
Abstract
Anterior gradient 2 (AGR2), a protein disulfide isomerase (PDI), is a multifunctional protein under physiological and pathological conditions. In this study we investigated the roles of AGR2 in regulating cholesterol biogenesis, lipid-lowering efficiency of lovastatin as well as in protection against hypercholesterolemia/statin-induced liver injury. We showed that AGR2 knockout significantly decreased hepatic and serum total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C) in mice with whole-body or hepatocyte-specific Agr2-null mutant, compared with the levels in their wild-type littermates fed a normal chow diet (NCD) or high-fat diet (HFD). In contrast, mice with AGR2 overexpression (Agr2/Tg) exhibited an increased cholesterol level. Mechanistic studies revealed that AGR2 affected cholesterol biogenesis via activation of AKT/sterol regulatory element-binding protein-2 (SREBP2), to some extent, in a PDI motif-dependent manner. Moreover, elevated AGR2 led to a significant decrease in the lipid-lowering efficacy of lovastatin (10 mg· kg−1· d−1, ip, for 2 weeks) in mice with hypercholesterolemia (hyperCho), which was validated by results obtained from clinical samples in statin-treated patients. We showed that lovastatin had limited effect on AGR2 expression, but AGR2 was inducible in Agr2/Tg mice fed a HFD. Further investigations demonstrated that drug-induced liver toxicity and inflammatory reactions were alleviated in hypercholesterolemic Agr2/Tg mice, suggesting the dual functions of AGR2 in lipid management and hyperCho/statin-induced liver injury. Importantly, the AGR2-reduced lipid-lowering efficacy of lovastatin was attenuated, at least partially, by co-administration of a sulfhydryl-reactive compound allicin (20 mg· kg−1· d−1, ip, for 2 weeks). These results demonstrate a novel role of AGR2 in cholesterol metabolism, drug resistance and liver protection, suggesting AGR2 as a potential predictor for selection of lipid-lowering drugs in clinic.
Similar content being viewed by others


Introduction
Cholesterol plays an important role in the fluidity and permeability of cell membranes, and is essential for bile acid and steroid hormone biosynthesis [1]. It has been well documented that the cholesterol pool is derived from de novo synthesis of cholesterol in the liver, absorption of dietary cholesterol in the gastrointestinal tract, reabsorption of biliary cholesterol, and transport of cholesterol in circulation [2]. Disrupted cholesterol homeostasis, particularly hypercholesterolemia, as evidenced by high levels of total cholesterol (TC) and/or low-density lipoprotein cholesterol (LDL-C) in the blood, acts as one of the main causes for the development of cardiovascular diseases, liver disorders, metabolic diseases, and cancers [3, 4]. Therefore, many investigations have revealed key factors and pathways that control cholesterol metabolism. For example, statins, which competitively inhibit the activity of cholesterol synthesis rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), as first-line treatment to reduce intracellular cholesterol synthesis and lower LDL-C levels [5]. Proprotein convertase subtilisin kexin 9 (PCSK9) is involved in the degradation of the LDL receptor (LDLR) in the liver. Inhibition of PCSK9 by monoclonal antibodies, including alirocumab and evolocumab, effectively enhances LDL-C clearance and results in lowering LDL-C levels in circulation [6]. Bile acid sequestrants, such as cholestamide, bind irreversibly with bile acids in the intestine, accelerating the conversion of cholesterol to bile acids and reducing cholesterol levels in the liver and plasma. Additionally, inhibition of cholesterol absorption in the intestine, such as ezetimibe that selectively targets the sterol transporter Niemann-Pick C1-Like 1 (NPC1L1) in the small intestine, was used as a combination therapy to effectively lower cholesterol levels. However, in clinical practice, many patients are unable to achieve the therapeutical LDL-C goals (<1.8 mmol/L or 1.4 mmol/L currently recommended). This may be resulted from inadequacy and/or variability in response to lipid-lowering agents [7, 8]. It has been observed that statin therapy has limitations of effectiveness in addition to their adverse effects in clinical treatments [5, 9]. Thus, alternative approaches to lipid management, and also specific biomarkers for suitable drug selection and patient stratification are urgently needed in clinic.
Anterior gradient 2 (AGR2), an endoplasmic reticulum (ER)-resident protein belonged to protein disulfide isomerase (PDI) family, is also a secret protein [10]. AGR2 functions as a chaperon/isomerase in a CPHS (81–84) motif-dependent manner, mutation in Cys81 in the motif results in loss its activity [11]. The roles of AGR2 in tumorigenesis and metastasis have been well-established by many studies [12]. Recent investigations including ours demonstrate that secreted AGR2 is a promoter to cancer invasion and metastasis by binding to the metastasis-associated proteins or modulating [13, 14]. Given the changes in phenotype of AGR2-null mice (Agr2−/−) mice, we demonstrated a novel role of AGR2, in the regulation of lipid metabolism via promoting long-chain fatty acid activation [15]. In the present study, we went a further step and revealed a novelty of AGR2 in affecting lipid-lowering efficiency of lovastatin and protection against hypercholesterolemia/statin-induced liver injury. This effect of AGR2 was ascribed to its regulatory activity on activation of the AKT/SREBP2 signaling that controls cholesterol biosynthesis, and anti-inflammation and apoptosis effects. We also provided an option to reverse AGR2-mediated drug resistance by combination therapy with allicin.
Materials and methods
Cell lines and cell culture
The human hepatocellular cancer lines (HepG2 and Huh-7) were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China) and were maintained in DMEM medium (Hyclone, Logan, UT, USA). All cells were supplemented at 37 °C in a humid atmosphere (5% CO2 and 95% air) with 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 100 U/mL of penicillin and 100 μg/mL of streptomycin. All cells were harvested by brief incubation in 0.25% (w/v) EDTA-PBS.
Animals
C57BL/6 male mice (6-week-old) were obtained from Animal Center of China Academy of Medical Sciences (Beijing, China). Agr2−/− mice were obtained from Jackson Lab (USA). We kept breeding by crossbreeding Agr2+/− heterozygote each other to obtain Agr2−/− homozygous mice [16]. TgTn (CAG-Agr2-3×Flag-WPRE-pA) overexpression transgenic mice and Agr2fl/fl mice were constructed in Shanghai Model Organisms Center (Shanghai, China). To obtain hepatocyte-specific conditional Agr2 knockout mice, we initially established AGR2 mice with flox sites flanking exon 2 and 3 of Agr2 (Agr2fl/fl) with those expressing albumin (Alb) promoter-driven Cre recombinase (Alb-Cre). Homozygous hepatocyte-specific conditional Agr2 knockout (Agr2fl/fl; Alb-Cre+) mice were generated by crossing Agr2fl/fl mice with the mice expressing Alb-Cre. The genotyping changes in mice were analyzed by PCR assays, and the data were shown in Supplementary Fig. S1. In animal experiments, three to five mice were included in each group. Mice were caged in an environment with controlled temperature and humidity with free access to water and food under a light-dark (12 h:12 h) cycle. At the end of experiments, mice were fasted overnight and then euthanized with an overdose of pentobarbital sodium (150 mg/kg, i.v.). All animal experiments were approved by Ethics Committee of School of Medicine of Shandong University (Ji-nan, China). The study was conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978).
