
Abstract
Oxidative stress is extensively involved in neurodegeneration. Clinical evidence shows that keeping the mind active through mentally-stimulating physical activities can effectively slow down the progression of neurodegeneration. With increased physical activities, more neurotransmitters would be released in the brain. In the present study, we investigated whether some of the released neurotransmitters might have a beneficial effect against oxidative neurodegeneration in vitro. Glutamate-induced, glutathione depletion-associated oxidative cytotoxicity in HT22 mouse hippocampal neuronal cells was used as an experimental model. We showed that norepinephrine (NE, 50 µM) or dopamine (DA, 50 µM) exerted potent protective effect against glutamate-induced cytotoxicity, but this effect was not observed when other neurotransmitters such as histamine, γ-aminobutyric acid, serotonin, glycine and acetylcholine were tested. In glutamate-treated HT22 cells, both NE and DA significantly suppressed glutathione depletion-associated mitochondrial dysfunction including mitochondrial superoxide accumulation, ATP depletion and mitochondrial AIF release. Moreover, both NE and DA inhibited glutathione depletion-associated MAPKs activation, p53 phosphorylation and GADD45α activation. Molecular docking analysis revealed that NE and DA could bind to protein disulfide isomerase (PDI). In biochemical enzymatic assay in vitro, NE and DA dose-dependently inhibited the reductive activity of PDI. We further revealed that the protective effect of NE and DA against glutamate-induced oxidative cytotoxicity was mediated through inhibition of PDI-catalyzed dimerization of the neuronal nitric oxide synthase. Collectively, the results of this study suggest that NE and DA may have a protective effect against oxidative neurodegeneration through inhibition of protein disulfide isomerase and the subsequent activation of the MAPKs‒p53‒GADD45α oxidative cascade.
Similar content being viewed by others


Introduction
Neuronal oxidative stress plays an important role in the long pathogenic process of neurodegeneration [1,2,3,4]. Glutamate, an excitatory neurotransmitter in the central nervous system (CNS), can induce neurotoxicity when it is present at high concentrations. This is believed to be a contributing factor in some neurodegenerative diseases (e.g., Alzheimer’s disease), occurring before the onset of significant clinical pathology and symptoms [5,6,7]. Earlier studies showed that glutamate induces oxidative cytotoxicity mostly through two different pathways, i.e., the receptor-mediated excitotoxicity and the non-receptor-mediated oxidative cytotoxicity [5, 8, 9]. In the former case, glutamate activates ionotropic glutamate receptor, subsequently resulting in transient Ca2+ flux, increased levels of reactive oxygen species (ROS), and ultimately cell death. In the latter case, glutamate inhibits the glutamate-cysteine antiporter, subsequently resulting in increased cysteine efflux. This leads to depletion of the intracellular glutathione (GSH) and ultimately oxidative cytotoxicity [10,11,12]. Glutamate-induced GSH depletion and oxidative cytotoxicity have been observed in different systems, including neuronal cell lines [10, 13], primary neuronal cultures [14], and oligodendrocytes [15].
Memory and cognitive function will decline during normal aging. Interestingly, clinical studies have shown that keeping the mind active through physical activities can help slow down this neurodegenerative process by improving working memory and brain health [16,17,18]. Moreover, it has also been shown that mentally-stimulating physical activities can improve the cognitive performance in children and adolescents with attention deficit hyperactivity disorder [19]. However, the mechanism by which these mentally-stimulating physical activities improve brain health or slow down the progression of neurodegeneration is not fully understood at present. During heightened physical activities, many neurotransmitters are released in the CNS, including the catecholamine neurotransmitters norepinephrine (NE) and dopamine (DA) [20, 21]. However, it is not known whether some of the released neurotransmitters may have a protective effect against oxidative neuronal death and thereby enhance neuronal survival. This intriguing possibility is tested in the present study by using immortalized HT22 mouse hippocampal neuronal cells as an in vitro model. The HT22 cells lack the glutamate receptor, and this cell line has been widely used as a model for selectively studying GSH depletion-associated oxidative cytotoxicity [12, 22,23,24]. Recent studies have shown that the glutamate-induced GSH depletion and subsequent oxidative cytotoxicity in HT22 cells has many of the characteristics of ferroptosis [25]. In addition, our earlier study showed that treatment of HT22 cells with glutamate results in ROS accumulation, JNK activation, p53 translocation into the nucleus, and subsequently, up-regulation of the transcription of the p53-response gene GADD45α [24]. Similar hippocampal GADD45α expression was also observed in vivo in the kainic acid-induced neurodegeneration and memory impairment rat model [24].
Protein disulfide isomerase (PDI) is an important member of the thioredoxin superfamily, and plays a critical role in catalyzing the oxidation and isomerization of disulfide bonds and facilitating nascent protein folding [26,27,28]. While PDI primarily resides in endoplasmic reticulum, it was also found to be present in cytosol [29,30,31], mitochondria [32] and cell surface [33]. Our earlier studies have shown that PDI can bind with several small molecules, such as endogenous estrogens [34, 35], which serve as effective inhibitors of PDI’s catalytic function. In addition, we have shown that cystamine, a small-molecule inhibitor of PDI’s isomerase activity [36], can effectively prevent GSH depletion-associated accumulation of NO and O2– as well as the oxidative cytotoxicity in cultured HT22 cells [37]. Furthermore, we have recently shown that some of the endogenously-formed estrogen derivatives (such as 4-hdyroxyestrone) have a powerful neuroprotective effect both in vitro and in vivo against oxidative cell death induced by glutamate or kainic acid, respectively [38].
In the present study, by using the glutamate-induced, GSH depletion-associated oxidative cytotoxicity in cultured HT22 cells as an in vitro model, we sought to test a hypothesis whether certain endogenous neurotransmitters and their metabolites might have a beneficial effect against oxidative neuronal death. We found that NE and DA, two well-known catecholamine neurotransmitters, exert a strong protective effect against GSH depletion-associated oxidative stress and cytotoxicity, but this neuroprotective effect is not shared by several other neurotransmitters. Following this interesting observation, we also investigated the mechanism underlying the neuroprotective effect of NE and DA with a focus on their regulation of mitochondrial dysfunction (including superoxide accumulation, ATP depletion and AIF release) and the MAPKs‒p53‒GADD45α oxidative cascade. To probe the direct target that mediates the neuroprotective actions of NE and DA, we examined the ability of these two neurotransmitters to bind to PDI protein and alter its catalytic functions as our earlier study showed that PDI plays a critical role in mediating glutamate-induced oxidative toxicity in HT22 cells.
