu XW, et al. Melittin ameliorates inflammation in mouse acute liver failure via inhibition of PKM2-mediated Warburg effect. Acta Pharmacol Sin. 2020;42:1256–66.
Kossatz E, Maldonado R, Robledo P. CB2 cannabinoid receptors modulate HIF-1alpha and TIM-3 expression in a hypoxia-ischemia mouse model. Eur Neuropsychopharmacol. 2016;26:1972–88.
Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84:1014–20.
Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–8.
Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44.
Zhan CY, Chen D, Luo JL, Shi YH, Zhang YP. Protective role of down-regulated microRNA-31 on intestinal barrier dysfunction through inhibition of NF-kappaB/HIF-1alpha pathway by binding to HMOX1 in rats with sepsis. Mol Med. 2018;24:55.
Martinez-Garcia JJ, Martinez-Banaclocha H, Angosto-Bazarra D, de Torre-Minguela C, Baroja-Mazo A, Alarcon-Vila C, et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat Commun. 2019;10:2711.
Sparkenbaugh EM, Saini Y, Greenwood KK, LaPres JJ, Luyendyk JP, Copple BL, et al. The role of hypoxia-inducible factor-1alpha in acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 2011;338:492–502.
Feng J, Li J, Wu L, Yu Q, Ji J, Wu J, et al. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2020;39:126.
Lee SH, Golinska M, Griffiths JR, HIF-1-independent mechanisms regulating metabolic adaptation in hypoxic cancer cells. Cells. 2021;10:2371.
Li Y, Lu L, Luo N, Wang YQ, Gao HM. Inhibition of PI3K/AKt/mTOR signaling pathway protects against d-galactosamine/lipopolysaccharide-induced acute liver failure by chaperone-mediated autophagy in rats. Biomed Pharmacother. 2017;92:544–53.
Zhong W, Qian K, Xiong J, Ma K, Wang A, Zou Y. Curcumin alleviates lipopolysaccharide induced sepsis and liver failure by suppression of oxidative stress-related inflammation via PI3K/AKT and NF-kappaB related signaling. Biomed Pharmacother. 2016;83:302–13.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81700561, 81970523), National Science and Technology Major Project (2018ZX10723203), National Natural Sciences Foundation of Hunan province (2019JJ40496, 2020JJ4877, 2020JJ4879). Sources of funding did not have any role in study design, data collection or analysis, interpretation of results, or manuscript preparation or submission. The corresponding author confirms that all the authors have full access to the full data in the study and accept responsibility to submit for publication.
Author information
Authors and Affiliations
Contributions
ZBH: Conceptualization, project design, and guidance, Funding acquisition, project administration. XGF, JW, YW: writing—review & editing. SLC: Writing—original draft, data curation, formal analysis, visualization. SYP, YXZ, JC: Methodology, investigation. XWH, YH, NL: Writing—review & editing, methodology. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cai, Sl., Fan, Xg., Wu, J. et al. CB2R agonist GW405833 alleviates acute liver failure in mice via inhibiting HIF-1α-mediated reprogramming of glycometabolism and macrophage proliferation. Acta Pharmacol Sin 44, 1391–1403 (2023). https://doi.org/10.1038/s41401-022-01037-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-01037-8