HFD-induced obese mice
Diets were purchased from Trophic Animal Feed High-tech Co. (Nantong, China). After 1 week of diet adaptation, mice at the age of 8 weeks were fed on a high-fat diet (HFD; fat 60%) for 8 weeks to induce obesity; mice fed a normal chow diet (NCD) were used as lean controls.
Hypercholesterolemia model food-induced hyperCho mice
After 1 week of diet adaptation, mice at the age of 8 weeks were fed on a hypercholesterolemia model food diet (sucrose 20.0%, fat 15%, cholesterol 1.2%, sodium cholate 0.2% calories) for 8 weeks to induce hypercholesterolemia (hyperCho). Mice fed a NCD were used as controls. Then mice were intraperitoneally injected with lovastatin (10 mg/kg) (Solarbio, Beijing, China) and allicin (20 mg/kg) (Solarbio, Beijing, China) every day for 2 weeks, while control groups of mice were administered the same volume of sterile saline.
Poloxamer 407-induced acute hyperlipidemia mice
Poloxamer 407 (P407; Solarbio, Beijing, China) was used to induce acute hyperlipidemia. Mice were daily intraperitoneally injected with P407 (0.5 g/kg) for 3 days, while control groups of mice were administered the same volume of sterile saline. Then mice were intraperitoneally injected with lovastatin (10 mg/kg) and allicin (20 mg/kg). Wortmannin (Selleck, Shanghai, China) were injected in 0.7 mg/kg and mice were sacrificed on the fourth day.
Human samples
All samples used in this study were obtained after approval by the Medicine Ethics Committee of the Second Hospital of Shandong University. The ethics approval number is KYLL-2022LW015, and informed consent paper was signed for each subject.
Transient transfection of plasmids and siRNAs
The cells were transiently transfected with pcDNA3.1, pcDNA3.1-Flag-AGR2 and pcDNA3.1-AGR2-C81A for 48 h using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instruction. For the siRNA assay, AGR2 siRNA (GenePharma, Shanghai, China) was transfected into the cells for 48 h. Scramble siRNA served as a control. After transfection, the cells were subjected to treatment. Full-length AGR2 expression plasmid and various AGR2 mutant plasmids, small interference sequences of AGR2 (siRNA) have been used and verified in our and other reports [15, 17]. At least three independent experiments were performed. The siRNA sequences were listed in Supplementary Table S1.
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted using a Trizol Kit (Takara, Dalian, China). cDNA was prepared using a PrimerScript RT Reagent Kit with gDNA Eraser (TOYOBO, Shanghai, China) according to the manufacturer’s protocol. Real-time PCR was performed with SYBR Green (Accurate Biology, Changsha, China). The cycling conditions for: 95 °C for 5 min; followed by 40 cycles of: 95 °C for 15 s; 55 °C for 15 s; 68 °C for 20 s. Changes in the transcript level were calculated using the ΔΔCt method. The expressions of genes were normalized against that of a housekeeping gene and plotted as relative change in the expression with respect to control. Each reaction was performed in triplicate. The primers of the target genes are listed in Supplementary Table S2.
Western blotting
Western blotting assays were performed to analyze the expressions of proteins in cells and liver tissues. Cells were washed with ice-cold PBS and cell lysates were prepared using SDS buffer containing fresh protease inhibitor mixture (50 μg/mL aprotinin, 0.5 mM phenylmethanesulfonyl fluoride (PMSF), 1 mM sodium orthovanadate, 10 mM sodium fluoride and 10 mM glycerolphosphate). Liver was weighed to 0.05 g and tissue lysates were subjected for protein analysis. Proteins were quantified using the BCA protein assay. Samples containing equal amounts of protein (40 μg protein per lane) from lysates were separated by 10%–12% SDS–PAGE and electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes. After being blocked with 5% fat-free dry milk in TBS (20 mM Tris-HCl (pH 7.6), 150 mM NaCl) for 1 h at room temperature, the membranes were immunoblotted with primary antibodies overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies for 1 h at room temperature. Membranes were visualized by enhanced chemiluminescence detection system (Millipore) and followed by exposure to X-ray films. The detail information of the primary antibody is listed in Supplementary Table S3. Triplicate experiments with triplicate samples were performed.
Hematoxylin and eosin (H&E) staining and immunohistochemical analysis
Tissue sections were prepared from organs fixed with 4% paraformaldehyde and embedded with paraffin using embedding machine (Sakura, Japan). The paraffin tissue specimens were sectioned at a 5 μm thickness for H&E staining and immunohistochemical analysis. For H&E staining the experiment was performed by routine methods. For immunohistochemical analysis, after antigen retrieval, the appropriate primary antibodies were added onto the sections and incubated overnight at 4 °C. After being stained with IgG conjugated HRP and DAB (Vector Laboratories), samples were counterstained with hematoxylin and subjected to capture images by Nanozoomer Digital Pathology scanner microscopy (NanoZoomer S60).
Immunofluorescence staining
Tissue sections were incubated with primary antibodies prior to being rinsed with phosphate-buffered saline, then immunostained with secondary antibodies and nuclei were stained with DAPI (Sigma-Aldrich, USA). Fluorescence images were captured using a confocal microscopy (Carl Zeiss, Germany).
ELISA
AGR2 ELISA Kit (Cusabio, Wuhan, China) was used for quantitate AGR2 from mice and human serum.
Measurement of lipid profiles
The biochemistry reagent discs for serum TG, TC, LDL-C, HDL-C, ALT and AST were bought from Chengdu Polytech Biological Technology Co., Ltd (Polytech, Chengdu, China).
Statistical analysis
The data are presented as the mean ± SD and analyzed with GraphPad Prism software (GraphPad 8.4.2, La Jolla, CA, USA). Student’s t test was used for comparison among different groups. All the experiments have been repeated at least three times. Values of P < 0.05 were considered to be statistically significant. The bands in Western blotting were densitometrically quantified with ImageJ (1.8.0.172) software, and normalized by internal controls.