Materials and methods
Chemicals and reagents
Glutamate (catalog No. 49621), epinephrine (catalog No. E4250), DA (catalog No. H8502), NE (catalog No. A7257), levodopa (catalog No. D9628), histamine (catalog No. H7250), GABA (catalog No. A2129), serotonin (catalog No. 14927), choline (catalog No. A2129), and inhibitors PD98059 (catalog No. P215), SP600125 (catalog No. S5567), and SB202190 (catalog No. S7067) were all purchased from Sigma-Aldrich (St. Louis, MO, USA). 2’,7’-Dichlorodihydrofluorescein diacetate (H2DCFDA) was purchased from Fluka (Carlsbad, CA, USA). The siRNA for mouse PDI was a commercial product containing three mixed siRNAs (sequence information not provided by the manufacturer), which were designed and prepared by RiboBio (Guangzhou, China) according to the mouse P4hb gene (ID18453); the control siRNA was also provided by the same company. Most of the other chemicals, unless specified, were also obtained from Sigma-Aldrich.
Cell culture
The HT22 cells, an immortalized mouse hippocampal neuronal cell line, were a generous gift from Prof. Dave Schubert at the Salk Institute, San Diego, CA, USA. The cells were grown in the DMEM medium (Sigma-Aldrich) containing fetal bovine serum (FBS, 10%), glucose (4.5 g/L), L-glutamine, sodium bicarbonate, and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin) as described earlier [24, 39]. The cells were maintained at 37 °C in 5% CO2 atmosphere, and usually sub-cultured once every 2 to 3 days.
Cytotoxicity assay
Cell viability was assayed by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. The HT22 cells were first seeded in 96-well plates at a density of 5000 cells/well, and 24 h after seeding, cells were treated with glutamate in the presence of different concentrations of neurotransmitters or their metabolic derivatives. Twenty-four hours later, MTT assay was performed by incubating cells with 10 μL MTT solution (at a final concentration of 500 µg/mL) for 1 h, and then the formazon formed was dissolved in 100 μL dimethyl sulfoxide (DMSO). Absorbance at 560 nm was measured using a microplate reader (Molecular Device, Palo Alto, CA, USA). The relative cell viability was expressed as a percentage of the control cells that were not treated with glutamate.
Analysis of DNA fragmentation by agarose gel electrophoresis
The HT22 cells were plated in 10 cm dishes at 1 × 106 cells per dish in complete medium and incubated for 24 h. NE and DA were added to cultured cells in the absence or presence of 5 mM glutamate and further incubated for additional 16 h. Cells were then harvested using a scraper. Total DNA was extracted with 500 μL of lysis buffer (containing 10 mM Tris-HCl, 10 mM EDTA and 0.5% Triton X-l00, pH 7.4) and incubated for 15 min at 4 °C. The lysates were centrifuged at 12,000 × g for 20 min, treated for 1 h with 0.1 mg/mL RNaseA at 37 °C, and followed by incubation for 2 h at 50 °C with 200 μg/mL proteinase K. The isolated DNA was precipitated with ethanol, and the pellet was analyzed by electrophoresis on a 1.5% agarose gel containing ethidium bromide.
Determination of intracellular ROS levels
The HT22 cells were plated in 24-well plates at a density of 25,000 cells per well. To determine intracellular ROS level, cells were washed once with phosphate-buffered saline (PBS) and incubated with H2DCFDA (1 μM in PBS, pH 7.4). Cells were further incubated for 10 min at 37 °C in the dark. The H2DCFDA fluorescence intensity was measured using a Sinergy-2 fluorescent plate reader at wavelengths of 485 and 538 nm for excitation and emission, respectively, under constant conditions to allow quantitative comparison of the relative fluorescence intensity in cells with different treatments. Fluorescent cell images were captured simultaneously with a fluorescent inverted microscope equipped with a digital camera (Carl Zeiss Corporation, Germany).
siRNA transfection
The HT22 cells were seeded in 96-well plates at a reduced density of 500 cells/well. After 24 h culture, cells were transfected with three mixed siRNAs for PDI with lipofectamine RNA iMAX. Twenty-four hours following PDI knockdown, cells were treated with respective drugs for 24 h, and then processed for MTT assay of cell viability. For immunoblot analysis of PDI protein levels in knockdown cells, cells were cultured in 6-well plates, while other experimental procedures were the same as described above for the MTT assay.
Flow cytometric analysis
The HT22 cells were treated with catecholamines or other neurotransmitters for the indicated length of time (usually 6 or 12 h) in the presence or absence of 5 mM glutamate. Cells were harvested using a scraper, and then washed twice with PBS. Flow cytometric analysis was performed to analyze cell cycles, exposed phosphatidylserine (PS), and mitochondrial membrane potential (MMP). For cell cycle analysis, cells were re-suspended in 0.9% NaCl (1 mL), and then ice-cold 90% ethanol (2.5 mL) was added. After incubation at room temperatures for 30 min, cells were undergone brief centrifugation to form a pellet. The cell pellet was re-suspended in 1 mL of propidium iodide (PI) solution (50 μg/mL in PBS) and RNase A (100 μg/mL), followed by incubation at 37 °C for 30 min. For MMP analysis, cells were re-suspended in 1 mL DiOC6 (50 nM in culture medium), and then incubated for 15 min at 37 °C. After centrifugation, the pellets were re-suspended in PBS. For Annexin V/PI double staining, the Annexin V-FITC apoptosis detection kit (BD Biosciences, San Jose, CA, USA) was used according to the manufacturer’s instructions. Briefly, cells (1 × 105) were re-suspended in 100 μL of the binding buffer (1×), and 5 μL of annexin V-FITC and PI were added to the suspended cells, and followed by incubation for 15 min at room temperature. The binding buffer was then added to the reaction mixture. The samples were analyzed using a flow cytometer (model BD LSRII, BD Bioscience, San Jose, CA, USA).
Western blotting
For Western blotting, cells were washed first, and then suspended in a lysis buffer (pH 7.4), containing 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mM sodium orthovanadate, 1 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 μM aprotinin, 1 μM leupeptin and 1 μM pepstatin A. The concentration of proteins was determined by the Bio-Rad protein assay (catalog No. 5000005; Bio-Rad, Hercules, CA, USA). Proteins (20‒30 μg) were separated using 8% or 10% SDS-polyacrylamide gel electrophoresis (PAGE), and then electrically transferred to a polyvinylidene difluoride (PVDF, 0.45 µm) membrane (Bio-Rad). The membrane was incubated in the blocking buffer (5% skim milk in the TBS-T solution, namely, Tris-buffered saline containing 0.1% Tween-20) for 1 h at room temperature, and then the membrane was incubated with different primary antibodies. All primary antibodies, including the antibodies against AIF (catalog No. 4642; MW 67 kDa), histone H3 (catalog No. 4499; MW 17 kDa), GAPDH (catalog No. 5174; MW 37 kDa), p-ERK (catalog No. 4370; MW 44 and 42 kDa), ERK (catalog No. 4695; MW 44 and42 kDa), JNK (catalog No. 9252; MW 54 and 46 kDa), p-p38 (catalog No. 4511; MW 43 kDa), p38 (catalog No. 8690; MW 40 kDa), p-p53 (catalog No. 12571; MW 53 kDa), PARP-1 (catalog No. 9532; MW 116 and 89 kDa) and caspase-3 (catalog No. 14220; MW 35, 19 and 17 kDa), were purchased from Cell Signaling Technology (Beverly, MA, USA), and the antibodies against phosphorylated JNK (catalog No. 44-682; MW 54 and 46 kDa) were purchased from Biosource (Camarillo, CA, USA). These primary antibodies were used at a concentration of 1:1000 dilutions according to the suppliers. After the membrane was washed with the TBS-T solution, it was further incubated with the horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-rabbit IgG, catalog No. 7074; ant-mouse IgG, catalog No. 7076) at a concentration of 1:1000 and 1:3000, respectively. The signal was detected using enhanced chemiluminescence (ECL) plus detection system (GE Healthcare, Piscataway, NJ, USA).