Results
AGR2 facilitates the de novo cholesterol synthesis
We have previously observed a reduction in blood lipid levels (TC and LDL-C) in whole-body AGR2-null mice (Agr2−/−) [15], and a hepatocyte-specific conditional Agr2 knockout (Agr2fl/fl; Alb-Cre+) mice (Agr2/LKO) was further generated by crossing mice with flox sites flanking exon 2 and 3 of Agr2 (Agr2fl/fl) with those expressing albumin promoter-driven Cre recombinase (Alb-Cre) to confirm the role of AGR2 in the regulation of lipid metabolism in the liver, a main site for de novo lipid synthesis. It was clearly shown that, in addition to a decreased TC level in the blood, TC in the liver was also significantly reduced in Agr2/LKO mice compared to the control Agr2fl/fl mice fed either a NCD or a HFD (Fig. 1a). Hepatic TC decreased by 19% and 14% in response to NCD and HFD feeding, respectively (Fig. 1a), implicating that AGR2 depletion impaired cholesterol biosynthesis in the liver. We analyzed the changes in gene expression involved in the cholesterol biogenesis mevalonate (MVA) pathway and lipid regulatory processes. Although the expression of most of genes tended to decline, no significance was shown between the hepatic samples obtained from knockout mice and their matched controls (Fig. 1b, c). However, the expression levels of HMGCR and squalene epoxidase (SQLE), two enzymes in the MVA pathway, were downregulated in hepatic samples either from Agr2/LKO and Agr2−/− mice (Fig. 1b, c). By contrast, upregulated HMGCR and SQLE were observed in Agr2/Tg mice (Fig. 1d). Protein abundance analysis demonstrated that AGR2 was positively correlated with the changes of HMGCR and SQLE in the liver tissues from Agr2−/− mice as well as Agr2/Tg mice (Fig. 1e, f). Furthermore, ectopic expression of AGR2 in HepG2 cells (AGR2 level is low) increased HMGCR and SQLE (Fig. 1g), while downregulation of AGR2 led to a significant decrease in HMGCR and SQLE in hepatocellular carcinoma Huh7 cells (Fig. 1h). Thus, AGR2 was able to facilitate cholesterol production through activating the MVA pathway.

a TC contents in circulating (left) and livers (right) of Agr2fl/fl and Agr2/LKO mice fed a NCD or a HFD (n = 5). b MVA pathway and Aacs expression in the livers of the 8-week-old Agr2fl/fl and Agr2/LKO mice fed a NCD (n = 3). c MVA pathway expression and Aacs in the livers of the 8-week-old Agr2+/+ and Agr2−/− mice fed a NCD (n = 3). d MVA pathway and Aacs expression in the livers of the 8-week-old WT and Agr2/Tg mice fed a NCD (n = 3). e Immunohistochemical staining of HMGCR in the livers of the 8-week-old Agr2+/+ and Agr2−/− mice fed a NCD (left) and WT and Agr2/Tg mice fed a NCD (right). Scale: 100 μm. f Western blotting analysis of HMGCR and SQLE in the livers of the 8-week-old Agr2+/+ and Agr2−/− mice (top), WT and Agr2/Tg mice (bottom) fed a NCD. Quantification was performed by normalizing proteins to β-actin. g Western blot analysis of HMGCR and SQLE in whole-cell lysates from HepG2 cells treated with AGR2 and AGR2-C81A expression plasmids. Quantification was performed by normalizing proteins to β-actin. h Western blotting analysis of HMGCR and SQLE in whole-cell lysates from Huh7 cells treated with siRNA targeting AGR2. Quantification was performed by normalizing proteins to β-actin. Representative figures were generated with data from at least three independent experiments. The data were presented as the mean ± SD values. *P < 0.05 by Student’s t test.
Activation of AKT/SREBP2 by AGR2 contributes to its effect on cholesterol synthesis
As upstream gene HMGCR and downstream SQLE in the MVA pathway appeared to respond to AGR2 depletion or overexpression at mRNA levels shown in Fig. 1, it is possible that AGR2 might exert a regulatory effect upstream of the MVA pathway, particularly on HMGCR. We therefore sought to determine whether some vital regulators of cholesterol, including insulin-induced gene 2 (INSIG2) and sterol regulatory element-binding protein 2 (SREBP2) are involved in AGR2-mediated regulation on cholesterol. Elevation of INSIG2 mRNA was displayed in the liver obtained from Agr2−/− mice, however no detectable changes were observed in Agr2/LKO or Agr2/Tg mice (Fig. 2a, b). Regarding of SREBP2, it was upregulated in Agr2/Tg mice, but unaltered in the liver of Agr2 deficient mice (Fig. 2a, b). A protein abundance analysis showed that INSIG2 did not show clear change pattern upon AGR2 depletion and overexpression (Fig. 2c, d), suggesting that INSIG2 might not be important to the AGR2-mediated effect. The production of mature SREBP2 (m-SREBP2) significantly dropped down in Agr2/LKO and Agr2−/− mice than their control mice Agr2fl/fl and Agr2+/+, respectively, while it was upregulated in Agr2/Tg mice compared with their wild-type (WT) littermates (Fig. 2c, d). The precursor SREBP2 (p-SREBP2) remained almost unchanged upon AGR2 depletion, although it increased, to some extent, in Agr2/Tg mice (Fig. 2d). The changes in p-SREBP2 and m-SREBP2 were validated in HepG2 and Huh7 cells upon overexpression or downregulation of AGR2 (Fig. 2e, f), highlighting the importance of SREBP2 processing in the AGR2-mediated regulatory effect. To determine the mechanism by which AGR2 affects SREBP2 activation, we examined the AKT/mTOR signaling pathway, which is critical in controlling intracellular cholesterol levels by modulating SREBP2 activation [18]. As shown in Fig. 2g, AGR2 depletion significantly suppressed AKT/mTOR activation in the liver of either specific Agr2/LKO mice or whole-body Agr2−/−, whereas enhancement of AGR2 marginally stimulated AKT signaling, as indicated by the changes of phosphor-AKT in Agr2/Tg mice when compared to their matched control mice (Fig. 2h). Similarly, overexpression of AGR2 slightly influenced the phosphorylation of AKT and mTOR, mutation of the thioredoxin motif AGR2 (C81A), impaired phospho-AKT and phospho-mTOR in HepG2 cells to some extent (Fig. 2i), suggesting a role of the thioredoxin motif of AGR2 in regulation of AKT signaling. By contrast, AGR2 depletion profoundly inhibited AKT/mTOR activation (Fig. 2j).

a Insig2 and Srebp2 expression in the livers of the 8-week-old Agr2fl/fl and Agr2/LKO fed a NCD (left), Agr2+/+ and Agr2−/− mice fed a NCD (right) (n = 3). b Insig2 and Srebp2 expression in the livers of the 8-week-old WT and Agr2/Tg mice fed a NCD (n = 3). c Western blotting analysis of INSIG2, p-SREBP2 and m-SREBP2 in the livers of the 8-week-old Agr2fl/fl and Agr2/LKO mice fed a NCD (left), Agr2+/+ and Agr2−/− mice fed a NCD (right). d Western blotting analysis of INSIG2, p-SREBP2 and m-SREBP2 in the livers of the 8-week-old WT and Agr2/Tg mice fed a NCD. e Western blotting analysis of p-SREBP2 and m-SREBP2 in whole-cell lysates from HepG2 cells treated with AGR2 and AGR2-C81A expression plasmids. f Western blotting analysis of p-SREBP2 and m-SREBP2 in whole-cell lysates from Huh7 cells treated with siRNA targeting AGR2. g Western blotting analysis of AKT/mTOR signal pathway in the livers of the 8-week-old Agr2fl/fl and Agr2/LKO mice fed a NCD (left), Agr2+/+ and Agr2−/− mice fed a NCD (right). h Western blotting analysis of AKT/mTOR signal pathway in the livers of the 8-week-old WT and Agr2/Tg mice fed a NCD. i Western blotting analysis of AKT/mTOR signal pathway in whole-cell lysates from HepG2 cells treated with AGR2 and AGR2-C81A expression plasmids. j Western blotting analysis of AKT/mTOR signal pathway in whole-cell lysates from Huh7 cells treated with siRNA targeting AGR2. Quantification for Western blotting was performed by normalizing proteins to β-actin. Representative figures were generated with data from at least three independent experiments. The data were presented as the mean ± SD values. *P < 0.05 by Student’s t test.
Additional experiments were performed to elucidate a role of AKT/mTOR signal in AGR2-mediated effect on cholesterol in vitro and in vivo. We transfected cells with pcDNA3.1-Flag-AGR2 to increase AGR2 expression and inactivated AKT with PI3K inhibitors (wortmannin) [19] to examine changes of cholesterol. As is shown in Fig. 3a, inactivation of PI3K by wortmannin obviously decreased phospho-AKT, accompanied with a reduction of HMGCR in cells with overexpression of AGR2. Suppression of phospho-AKT by wortmannin was somehow restored when AGR2 expression was enhanced by transfection, with a slight increase in HMGCR (Fig. 3a). We also examined total cholesterol (TC) production in cells treated with wortmannin. The results shown in Fig. 3b, addition of wortmannin had limited effect on TC in HepG2 cells, but reduced the content of TC in cells overexpressing AGR2 (wortmannin at 5 μM). Animal study was carried out to further confirm the role of AKT signal pathway in AGR2-mediated effect on cholesterol. Agr2/Tg mice under P407-induced acute hyperlipidemia model [20] was used for the examination of the changes of HMGCR and TC upon wortmannin treatment. As shown in Fig. 3c, the results indicated that administration of wortmannin, significantly suppressed AKT activity in WT mice but had less effect in Agr2/Tg mice. Importantly, induction of HMGCR was evidenced in Agr2/Tg mice under P407 treatment, but noticeably reduced in Agr2/Tg hyperlipidemic mice in the presence of wortmannin (Fig. 3c), implicating that AKT inactivation was able to block AGR2-promoted HMGCR under hyperlipidemic conditions. Furthermore, as is shown in Fig. 3d, the content of TC in the serum and the livers of P407-induced hyperlipidemic Agr2/Tg mice was higher than that of WT littermates. Inactivation of AKT by wortmannin partially decreased AGR2-induced TC production in Agr2/Tg hyperlipidemic mice, indicating that activation of AKT signaling by AGR2 partially contributed to AGR2-promoted enhancement in cholesterol synthesis.

a Western blotting analysis of AKT/p-AKT and HMGCR in whole-cell lysates from HepG2 cells treated with AGR2 expression plasmids and wortmannin. b TC contents in HepG2 cells treated with AGR2 expression plasmids and wortmannin. c Western blotting analysis of AKT/p-AKT and HMGCR in the livers of WT and Agr2/Tg mice treated with P407 and wortmannin. d TC contents in serum and livers of WT and Agr2/Tg mice treated with P407 and wortmannin. Quantification for Western blotting was performed by normalizing proteins to β-actin. Representative figures were generated with data from at least three independent experiments. The data were presented as the mean ± SD values. *P < 0.05; **P < 0.01 by Student’s t test.
Thus, these results indicated that AGR2 is essential for maintaining AKT/mTOR activation, leading to boosted cholesterol synthesis by stimulating SREBP2 activation.
AGR2 impairs the lipid-lowering efficacy of lovastatin
Given the role of AGR2 in cholesterol synthesis, we evaluated whether AGR2 impacts the efficiency of statins, which are clinically used antihyperlipidemic agents. To this end, a hypercholesterolemic (hyperCho) mouse model was developed by feeding a special food diet containing high-cholesterol for 8 weeks [21, 22], and the mice were treated with lovastatin by intraperitoneal injection for an additional 2 weeks (Fig. 4a). Serum lipid analysis revealed that the increased triglyceride (TG), TC and LDL-C levels were significantly reduced in both WT and Agr2−/− mice with hyperCho in response to lovastatin (Fig. 4b). TC and LDL-C were decreased by 33% and 43% in the WT control mice, respectively, after lovastatin treatment (Fig. 4b). However, these parameters were reduced by 41% and 62% in Agr2−/− hyperCho mice, more than that of WT control group (Fig. 4b). These results suggested that Agr2−/− mice with hyperCho were more sensitive to lovastatin treatment than that of WT controls. We were prompted to examine whether the enhancement of AGR2 in mice is developed against statin therapy. It was clearly shown that serum lipid parameters were significantly increased in the hyperCho group and were even further increased in the Agr2/Tg model when compared to those in control mice (Fig. 4c). Importantly, lovastatin exerted a lipid-lowering effect on WT mice with hyperCho, but this efficacy was significantly dampened in the hyperCho Agr2/Tg mice, as indicated by the noticeable restoration of TG, TC, and LDL-C levels (Fig. 4c). It was noted that AGR2 secretion was not affected upon lovastatin treatment (Fig. 4d), and AGR2 expressions remained unchanged in cells challenged with lovastatin (Supplementary Fig. S1b, c). However, elevated secretion of AGR2 was clearly shown in Agr2/Tg mice, rather than in WT mice, under hyperCho conditions (Fig. 4d). Consistent with the results shown in Fig. 4d, expression levels of AGR2 in the liver of WT mice were slightly increased upon HFD treatment, and robustly induced in Agr2/Tg mice under same conditions (Fig. 4e, f). These results implicated that high level of AGR2 was more inducible than that of mice with normal level AGR2 in response to lipid metabolism stress.