Molecular docking analysis
The binding interaction of NE and DA with PDI protein was predicted using the protein-ligand docking method. The human PDI structure (PDB id: 6I7S) was processed with the Protein Preparation Wizard in the Schrödinger Suite (Maestro 11.9, 2019; Schrödinger LLC, New York, NY, USA). Hydrogen atoms were added and then adjusted for bond orders. The protonation and tautomeric states for residues were adjusted according to a theoretical pH at 7.0. Missing residues and loop segments close to the active site were added back using the Prime (Prime 2.1, 2019; Schrödinger LLC). Water molecules were deleted. Proteins were subjected to geometry optimization using the OPLS3e force field.
The docking method Glide-XP (extra precision) in Schrodinger Glide software was used with default docking parameters. The docking grids for protein structures were generated using Maestro. The grid box was centered at the binding site in the crystal structures, and the grid box was set at 20 Å × 20 Å × 20 Å. Glide-XP docking uses hierarchical filters to find the best ligand binding poses in the defined grid space for a given target protein. The filters include the positional, conformational and orientational sampling of the ligand. Afterwards, the lowest energy poses were subjected to the Monte Carlo (MC) procedure that sampled the nearby torsional minima. The best poses for the given ligand were determined according to the GlideScore.
Purification of PDI
The cDNA for human PDI protein (NM_000918.4) was cloned into the pET-28a vector at the Hind III/BamH I restriction sites. PDI was expressed in BL-21 (DE3) plysS Escherichia coli host cells following induction with 0.5 mM IPTG at 22 °C for 8 h. Cell pellets were dissolved in 0.1 M Tris-HCl (containing 0.15 M NaCl, 2% Triton X-100, 2 mM DTT, and 1 mM PMSF, pH 8.0), and sonicated for 20 min (on 4 s/off 8 s in each cycle). Then the cell lysates were centrifuged at 18,000 × g for 20 min at 4 °C. The supernatant was incubated with Ni-NTA agarose (QIAGEN) at 4 °C for 1 h, and the unbound proteins were removed by washing three times with 0.1 M Tris-HCl (containing 0.15 M NaCl and 20 mM imidazole, pH 8.0). After elution with 0.2 M imidazole–Tris-HCl (containing 0.15 M NaCl, pH 8.0), fractions containing the target protein (examined by SDS-PAGE analysis) were combined, and concentrated with an Amicon Ultra-4 tube. Then the proteins were further separated on a ÄKTA FPLC system using the Superdex 200 10/ 300 GL affinity chromatography, and collected protein fraction was stored at −80 °C. Protein concentration was determined by Bio-Rad Protein Assay using BSA as standard.
Inhibition of PDI’s reductase activity by NE and DA
To determine whether NE and DA can directly bind to PDI protein, we chose to determine the ability of NE and DA to directly inhibit PDI’s reductase activity in an in vitro assay. The reductase activity of PDI was measured by recording the PDI-induced aggregation of the insulin B chain as described in our earlier study [40]. Insulin (125 μM) was reduced by 5 mM DTT in a 96-well plate in 10 mM sodium phosphate buffer (pH 7.4), in the absence or presence of PDI (at 3.5 μM). The aggregation was monitored at 37 °C using a Synergy plate reader (BioTek), with wavelength (λ) set at 650 nm.
Statistical analysis
The quantitative data were expressed as means ± SD. Comparisons between two groups were analyzed using one-way ANOVA. Individual differences among groups were analyzed using the Dunnett’s test (SPSS 11.5 software). P < 0.05 is considered statistically significant.
Results
NE and DA protect against glutamate-induced oxidative cytotoxicity
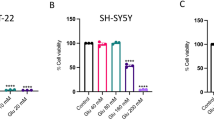
Earlier studies showed that glutamate could induce GSH depletion, which subsequently resulted in oxidative cytotoxicity in cultured HT22 neuronal cells [9, 23, 24, 41]. The concentration-dependent cytotoxic effect of glutamate on cell viability in this in vitro neuronal cell model is shown in Fig. 1a. Using this in vitro model, we tested the protective effect of several CNS neurotransmitters and their metabolites. As shown in Fig. 1b, NE and DA exerted concentration-dependent protection against glutamate-induced oxidative cytotoxicity, but 3-methyl-NE and 3-methyl-DA (the respective endogenous O-methylated metabolites of NE and DA) did not have any protective effect. Epinephrine, an endogenous neurohormone which shares a very similar structure with NE and DA, also did not have a protective effect, and so was 3-methyl-epinephrine, an endogenous O-methylated metabolite of epinephrine (Fig. 1b). Levodopa (DOPA), an endogenous precursor in the biosynthesis of NE and DA in the brain, showed a protective effect against glutamate-induced cytotoxicity, but its potency was weaker than NE and DA, but 3-methyl-levodopa did not have a similar protective effect (Fig. 1c).

a Cell viability change (based on MTT assay) after the cells were treated for 24 h with increasing concentrations of glutamate. Each data point was the mean ± S.D. (n = 6). b The protective effect of NE, DA, epinephrine (EPI) and their respective 3-methyl derivatives against glutamate-induced cytotoxicity. c The protective effect of L-DOPA and its 3-methyl derivative against glutamate-induced cytotoxicity. The glutamate treatment (at 5 mM) lasted 24 h. Each value is a mean ± S.D. (n = 6). The inset in each panel depicts the chemical structure of the respective chemical tested.
For comparison, we have tested the neuroprotective effect of several other non-catecholic neurotransmitters, including histamine, γ-aminobutyric acid (GABA), serotonin, glycine, and acetylcholine (some of the data are summarized in Fig. 2). Except for serotonin, which had a weak protective effect, the other neurotransmitters did not show any meaningful protection.

The cells were jointly treated for 24 h with 5 mM glutamate alone or in combination with each of the neurotransmitters as shown at three different concentrations (10, 25, and 50 µM). The control group was exposed to vehicle only. Cell viability was determined by the MTT assay. Each data point was the mean ± S.D. (n = 6). The inset in each panel depicts the chemical structure of the respective chemical tested.
Next, we conducted additional experiments using different methods to further study the protective effect of NE and DA against glutamate-induced neuronal death. Using PI staining, we found that glutamate significantly increased sub-G1 cell population compared to control, and NE and DA strongly reduced glutamate-induced increase in the sub-G1 cell population (from 52.7% to 10.9% and 1.0%, respectively) (Fig. 3a). Similarly, glutamate induced the populations of annexin V positive cells compared to control, and NE and DA significantly reduced the populations of glutamate-induced annexin V positive cells, which include either single-positive for annexin V or double-positive for both annexin V and PI cells (Fig. 3b). In addition, glutamate alone caused significant reduction in nuclear size yet without causing significant change in chromatin distribution, and joint treatment with NE or DA restored normal nuclear morphology (Fig. 3c).

a Flow cytometric analysis of PI-stained cells following their treatment with 5 mM glutamate alone or in combination with NE (50 µM) or DA (50 µM) for 24 h. b Flow cytometric analysis of cells that were double-stained with annexin V-FITC and PI. The cells were treated with 5 mM glutamate alone or jointly with NE (50 µM) or DA (50 µM) for 24 h before analysis. For comparison, cells that were treated with vehicle, NE or DA alone were also included. c Morphological analysis of the nuclei of cells (stained with Hoechst-33342) after they were treated with 5 mM glutamate alone or in combination with NE (50 µM) or DA (50 µM) for 24 h. All the experiments shown here were repeated multiple times, and only one set of representative data is presented.
NE and DA inhibit ROS accumulation and restore glutamate-induced mitochondrial dysfunction
To probe the mechanism by which NE and DA exerted their neuroprotection in HT22 cells, we chose to determine their effect on glutamate-induced intracellular ROS accumulation. ROS accumulation in HT22 cells was elevated at 8 h after glutamate treatment, but joint treatment with NE or DA abrogated glutamate-induced ROS accumulation (Fig. 4a). The number of H2DCFDA-positive cells (quantified by flow cytometry) was significantly reduced by joint treatment with glutamate plus NE or DA (Fig. 4b).

a The cells were treated with glutamate (5 mM) alone or in combination with NE (50 µM) or DA (50 µM) for 8 h. The ROS levels were determined using a fluorescence microscope (original magnification, ×200) with H2DCFDA staining. Data shown was a representative set from three experiments. b The relative fluorescence intensity was presented as fold of the control group (mean ± S.D. of triplicate determinations). *P < 0.05 compared with cells treated with glutamate alone. c MitoSOX Red staining of cells after treatment with glutamate (5 mM) alone or in combination with N-acetylcysteine (NAC, 500 µM), NE (50 µM) or DA (50 µM) for 12 h. Digital images were captured using a fluorescence microscope (original magnification, ×200). Data shown was a representative set from three experiments. d Flow cytometric analysis of mitochondrial membrane potential change. The cells were treated with glutamate (5 mM) alone or in combination with N-acetylcysteine (NAC 500 µM), NE (50 µM) or DA (50 µM) for 12 h before the analysis. e Changes in intracellular ATP levels in different treatment groups (as d) at 12 h. Each value is the mean ± S.D. (n = 6). *P < 0.05 compared to cells treated with glutamate alone.
In addition, we examined the effect of NE and DA on glutamate-induced mitochondrial superoxide accumulation. Mitochondrial superoxide accumulation was increased following glutamate treatment, and joint treatment with NE or DA strongly reduced glutamate-induced accumulation of mitochondrial superoxide (Fig. 4c). Notably, accumulation of mitochondrial superoxide was abrogated by joint treatment with N-acetylcysteine (NAC), an antioxidant (Fig. 4c). To probe the extent of oxidative mitochondrial damage, we determined the change in mitochondrial membrane potential (MMP) using flow cytometric analysis with the aid of a chemical probe DiOC6(3). Treatment with glutamate alone strongly reduced the mitochondrial membrane potential (MMP), whereas joint treatment with NE or DA plus glutamate abrogated glutamate-induced MMP reduction (Fig. 4d).
Next, we sought to determine the intracellular ATP levels in cells treated with glutamate alone or jointly with NE, DA or NAC. As shown in Fig. 4e, intracellular ATP level was decreased at 12 h after glutamate treatment, but joint treatment with NE or DA plus glutamate restored the intracellular ATP level. The protective effect of NE and DA against glutamate-induced ATP depletion was comparable to that of NAC (Fig. 4e).
AIF translocation is abrogated by treatment with NE or DA
Cellular immunostaining of AIF protein showed that AIF was translocated from the cytoplasmic compartment to the nucleus following glutamate treatment (Fig. 5a). Joint treatment with 50 µM NE or DA significantly inhibited AIF nuclear translocation in glutamate-treated cells, but z-VAD-fmk did not have a significant effect on AIF nuclear translocation (Fig. 5a). AIF protein level was increased in the nuclear fraction in a time-dependent manner following glutamate treatment, whereas its level was decreased in cytosolic fraction (Fig. 5b), and this pattern of change was abrogated by joint treatment of cells with NE or DA (Fig. 5c).

a Subcellular localization of the AIF protein. After the cells were treated with glutamate (5 mM) alone or in combination with NE (50 µM), DA (50 µM) or z-VAD-fmk (z-VAD, 20 µM) for 24 h, they were analyzed for immunofluorescence staining of AIF (original magnification, ×200). b, c The cells were treated with 5 mM glutamate for 6, 12, and 24 h (b), or treated with glutamate (5 mM) alone or in combination with NE (50 µM) or DA (50 µM) for 24 h (c). The cytosolic and nuclear fractions were isolated and subjected to Western blotting of AIF. Histone H3 (H3) was used an internal control for the nuclear fraction, and GAPDH was used an internal control for the cytosolic fraction and the whole cell lysate. d Effect of siAIF and z-VAD-fmk (z-VAD) on glutamate-induced AIF change. The cells were transfected with either the negative control siRNAs (siCon) or AIF siRNAs (siAIF), and 24 h later, cells were exposed to 5 mM glutamate for additional 24 h before analysis of the AIF protein level in the whole cell lysate by Western blotting. e Changes in PARP-1, pro-caspase-3 and its fragment(s) in cells treated with 5 mM glutamate alone or in combination with NE (50 µM) or DA (50 µM) for 24 h. f Effect of siAIF on cell viability change (MTT assay). The cell treatment protocol was the same as panel d. Each value is the mean ± SD. (n = 6). *P < 0.05 compared to cells treated with glutamate alone. g Flow cytometric analysis of the effect of z-VAD-fmk (z-VAD) on glutamate-induced cell death. The cells were pretreated alone with 5 mM glutamate or in combination with 20 µM z-VAD for 24 h and then were stained with annexin V-FITC and PI staining. Data shown was a representative set from two experiments.
Next, we employed the siRNAs to determine whether AIF knockdown could effectively suppress glutamate-induced AIF translocation and cell death. As shown in Fig. 5d, the level of AIF protein expression was reduced following transfection with AIF-siRNAs. AIF knockdown afforded a significant protection against glutamate-induced cell death (Fig. 5f). Joint treatment with z-VAD-fmk plus glutamate did not significantly alter AIF protein expression. Moreover, caspase-3 and PARP-1 were not activated during glutamate-induced cell death (Fig. 5e). As expected, pretreatment of cells with z-VAD-fmk did not confer appreciable protection (Fig. 5f) or affect the number of glutamate-induced annexin V-positive cells (Fig. 5g).