a Schedule of treatment for food-induced hyperCho. Mice were fed on a hyperCho model food diet for 8 weeks and mice fed a normal chow diet (NCD) were used as controls. At the 8th week, mice were intraperitoneally injected with lovastatin for 2 weeks. b Circulating lipid profiles of Agr2+/+ and Agr2−/− mice fed a NCD, treated with hyperCho and lovastatin (n = 5). c Circulating lipid profiles of WT and Agr2/Tg mice fed a NCD, treated with hyperCho and lovastatin (n = 5). d Circulating AGR2 of WT and Agr2/Tg mice fed a NCD, treated with hyperCho and lovastatin (n = 5). e Western blotting analysis of AGR2 in the livers of WT and Agr2/Tg mice fed a NCD or HFD. Quantification for Western blotting was performed by normalizing proteins to β-actin. f Immunohistochemical staining of AGR2 in the livers of WT mice (left) and Agr2/Tg mice (right) fed a NCD or HFD. g Correlation analysis of serum AGR2 with ΔTG, ΔTC, ΔLDL-C and ΔHDL-C in clinical statin-treated patients (n = 40). Representative figures were generated with data from at least three independent experiments. The data were presented as the mean ± SD values. *P < 0.05; **P < 0.01 by Student’s t test.
To confirm the role of AGR2 in conferring resistance to statin therapy, we further collected blood samples from 40 patients who received statin therapy for 2 months due to hyperCho. The lipid-lowering efficacy of statins, including atorvastatin, fluvastatin and rosuvastatin was evaluated by the changes in the content of plasma lipids before and after treatment, namely, the ΔTC (ΔTG, ΔLDL-C, or ΔHDL-C) values were different from TC (TG, LDL-C, or HDL-C) before and after treatment, to indicate the ability of AGR2 on lipid-lowering efficacy of statins. Correlation analysis indicated that high levels of plasma AGR2 resulted in lower efficacy of statins, as evidenced by lower ΔTC in circulation (P < 0.05) (Fig. 4g), displaying a negative correlation between AGR2 levels and the efficacy of statin treatments. Similar correlation patterns of AGR2 and ΔLDL-C were observed, as shown in Fig. 4g. It was hard to conclude that AGR2 had relevance with ΔTG or ΔHDL-C, although a negative trend between AGR2 levels and ΔTG was observed after statin treatments (Fig. 4g). These results provided solid evidence that hypercholesterolemic individuals with low levels of AGR2 would benefit from statin therapy than those patients with higher AGR2. Taken together, these data showed that high levels of AGR2 can effectively suppress the lipid-lowering activity of statins, and plasma AGR2 may act as a biomarker for the selection of lipid-lowering drugs in the clinical treatment of hypercholesterolemia.
AGR2 alleviates liver toxicity induced by statins
Considering the hepatic toxicity of statins and multifunction of AGR2 in physiopathology [23,24,25,26], we examined the changes in alanine aminotransferase (ALT) and aspartate aminotransferase (AST), which are typical markers for the evaluation of liver functions. As shown in Fig. 5a, in contrast to plasma AST, which was slightly increased in the WT mice, ALT was significantly elevated in the WT mice in response to lovastatin treatment or fed a hyperCho model food diet. Notably, elevated ALT profoundly declined in the Agr2/Tg mice under the same conditions. The AGR2-mediated protective effect on ALT release in the Agr2/Tg mice was comparable to that of the control mice treated with saline (Fig. 5a). These results suggested that AGR2 might exhibit protection from hyperCho- and/or statin-induced liver injury. We performed H&E staining assays to validate the effect of AGR2 on liver functions. As shown in Fig. 5b, there were no detectable morphological changes between the WT control and Agr2/Tg mice under a normal diet or hyperCho model food feeding. However, lipid accumulation was more evident in the liver of Agr2/Tg mice than that of the WT mice under hyperCho conditions, as indicated by changes in lipid droplets in the liver sections of the mice (Fig. 5b). Lovastatin treatment in mice with hyperCho induced punctate necrosis in the WT mice but did not cause significant hepatic changes in the Agr2/Tg mice, which was associated with reduced lipid depositions in both the WT and Agr2/Tg groups (Fig. 5b). We further examined alterations in inflammation and cell death due to increased ALT secretion in response to lovastatin. It was obvious that IL-6, served as a typical pro-inflammatory marker, was somehow induced in WT mice with hypercholesterolemia, and this change was more pronounced in hypercholesterolemic mice treated with lovastatin (Fig. 5c). However, the enhanced IL-6 significantly declined in hyperCho Agr2/Tg mice upon lovastatin treatment (Fig. 5c), implicating that AGR2 had anti-inflammatory effect. Consistently, F4/80, a predictive marker for macrophage cells, was evident in WT mice compared with Agr2/Tg mice challenged with lovastatin (Fig. 5c). Given these observations, we further examined whether increased ALT due to induction of apoptosis upon hyperCho and drug treatments. Apoptotic markers, Caspase 3 and poly (ADP-ribose) polymerase (PARP), were used to evaluate the effect of lovastatin on apoptosis by Western blotting assays. As shown in Fig. 5d, hyperCho somehow induced apoptosis with or without lovastatin in WT mice, as indicated by slightly increased cleavage of PARP, a substrate of activated caspase-3. It appeared that high levels of AGR2 were able to attenuate apoptosis induced by hyperCho and lovastatin as observed in Agr2/Tg mice, indicating that AGR2 may exert a protective function in response to stimuli. Therefore, AGR2 displayed an anti-inflammatory and anti-apoptotic role in alleviating liver injury induced by hypercholesterolemia/lovastatin.