NE and DA suppress glutamate-induced activation of MAPKs
As shown in Fig. 6a, after 12-h treatment with glutamate alone, phosphorylation of JNK1, ERK and p38 was significantly increased. When cells were jointly treated with glutamate plus 50 µM NE or DA, activation of these MAPKs was abrogated. Notably, glutamate-induced loss of cell viability was also strongly protected by joint treatment with the pharmacological inhibitors of JNK, ERK, and p38 (Fig. 6b).

a Changes in protein levels of p-JNK, total JNK, p-ERK, total ERK, p-p38 and total p38 in cells treated with 5 mM glutamate alone or in combination with NE (50 µM) or DA (50 µM) for 24 h. b Effect of MAPK inhibitors on cell viability. Cells were pretreated for 1 h with 20 μM of PD98059 (MEK inhibitor), SP600125 (JNK1/2 inhibitor) or SB202190 (p38 inhibitor), and then exposed to 5 mM glutamate for additional 24 h. Cell viability was measured by the MTT assay. Each value is a mean ± SD. (n = 6). *P < 0.05 compared to cells treated with glutamate alone.
NE and DA suppress glutamate-induced activation of the p53-GADD45α signaling cascade
As shown in Fig. 7a, after treatment with glutamate alone, there was a marked increase in p53 phosphorylation, but joint treatment of cells with glutamate and NE or DA significantly suppressed nuclear p53 phosphorylation. Immunofluorescence microscopy results showed that while >90% of glutamate-treated cells displayed p53 nuclear translocation at 24 h, only <10% of the cells jointly treated with glutamate + NE or DA displayed p53 nuclear translocation (Fig. 7b). In addition, the increase in mRNA level of GADD45α, a p53-regulated gene, was abrogated by joint treatment with glutamate + NE or DA or NAC (Fig. 7c).

a Changes in nuclear and cytosolic phospho-p53(Ser15) (p-p53) in cells treated with 5 mM glutamate alone or in combination with NE (50 µM) or DA (50 µM) for 24 h. b Immunofluorescence microscopy analysis of the subcellular localization of p-p53 in cells treated with 5 mM glutamate alone or jointly with 50 µM NE or DA for 24 h. c Real-time PCR analysis of the GADD45α mRNA levels in cells treated with glutamate alone or in combination with NE (50 µM), DA (50 µM) or NAC (500 µM) for 24 h. *P < 0.05 compared to cells treated with glutamate alone.
NE and DA can bind to PDI and inhibit its catalytic activity
Molecular docking analysis predicted that both NE and DA each could bind quite deep inside PDI’s b’ domain (Fig. 8). NE formed two hydrogen bonds with PDI’s His256 (bond length = 2.03 Å and 2.04 Å), and its aromatic ring also formed a hydrophobic Pi-alkyl interaction with PDI’s Leu258. Similarly, DA also formed two hydrogen bonds with PDI’s His256 (bond length = 2.05 Å and 2.34 Å), plus a hydrophobic Pi-alkyl interaction between its aromatic ring and PDI’s Leu258. Based on the binding energy values (Table 1), it is predicted that NE and DA may have similar binding affinities for PDI (Table 1).

a, e The binding of PDI with NE and DA, respectively. b, f The 2D illustration of the interactions of key amino acid residues of PDI with NE and DA, respectively. c, g The 3D illustration of the interactions of key amino acid residues of PDI with NE and DA, respectively. d, h The surface of PDI’s binding pocket with NE and DA bound inside, respectively. Note: The PDI protein structure is shown as ribbon and colored rainbow. NE and DA are shown in balls and sticks (NE: yellow for carbon, red for oxygen, blue for nitrogen, and white for hydrogen; DA: cyan for carbon, red for oxygen, blue for nitrogen, and white for hydrogen). Green dashed lines indicate hydrogen bonds. Amino acid residues that interact with NE or DA are shown in lines (c and g).
Based on the results of computational docking analysis, next we chose to experimentally determine whether NE and DA could dose-dependently inhibit the reductase activity of PDI in the in vitro biochemical enzymatic activity assay (Fig. 9). First, we determined the effect of varying the preincubation time with NE and DA on PDI’s reductase activity. We found that preincubation of PDI with NE for 1 h did not have a meaningful effect on PDI activity, but preincubation for 2 to 4 h showed a time-dependent inhibition of PDI’s activity (Fig. 9a). Similarly, DA also showed a preincubation time-dependent inhibition of PDI’s activity, but the preincubation time required for DA was significantly shorter than NE (Fig. 9d). In this study, we selected two suitable preincubation times for each neurotransmitter to evaluate their ability to inhibit the PDI activity. We found that NE could inhibit PDI’s catalytic activity in a dose-dependent manner (at 25, 50, and 100 µM), and it is evident that with a longer preincubation, a stronger inhibition was consistently observed with NE (Fig. 9b, c). Similarly, DA in the same concentration range also showed a dose-dependent inhibition of PDI’s activity, and a stronger inhibition was observed with a longer preincubation with the inhibitor (Fig. 9e, f).

a, d Inhibition of PDI’s catalytic activity by 100 µM NE and DA at different preincubation time points for each chemical. b, c, e, f Inhibition of PDI’s catalytic activity by three different concentrations (25, 50 and 100 µM) of NE or DA at two selected suitable preincubation times (as indicated) for each chemical.
PDI mediates the protective effect of NE and DA against glutamate-induced cytotoxicity
Next we sought to determine whether PDI mediates the protective effect of NE and DA against glutamate-induced oxidative cytotoxicity in HT22 cells. We sued the siRNA approach to selectively knockdown PDI expression in HT22 cells. We found that treatment of cells with siPDIs could significantly reduce PDI protein level by approximately 80% compared to the control siRNAs (data not shown). It is found that PDI knockdown did not significantly alter cell viability (Fig. 10a). However, the oxidative cytotoxicity induced by two different concentrations (10 or 20 mM) of glutamate was similarly abrogated by PDI knockdown (Fig. 10b), indicating that glutamate-induced cytotoxicity is related to PDI functions. This result is consistent with our earlier study [37]. Moreover, we found that the protective effect of NE and DA against glutamate-induced oxidative cytotoxicity was abrogated by PDI knockdown (Fig. 10c, d).

a Cell viability (by MTT assay) was measured in HT22 cells transfected with siCon and siPDI for 24 h. b Cell viability of siCon or siPDI-treated cells following exposure to 10 or 20 mM glutamate for 24 h. c, d Relative cell viability of cells transfected with siCon (c) and siPDI (d) and treated with 20 mM glutamate with or without NE (50 µM) or DA (50 µM) for 24 h. Cells treated with 20 mM glutamate alone were set to be 1. Mean ± SD. e Dimer nNOS in cells treated with 20 mM glutamate alone or in combination with NE (50 µM) or DA (50 µM) for 8 h and the proteins were separated by non-reducing SDS-PAGE and followed by immunoblot analysis of nNOS, PDI and β-actin. Data shown is a representative data set from two experiments. **P < 0.01; n.s., non-significant compared to cells transfected with siCon (a) or cells treated with glutamate alone (d). It is of note that the sensitivity of HT22 cells to glutamate had marked batch-to-batch variations over time, as reflected by the differences in Fig. 1a and Fig. 10b.
Our earlier study showed that dimerization of the neuronal nitric oxide synthase (nNOS) is catalyzed by PDI, which plays a critical role in mediating glutamate-induced oxidative cytotoxicity [37]. In this study, we also further determined whether NE and DA could inhibit PDI-mediated nNOS dimerization in glutamate-treated HT22 cells. We found that while treatment of cells with glutamate strongly induced nNOS dimerization, joint treatment with NE and DA significantly suppressed nNOS dimerization (Fig. 10e). Together, these results indicate that NE and DA protect against glutamate-induced oxidative cytotoxicity through inhibition of PDI-mediated nNOS dimerization.
Discussion
In neuronal cells, GSH depletion is a rather common pathogenic event, which is often associated with oxidative stress and cytotoxicity, and plays an important role in the etiology of neurodegeneration [12, 42]. Earlier studies have shown that mitochondrial superoxide is a key mediator of glutamate-induced GSH depletion and oxidative cytotoxicity in HT22 cells [23, 24, 39], which is accompanied by rapid depolarization of the inner mitochondrial membrane potential (MMP), impairment of oxidative phosphorylation [39], and ultimately ATP depletion [23]. These observations were confirmed in our present study. Furthermore, we found that NE and DA could inhibit glutamate-induced mitochondrial superoxide accumulation and oxidative stress, along with a restoration of mitochondrial membrane potential and cellular ATP levels.
ATP depletion would lead to inactivation of the ATP-binding caspase activator Apaf-1, thereby preventing caspase activation [43, 44]. In HT22 neuronal culture model, earlier studies showed that the oxidative cytotoxicity involves AIF [23, 45, 46], which is a death effector released from mitochondria and can induce DNA fragmentation and chromatin condensation in a caspase-independent manner [47]. Earlier we also showed that glutamate treatment induces AIF translocation from the mitochondria to the cytoplasm and then to the nucleus [45], and this observation is confirmed in the present study. Moreover, we found that glutamate-induced AIF translocation is suppressed by joint treatment with NE or DA, but not by z-VAD-fmk, a pan-caspase inhibitor.
Our earlier studies have reported that MAPKs (JNK1, ERK, and p38) are activated in HT22 cells following glutamate treatment [23, 24, 48]. Further, glutamate-induced JNK1, ERK and p38 phosphorylation and ROS accumulation can be effectively suppressed by the presence of antioxidants [23, 24, 49]. The results of our present study confirmed our early finding that glutamate-induced ROS accumulation could lead to activation of MAPKs (JNK1, ERK, and p38) in HT22 cells. Moreover, we found that their activation was abrogated by the presence of NE or DA.
Earlier we also showed that activation of the MAPKs could lead to p53 phosphorylation and subsequently GADD45α activation [24]. In the present study, we confirmed that glutamate increased p53 phosphorylation as well as GADD45α mRNA levels in HT22 cells. Moreover, we found that the increase in p53 phosphorylation and GADD45α expression was abrogated by joint treatment with NE or DA. These observations support the notion that suppression of glutamate-induced activation of the MAPKs-p53-GADD45α signaling pathway is a mechanism for the protection by NE and DA against glutamate-induced oxidative cytotoxicity.
To investigate the potential cellular target of NE and DA that mediates their observed neuroprotective action, we sought to determine whether NE and DA could directly bind to the PDI protein and inhibit its catalytic activity, based on our earlier study showing that the glutamate-induced oxidative cytotoxicity in HT22 cells is mediated by PDI [37]. Here it is of note that the possibility of NE and DA receptors as potential targets of their action is deemed unlikely since no appreciable protection was seen when NE or DA was present at 5 µM (a concentration which can readily occupy nearly all NE or DA receptors present on cell surface). Results from the molecular docking analysis indicated that NE and DA each could bind inside a rather deep pocket in PDI’s b’ domain, and the two phenolic hydroxyl groups of both chemicals could form two hydrogen bonds with PDI’s His256 plus a hydrophobic Pi-alkyl interaction with Leu258. Notably, the binding pocket of NE and DA and also the PDI’s key amino acid residue with which they form interaction are nearly identical with 17β-estradiol as described in our earlier study [35].
In agreement with the predictions from the molecular docking analysis, we found that both NE and DA can directly inhibit PDI’s reductase activity in a time- and concentration-dependent manner. It is of note that the inhibition of PDI’s reductase activity was only observed when PDI had been preincubated with NE and DA for a sufficient length of time. Without the pre-incubation, NE and DA would not be able to exert a significant inhibition of PDI’s catalytic activity. In addition, we found that a longer preincubation would produce a markedly stronger inhibition of PDI’s activity. The unusual long preincubation time needed for NE and DA to exert a detectable inhibition of PDI’s activity in vitro might be due to the fact that PDI is a highly hydrophobic protein, and under the in-vitro aqueous conditions, it would be extremely difficult for NE and DA (both are highly water-soluble molecules) to gain access to the binding pocket that resides quite deep inside PDI’s b’ domain. If this is indeed the cause, then it is predicted, based on our experimental data, that when NE or DA is bound inside PDI’s binding pocket, the binding interaction would be exceptionally tight (i.e., high-affinity binding) and the bound complexes would have an ultra-slow dissociation rate. Otherwise, it would be kinetically impossible to observe a preincubation time-dependent inhibition of the PDI’s enzymatic activity by NE or DA which binds to PDI so slowly under the in vitro assay conditions. As for the mechanism of the tight-binding interactions between PDI and the two neurotransmitters, there are two likely possibilities: One is that NE and DA may bind inside the PDI’s binding pocket non-covalently but with ultra-high affinities. The other possibility is that NE and DA might bind to PDI in a covalent manner. This possibility does exist since both NE and DA can readily undergo auto-oxidation to form semiquinone/quinone intermediates, which are chemically reactive and are capable of forming a covalent bond with one of the cysteine residues at or near PDI’s active site. These possibilities merit further investigation.
Our earlier study showed that cystamine, a small-molecule inhibitor of PDI’s isomerase activity, could effectively prevent GSH depletion-induced accumulation of NO and superoxide as well as oxidative cytotoxicity in HT22 cells [37]. Mechanistically, it was reported that PDI can catalyze the dimerization of nNOS, which results in catalytic activation of nNOS and subsequently increased formation of NO [37]. Increased cellular NO levels would then lead to accumulation of mitochondrial ROS [37]. In this study, we also provided evidence showing that NE and DA can inhibit glutamate-induced, PDI-mediated dimerization of the nNOS in HT22 cells, thereby confirming that NE and DA indeed can inhibit PDI activity. Moreover, we found that the protective effect of NE and DA against glutamate-induced oxidative cytotoxicity is mostly abrogated by PDI knockdown. Based on these findings, it is clear that the neuroprotective effect of NE and DA against glutamate-induced oxidative cytotoxicity is mediated through their inhibition of PDI-mediated nNOS dimerization, which abrogates cellular ROS accumulation, and this effect would subsequently attenuate MAPKs activation, p53 phosphorylation, and GADD45α activation, which were all downstream mediators activated by ROS accumulation [24].