a Serum AST (left) and ALT (right) in WT and Agr2/Tg mice fed a NCD, treated with hyperCho and lovastatin (n = 5). b H&E staining in the livers of WT and Agr2/Tg mice fed a NCD, treated with hyperCho and lovastatin. Scale: 100 μm. c Immunohistochemical staining of IL-6, and immunofluorescence staining of F4/80 in the livers of WT mice with hyperCho and lovastatin, livers of Agr2/Tg mice with hyperCho and lovastatin. Scale: 100 μm. d Western blotting analysis of PARP and Caspase 3 in the livers of WT and Agr2/Tg mice fed a NCD, with hyperCho and lovastatin. Quantification for Western blotting was performed by normalizing proteins to β-actin. Representative figures were generated with data from at least three independent experiments. The data were presented as the mean ± SD values. *P < 0.05; **P < 0.01 by Student’s t test.
AGR2-mediated resistance to lovastatin is ameliorated by allicin
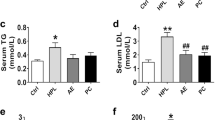
Given the dual roles of AGR2 in liver function and lovastatin efficacy, we aimed to develop an approach to safely alleviate AGR2-mediated resistance to lovastatin. Considering that the CPHS active motif in AGR2 is vital for stabilizing substrate proteins in a manner dependent on disulfide bond formation/isomerization, allicin, a naturally occurring compound mainly found in garlic, has received our attention. In addition to its slight lipid-lowering effects, allicin is a reactive sulfur-containing compound [27] that may impair the PDI function of AGR2 on lovastatin efficacy. The results of animal studies showed that allicin moderately decreased serum TC and LDL-C levels in both hyperCho WT and Agr2/Tg mice when administered as a single-agent treatment (Fig. 6a). Remarkably, the AGR2-mediated reduction in the effectiveness of lovastatin was almost completely reversed in the presence of allicin in hyperCho Agr2/Tg mice, and the anti-hypercholesterolemic efficiency was comparable to that of co-treatment with lovastatin and allicin in WT control groups, as evidenced by reduced content of TC and LDL-C (Fig. 6a). HDL-C levels remained undetectable changes (Fig. 6a). We further examined the changes of hepatic cholesterol in response to treatments. As shown in Fig. 6b, although allicin was intended to reduce cholesterol in the liver from either WT or Agr2/Tg mice with hyperCho, there was no significant difference between placebo treatment and allicin-treated mice. However, combined treatment with allicin and lovastatin synergistically enhanced the lipid-lowering effect of lovastatin in the hyperCho Agr2/Tg mice as noted by a significant reduction in cholesterol (Fig. 6b), consistent with the observations shown in Fig. 6a. We then tested if allicin has ability to affect AGR2 functions. As is shown in Fig. 6c, in response to allicin, the level of cholesterol was decreased in cells transfected with an AGR2 expression plasmid, but not in AGR2 mutant plasmid (C81A), As the dimerization of AGR2 acts as an important role in mediating the PDI function of AGR2 [26], indicating that allicin may exert interference to PDI function of AGR2 when the levels of AGR2 was high. This effect of allicin on reversing the suppressive effect of AGR2 on lovastatin efficacy was confirmed in the Agr2/Tg mice with poloxamer 407-induced hyperlipidemia (Fig. 6d). Furthermore, the results in Fig. 6e showed that lovastatin, or plus allicin, to some extent, was able to activate SREBP2 in the hyperCho Agr2/Tg mice compared to the matched control mice, probably due to the feedback regulation of SREBP2 by low cholesterol levels. In case of HMGCR, allicin alone had limited effect on HMGCR expression in both WT and Agr2/Tg mice, but augment of HMGCR by AGR2 was not sustained when hyperCho Agr2/Tg mice were challenged with lovastatin plus allicin (Fig. 6e). Thus, high levels of AGR2 reduced the efficiency of lovastatin, but this reduction could be overcome by combined treatment with lovastatin and allicin.

a Circulating lipid profiles of WT and Agr2/Tg mice treated with hyperCho and lovastatin and/or allicin (n = 5). b TC contents in the livers of WT and Agr2/Tg mice treated with hyperCho and lovastatin and/or allicin (n = 5). c TC contents in HepG2 cells treated with AGR2, AGR2-C81A expression plasmids and allicin. d Circulating lipid profiles of WT and Agr2/Tg mice treated with P407 and lovastatin and/or allicin (n = 5). e Western blotting analysis of HMGCR, p-SREBP2 and m-SREBP2 in the livers of WT and Agr2/Tg mice treated with hyperCho and lovastatin and/or allicin. Quantification for Western blotting was performed by normalizing proteins to β-actin. Representative figures were generated with data from at least three independent experiments. The data were presented as the mean ± SD values. *P < 0.05; **P < 0.01; ***P < 0.001 by Student’s t test.
Discussion
In the present study, we discovered a novel role of AGR2 in mediating resistance to the lipid-lowering agent lovastatin in the management of hypercholesterolemia. This effect of AGR2 was ascribed to its activity in the regulation of AKT/SREBP2, an important signaling axis that controls the cholesterol biogenesis MVA pathway. We also uncovered a combination approach, lovastatin plus allicin, is more effective than a statin-only treatment of AGR2-high hypercholesterolemia. Statins act as inhibitors of HMGCR, a rate-limited enzyme in controlling the MVA pathway, leading to inhibited cholesterol synthesis. Guideline committees and experts almost uniformly recommend statins as a first-line treatment for reducing cardiovascular events by lowering cholesterol levels [28].
However, questions have been raised with respect to some individuals whose LDL-C levels remained above certain thresholds after treatment [5, 7, 9]. Other patients exhibit intolerance and/or statin-associated side effects, presenting with muscle-related symptoms, liver injury, with a risk of new-onset diabetes and recurrent ischemic events [24, 25]. There are many genetic mechanisms and factors that are associated with statin efficiency and resistance and intolerance. Since uptake of statins in the liver largely depends on transmembrane organic anion transporting protein 1-B1 (OATP1B1), encoded by a solute carrier organic anion transporter family member 1B1 (SLCO1-B1) gene, variants of this gene, such as the c.521T>C variant, lead to decreased transport activity for hepatic uptake of statins [29, 30]. Pathogenic mutations in the hepatic LDL receptor, ApoB and PCSK9, cause deficient clearance of circulating LDL-C via the LDL receptor, leading to decreased statin efficacy [31,32,33]. Statin-mediated activation of PCSK9 and SREBP2, which in turn limits their efficacy in cholesterol-lowering treatments, has been noticed to be a mechanism conferring statin resistance [34, 35]. Our results first place AGR2 in a way that is related to lipid-lowering management. AGR2 is involved in peptide maturation and protein folding in the ER, can be secreted and function as a promotor and/or predictor for tumor aggressiveness. Like other PDI family members, a catalytic cysteine (C81) homologous to the (C/SXXC/S) motif found in PDI proteins is essential for AGR2-mediated functions [11, 12]. Additionally, the monomeric and dimeric forms of AGR2 and localization appear to be important in regulating the activities of this protein [26, 36]. In this study, we revealed a role of AGR2 in participating in the development of statin resistance. Although AGR2-mediated activation on AKT/mTOR/SREBP2 axis was demonstrated to contribute to its function against statin efficacy, the detailed mechanism underlying the regulation of AGR2 on cholesterol synthesis, particularly for HMGCR and/or others remained unclear. We found that CPHS motif was required for AGR2-promoted cholesterol synthesis, which subsequently leading to statin resistance. Whether localization and/or dimeric form of AGR2 play a role in the regulation of lipid metabolism is required for further investigations. As the efficiency and resistance in lipid management, investigations have focused on the development of alternative therapeutic strategies to improve treatment efficiency and identify biomarkers to tailor population stratification [37, 38]. Several clinical studies have demonstrated that combination therapies of statins with cholesterol absorption inhibitors, bile acid sequestrants, and PCSK9 inhibitors appear to be more effective in some patients [39]. For example, combination therapy with ezetimibe and simvastatin lowers LDL-C levels more potently than those achieved with simvastatin monotherapies, and the safety of the combination therapy displays similarity to that of low-dose statin monotherapies in the Chinese population [40].