It should be noted that when HT22 cells are treated with NE or DA alone, we observed a small but significant increase in nuclear p53 phosphorylation. The reason likely is because NE and DA are both catecholic compounds, and they can readily undergo chemical or metabolic redox cycling between the catechols and their reactive catechol semiquinone/quinone intermediates [50], thereby increasing the level of intracellular oxygen free radicals. However, when they were used in combination with glutamate, they still exerted a strong overall suppression of glutamate-induced nuclear p53 phosphorylation. A very similar phenomenon was also observed with 4-hydroxyestrone, an endogenous catechol estrogen metabolite, which was recently reported by us to have a very strong neuroprotective effect both in vitro and in vivo [51]. We observed that 4-hydroxyestrone could also cause a small increase in promoting nuclear p53 phosphorylation when present alone, but it could strongly suppress glutamate-induced nuclear p53 phosphorylation when used in combination [51].
The normal range of NE and DA in human blood samples is usually very low, around 0.41‒10.0 nM and 0‒0.20 nM, respectively [52], which is much lower than the effective concentrations observed in vitro for the neuroprotective effect. However, it is of note that this discrepancy does not necessarily mean that the observed in vitro protective effect is physiologically irrelevant, and this view is partially supported by two recent observations made in our laboratory [51, 53]. First, our earlier study with tea polyphenols showed that the effective concentrations of tea compounds that exerted a neuroprotective effect against glutamate-induced death in cultured HT22 cells were around 40 μM [53], and this concentration would not be achieved in vivo when the animals were administered orally with tea compounds (the blood concentration usually is below 1 μM). However, when tested in vivo using the kainic acid-induced brain damage model (through intracerebral injection), oral administration of tea compound at 100 and 400 mg/kg body weight produces very strong neuroprotection [53]. Second, our recent study with endogenously-formed estrogen metabolites showed that the effective concentration of 4-hydroxyestrone that exerted a neuroprotective effect against glutamate-induced death in cultured HT22 cells was around 5‒10 μM [53], and this concentration would not be achieved in vivo when the animals were administered with an estrogen (the blood concentration usually is in the pM to nM range). However, when 4-hydroxyestrone was tested in vivo using the kainic acid-induced brain damage model, i.p. injection of this estrogen derivative at only 10 μg/rat produced a very strong neuroprotection in vivo [51].
In this study, a similar in vivo study by using the kainic acid-induced neuronal damage model was not conducted because NE and DA are richly present in vivo (particularly in the central nervous system) in both male and female animals, and it would be difficult to determine the neuroprotective effect of an administered catecholamine. In addition, the administered catecholamine would not be able to cross the blood-brain barrier to reach neuronal cells. Because of these considerations, a similar in vivo experiment testing the neuroprotective pharmacological effect of NE and DA was not deemed feasible and thus not conducted in this study.
Here it is of interest to note that an earlier study by Fuller et al. [54] reported that the tissue levels of NE, DA and some of their metabolites in certain brain regions of animals are actually very high. For instance, the rat hypothalamus contains approximately 6.9 and 1.2 nmol of NE and DA, respectively, in every gram of wet brain tissue [54]. If we assume that roughly 50% of the tissue weight is water, then their estimated tissue concentrations might be around 13.8 and 2.4 μM, respectively. Similarly, cats, hamsters and mice also contain high hypothalamic levels of NE and DA [54]. While these literature values might provide some reference regarding the high tissue content of NE and DA in certain brain regions, they do not necessarily reflect the actual concentrations of NE and DA in the extracellular or intracellular space of the neuronal cells in vivo as a significant fraction is sequestered (stored) inside the intracellular vesicles as neurotransmitters.
There are animal studies showing that physical exercises can significantly increase the levels of endogenous neurotransmitters NE, DA and their metabolites in certain regions of the brain [20, 21]. For instance, an earlier study by Dunn et al. [20] investigated the NE release and metabolism in hippocampus and frontal cortex of rats performing treadmill or wheel running exercise. It was shown that these physical activities can significantly increase the turnover, release and concentration of NE in the central nervous system. If the combined concentrations of endogenous neurotransmitters NE and DA in certain brain regions are indeed capable of exerting a neuroprotective effect on the oxidative cytotoxicity as observed in vitro as observed in the present study, then it is reasonable to suggest that this protective effect likely would be enhanced by physical activities or exercises. Besides, it is also known that the exercise-induced release of central NE would have another beneficial effect by alleviating neuroinflammation [55,56,57], which is a well-known aggravating condition in the progression of neurodegeneration.
In summary, the results of our present study demonstrate that NE and DA, two catecholamine neurotransmitters, have a strong protective effect against glutamate-induced, glutathione depletion-associated oxidative stress and cytotoxicity, whereas other neurotransmitters do not share this neuroprotective effect. Mechanistically, we found that NE and DA suppress glutathione depletion-induced activation of the MAPKs‒p53‒GADD45α apoptotic cascade and restore mitochondrial dysfunctions, including mitochondrial superoxide accumulation, ATP depletion, and mitochondrial AIF release. Experimental evidence is also presented which shows that PDI is a direct target protein that is bound and inhibited by NE and DA. It is suggested that inhibition of PDI-mediated nNOS dimerization by NE and DA may mediate their neuroprotective actions. Undoubtedly, more studies are needed to determine whether the findings made in the present study in an in vitro neurodegeneration model might offer a mechanistic explanation for the clinical observations that keeping the mind active through physical activities or exercises would improve the cognitive functions of the brain and slow down the progression of neurodegeneration.
References
Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222:236–45.
Andersen JK. Oxidative stress in neurodegeneration: Cause or consequence? Nat Med. 2004;10:S18–S25.
Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21:172–88.
Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem. 2001;8:721–38.
Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95.
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–67.
Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int. 2004;45:583–95.
Choi DW. 1990. Methods for antagonizing glutamate neurotoxicity. Cerebrovasc Brain Metab Rev. 1990;2:105–47.
Tan S, Wood M, Maher P. Oxidative stress induces a form of programmed cell death with characteristics of both apoptosis and necrosis in neuronal cells. J Neurochem. 1998;71:95–105.
Murphy SN, Miller RJ. Two distinct quisqualate receptors regulate Ca2+ homeostasis in hippocampal neurons in vitro. Mol Pharmacol. 1989;35:671–80.
Pereira C, Paveto C, Espinosa J, Alonso G, Flawiá MM, Torres HN. Control of Trypanosoma cruzi epimastigote motility through the nitric oxide pathway. J Eukaryot Microbiol. 1997;44:155–6.