Given the advantage of combination therapy in lipid management, we are prompted to find a naturally occurring compound that possesses the ability to alleviate AGR2-mediated resistance for use in combination with statin therapy. Considering the association of statin/allicin and AGR2 in cholesterol metabolism, we tried to figure out if lovastatin and allicin affect AGR2 expression and functions. The results showed that both agents had limited influence on AGR2 expression and secretion, but allicin reduced AGR2-promoted cholesterol synthesis. A chemical docking analysis indicates that lovastatin may be unable to bind to AGR2 (data not shown). Like some nutraceuticals have been recommended as an alternative or add-on therapy to statins, the action mechanisms are still insufficient available [41], the molecular mechanisms by which allicin affects AGR2-mediated drug resistance remain to be elucidated.
It is believed that hyperlipidemia-specific biomarkers would improve lipid therapeutic treatment on the basis of patient stratification, drug selection, and dose use. As a secrete protein, high levels of serum AGR2 is ascribed to the elevated intracellular expression under stress conditions [26, 42]. Our study provides evidence that AGR2 might be a potential predictor for the selection of a subgroup patients during lipid-lowering therapy, and supplementation with garlic may be helpful for improving the efficiency of statins in patients with a high level of AGR2 in blood. Whether AGR2 acts as a predictor in lipid-lowering therapy needs further investigations in a large-scale population study.
Hepatotoxicity associated with statins occurs in some patients, but not all, as evidenced by an increase in transaminase [23, 24]. We noticed that increases in the serum ALT and AST, more potent in ALT, were obvious in wild-type mice, rather than in Agr2/Tg mice, treated with hypercholesterolemia model food or lovastatin. Although AGR2 has a negative effect in statin therapy, it tends to act as a protector against statin- and/or hyperCho-induced liver injury. It has been reported that AGR2 exerts a regulatory effect on inflammation, oxidative stress, stem cell proliferation and differentiation, and maintains ER homeostasis [26], which might contribute to AGR2-mediated protection in liver function. Hence, AGR2 plays double-side functions in statin therapy and drug-induced hepatotoxic effects. Statins are usually avoided to be used in patients with liver failure, acute liver injury, and cirrhosis. The protective effect of AGR2 might increase the safety of statin in some patients. Clinical studies need to investigate whether combined statins and allicin would offer an effective option for lipid control and for reducing side events induced by drugs.
In summary, the present study reveals that AGR2 may emerge as a specific biomarker for lipid-lowering drug selection and patient stratification because it facilitates cholesterol synthesis yielding statin resistance. The combination of allicin with lovastatin may pave a way for improving statin efficiency in dyslipidemia with high AGR2 expression.
References
Espinosa G, Lopez-Montero I, Monroy F, Langevin D. Shear rheology of lipid monolayers and insights on membrane fluidity. Proc Natl Acad Sci USA. 2011;108:6008–13.
Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–57.
Sozen E, Ozer NK. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: an updated mini-review. Redox Biol. 2017;12:456–61.
Chua NK, Coates HW, Brown AJ. Cholesterol, cancer, and rebooting a treatment for athlete’s foot. Sci Transl Med. 2018;10:eaat3741.
Adhyaru BB, Jacobson TA. Safety and efficacy of statin therapy. Nat Rev Cardiol. 2018;15:757–69.
Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol. 2019;16:155–65.
Gitt AK, Drexel H, Feely J, Ferrieres J, Gonzalez-Juanatey JR, Thomsen KK, et al. Persistent lipid abnormalities in statin-treated patients and predictors of LDL-cholesterol goal achievement in clinical practice in Europe and Canada. Eur J Prev Cardiol. 2012;19:221–30.
Newman CB, Preiss D, Tobert JA, Jacobson TA, Page RL 2nd, Goldstein LB, et al. Statin safety and associated adverse events: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2019;39:e38–81.
Jones PH, Nair R, Thakker KM. Prevalence of dyslipidemia and lipid goal attainment in statin-treated subjects from 3 data sources: a retrospective analysis. J Am Heart Assoc. 2012;1:e001800.
Persson S, Rosenquist M, Knoblach B, Khosravi-Far R, Sommarin M, Michalak M. Diversity of the protein disulfide isomerase family: identification of breast tumor induced Hag2 and Hag3 as novel members of the protein family. Mol Phylogenet Evol. 2005;36:734–40.
Higa A, Mulot A, Delom F, Bouchecareilh M, Nguyen DT, Boismenu D, et al. Role of pro-oncogenic protein disulfide isomerase (PDI) family member anterior gradient 2 (AGR2) in the control of endoplasmic reticulum homeostasis. J Biol Chem. 2011;286:44855–68.
Brychtova V, Mohtar A, Vojtesek B, Hupp TR. Mechanisms of anterior gradient-2 regulation and function in cancer. Semin Cancer Biol. 2015;33:16–24.
Fessart D, Domblides C, Avril T, Eriksson LA, Begueret H, Pineau R, et al. Secretion of protein disulphide isomerase AGR2 confers tumorigenic properties. Elife. 2016;5:e13887.
Jia M, Guo Y, Zhu D, Zhang N, Li L, Jiang J, et al. Pro-metastatic activity of AGR2 interrupts angiogenesis target bevacizumab efficiency via direct interaction with VEGFA and activation of NF-kappaB pathway. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1622–33.
Wang Y, Jia M, Liang C, Sheng N, Wang X, Wang F, et al. Anterior gradient 2 increases long-chain fatty acid uptake via stabilizing FABP1 and facilitates lipid accumulation. Int J Biol Sci. 2021;17:834–47.
Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, et al. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci USA. 2009;106:6950–5.
Wang D, Xu Q, Yuan Q, Jia M, Niu H, Liu X, et al. Proteasome inhibition boosts autophagic degradation of ubiquitinated-AGR2 and enhances the antitumor efficiency of bevacizumab. Oncogene. 2019;38:3458–74.
Eid W, Dauner K, Courtney KC, Gagnon A, Parks RJ, Sorisky A, et al. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc Natl Acad Sci USA. 2017;114:7999–8004.
Johnson C, Kastelic J, Thundathil J. Role of Akt and mammalian target of rapamycin signalling in insulin-like growth factor 1-mediated cell proliferation in porcine Sertoli cells. Reprod Fertil Dev. 2020;32:929–40.
Chaudhary HR, Brocks DR. The single dose poloxamer 407 model of hyperlipidemia; systemic effects on lipids assessed using pharmacokinetic methods, and its effects on adipokines. J Pharmacol Pharm Sci. 2013;16:65–73.
Zhang Q, Qian ZY, Zhou PH, Zhou XL, Zhang DL, He N, et al. Effects of oral selenium and magnesium co-supplementation on lipid metabolism, antioxidative status, histopathological lesions, and related gene expression in rats fed a high-fat diet. Lipids Health Dis. 2018;17:165.
Busch CJ, Hendrikx T, Weismann D, Jackel S, Walenbergh SM, Rendeiro AF, et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology. 2017;65:1181–95.
Meurer L, Cohen SM. Drug-induced liver injury from statins. Clin Liver Dis. 2020;24:107–19.
Bjornsson ES. Hepatotoxicity of statins and other lipid-lowering agents. Liver Int. 2017;37:173–8.
Liu A, Wu Q, Guo J, Ares I, Rodriguez JL, Martinez-Larranaga MR, et al. Statins: adverse reactions, oxidative stress and metabolic interactions. Pharmacol Ther. 2019;195:54–84.
Maurel M, Obacz J, Avril T, Ding YP, Papadodima O, Treton X, et al. Control of anterior GRadient 2 (AGR2) dimerization links endoplasmic reticulum proteostasis to inflammation. EMBO Mol Med. 2019;11:e10120.
Rana SV, Pal R, Vaiphei K, Sharma SK, Ola RP. Garlic in health and disease. Nutr Res Rev. 2011;24:60–71.
Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–88.
Thompson JF, Man M, Johnson KJ, Wood LS, Lira ME, Lloyd DB, et al. An association study of 43 SNPs in 16 candidate genes with atorvastatin response. Pharmacogenomics J. 2005;5:352–8.
Du Y, Wang S, Chen Z, Sun S, Zhao Z, Li X. Association of SLCO1B1 polymorphisms and atorvastatin safety and efficacy: a meta-analysis. Curr Pharm Des. 2018;24:4044–50.
Ten Kate GJ, Neefjes LA, Dedic A, Nieman K, Langendonk JG, Galema-Boers AJ, et al. The effect of LDLR-negative genotype on CT coronary atherosclerosis in asymptomatic statin treated patients with heterozygous familial hypercholesterolemia. Atherosclerosis. 2013;227:334–41.
Ahangari N, Ghayour Mobarhan M, Sahebkar A, Pasdar A. Molecular aspects of hypercholesterolemia treatment: current perspectives and hopes. Ann Med. 2018;50:303–11.
Tiwari V, Khokhar M. Mechanism of action of anti-hypercholesterolemia drugs and their resistance. Eur J Pharmacol. 2014;741:156–70.
Ma K, Malhotra P, Soni V, Hedroug O, Annaba F, Dudeja A, et al. Overactivation of intestinal SREBP2 in mice increases serum cholesterol. PLoS ONE. 2014;9:e84221.
Kim HJ, Lee J, Chung MY, Hong S, Park JH, Lee SH, et al. Piceatannol reduces resistance to statins in hypercholesterolemia by reducing PCSK9 expression through p300 acetyltransferase inhibition. Pharmacol Res. 2020;161:105205.
Tiemann K, Garri C, Lee SB, Malihi PD, Park M, Alvarez RM, et al. Loss of ER retention motif of AGR2 can impact mTORC signaling and promote cancer metastasis. Oncogene. 2019;38:3003–18.
Benincasa G, de Candia P, Costa D, Faenza M, Mansueto G, Ambrosio G, et al. Network medicine approach in prevention and personalized treatment of dyslipidemias. Lipids. 2021;56:259–68.
Su X, Cheng Y, Chang D. Lipid-lowering therapy: guidelines to precision medicine. Clin Chim Acta. 2021;514:66–73.
Crismaru I, Pantea Stoian A, Bratu OG, Gaman MA, Stanescu AMA, Bacalbasa N, et al. Low-density lipoprotein cholesterol lowering treatment: the current approach. Lipids Health Dis. 2020;19:85.
Han SN, Yang WH, Yin JJ, Tao HL, Zhang LR. Drug treatment of hyperlipidemia in Chinese patients: focus on the use of simvastatin and ezetimibe alone and in combination. Am J Cardiovasc Drugs. 2019;19:237–47.
Banach M, Patti AM, Giglio RV, Cicero AFG, Atanasov AG, Bajraktari G, et al. The role of nutraceuticals in statin intolerant patients. J Am Coll Cardiol. 2018;72:96–118.
Gupta A, Dong A, Lowe AW. AGR2 gene function requires a unique endoplasmic reticulum localization motif. J Biol Chem. 2012;287:4773–82.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81872896, 82173839 and 32000891), and Natural Science Foundation of Shandong Province (ZR2020QH363).
Author information
Authors and Affiliations
Contributions
NS and YQW performed the experiments, analyzed and interpreted the results, and wrote the paper. CFW collected clinical samples. MQJ and HMN analyzed and interpreted the results. QQL, YNW and XXZ assisted in animal experiments. DF was involved in data analysis. HQY designed the research, analyzed and interpreted the results, and wrote the paper. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Sheng, N., Wang, Yq., Wang, Cf. et al. AGR2-induced cholesterol synthesis drives lovastatin resistance that is overcome by combination therapy with allicin. Acta Pharmacol Sin 43, 2905–2916 (2022). https://doi.org/10.1038/s41401-022-00909-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00909-3