Okada K, Fukui M, Zhu BT. Protein disulfide isomerase mediates glutathione depletion-induced cytotoxicity. Biochem Biophys Res Commun. 2016;477:495–502.
Froissard P, Duval D. Cytotoxic effects of glutamic acid on PC12 cells. Neurochem Int. 1994;24:485–93.
Erdö SL, Michler A, Wolff JR, Tytko H. Lack of excitotoxic cell death in serum-free cultures of rat cerebral cortex. Brain Res. 1990;526:328–32.
Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: Pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–53.
Yamazaki Y, Sato D, Yamashiro K, Tsubaki A, Takehara N, Uetake Y, et al. Inter-individual differences in working memory improvement after acute mild and moderate aerobic exercise. PLoS One. 2018;13:e0210053.
Vecchio LM, Meng Y, Xhima K, Lipsman N, Hamani C, Aubert I. The neuroprotective effects of exercise: Maintaining a healthy brain throughout aging. Brain Plast. 2018;4:17–52.
Middleton LE, Mitnitski A, Fallah N, Kirkland SA, Rockwood K. Changes in cognition and mortality in relation to exercise in late life: a population based study. PLoS ONE. 2008;3:e3124.
Christiansen L, Beck MM, Bilenberg N, Wienecke J, Astrup A, Lundbye-Jensen J. Effects of exercise on cognitive performance in children and adolescents with ADHD: potential mechanisms and evidence-based recommendations. J Clin Med. 2019;8:841.
Dunn AL, Reigle TG, Youngstedt SD, Armstrong RB, Dishman RK. Brain norepinephrine and metabolites after treadmill training and wheel running in rats. Med Sci Sports Exerc. 1996;28:204–9.
Pagliari R, Peyrin L. Norepinephrine release in the rat frontal cortex under treadmill exercise: A study with microdialysis. J Appl Physiol. 1995;78:2121–30.
Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–73.
Fukui M, Song JH, Choi JY, Choi HJ, Zhu BT. Mechanism of glutamate-induced neurotoxicity in culture HT22 cells. Eur J Pharmacol. 2009;617:1–11.
Choi HJ, Kang KS, Fukui M, Zhu BT. Role of JNK‒p53‒GADD45α apoptotic cascade in oxidative stress-induced neuronal death. Br J Pharmacol. 2010;162:175–92.
Tang HM, Tang HL. Cell recovery by reversal of ferroptosis. Biol Open. 2019;8:bio043182.
Wilkinson B, Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta. 2004;1699:35–44.
Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: The structure of oxidative folding. Trends Biochem Sci. 2006;31:455–64.
Tu BP, Ho-Schleyer SC, Travers KJ, Weissman JS. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science. 2000;290:1571–4.
Parakh S, Atkin JD. Novel roles for protein disulphide isomerase in disease states: A double edged sword? Front Cell Dev Biol. 2015;3:30.
Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: Unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–63.
Wroblewski VJ, Masnyk M, Khambatta SS, Becker GW. Mechanisms involved in degradation of human insulin by cytosolic fractions of human, monkey, and rat liver. Diabetes. 1992;41:539–47.
El Hindy M, Hezwani M, Corry D, Hull J, El Amraoui F, Harris M, et al. The branched-chain aminotransferase proteins: Novel redox chaperones for protein disulfide isomerase—implications in Alzheimer’s disease. Antioxid Redox Signal. 2014;20:2497–513.
Soares Moretti AI, Martins, Laurindo FR. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch Biochem Biophys. 2017;617:106–19.
Fu X, Wang P, Zhu BT. Protein disulfide isomerase is a multifunctional regulator of estrogenic status in target cells. J Steroid Biochem Mol Biol. 2008;112:127–37.
Fu XM, Wang P, Zhu BT. Characterization of the estradiol-binding site structure of human pancreas-specific protein disulfide isomerase: Indispensable role of the hydrogen bond between His278 and the estradiol 3-hydroxyl group. Biochemistry. 2011;50:106e115.
Hoffstrom BG, Kaplan A, Letso R, Schmid RS, Turmel GJ, Lo DC, et al. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nat Chem Biol. 2010;6:900e906.
Okada K, Fukui M, Zhu BT. Protein disulfide isomerase mediates glutathione depletion-induced cytotoxicity. Biochem Biophys Res Commun. 2016;477:495–502.
Choi HJ, Lee AJ, Kang KS, Song JH, Zhu BT. 4-Hydroxyestrone, an endogenous estrogen metabolite, can strongly protect neuronal cells against oxidative damage. Sci Rep. 2020;10:7283.
Fukui M, Zhu BT. Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Rad Biol Med. 2010;48:821–30.
Fu XM, Zhu BT. Human pancreas-specific protein disulfide-isomerase (PDIp) can function as a chaperone independently of its enzymatic activity by forming stable complexes with denatured substrate proteins. Biochem J. 2010;429:157–69.
Herrera F, Martin V, García-Santos G, Rodriguez-Blanco J, Antolín I, Rodriguez C. Melatonin prevents glutamate-induced oxytosis in the HT22 mouse hippocampal cell line through an antioxidant effect specifically targeting mitochondria. J Neurochem. 2007;100:736–46.
Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–60.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13.
Leist M, Single B, Castoldi AF, Kühnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–6.
Fukui M, Choi HJ, Zhu BT. Rapid generation of mitochondrial superoxide induces mitochondron-dependent but caspase-independent cell death in hippocampal neuronal cells that morphologically resembles necroptosis. Toxicol Appl Pharmacol. 2012;262:156–66.
Zhang Y, Bhavnani BR. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006;7:49.
Nagata S, Nagase H, Kawane K, Mukae N, Fukuyama H. Degradation of chromosomal DNA during apoptosis. Cell Death Differ. 2003;10:108–16.
Fukui M, Choi HJ, Zhu BT. Mechanism for the protective effect of resveratrol against glutamate-induced neuronal death in HT22 cells. Free Rad Biol Med. 2010;49:800–13.
Haddad JJ, Land SC. Redox/ROS regulation of lipopolysaccharide-induced mitogen-activated protein kinase (MAPK) activation and MAPK-mediated TNF-α biosynthesis. Br J Pharmacol. 2002;135:520–36.
Zhu BT. CNS dopamine oxidation and catechol-O-methyltransferase: Importance in the etiology, pharmacotherapy, and dietary prevention of Parkinson’s disease. Int J Mol Med. 2004;13:343–53.
Choi HJ, Lee AJ, Song JH, Kang KS, Zhu BT. 4-Hydroxyestrogens are endogenous neuroestrogens that can strongly protect neurons against oxidative death by promoting p53 cytoplasmic localization. Sci Rep. 2020;10:7283.
MedlinePlus Website. Available from: https://medlineplus.gov/ency/article/003561.htm.
Kang KS, Yamabe N, Wen Y, Fukui M, Zhu BT. Beneficial effects of natural phenolics on levodopa methylation and oxidative neurodegeneration. Brain Res. 2013;1497:1–14.
Fuller RW, Hemrick-Luecke SK. Specific differences in epinephrine concentration and norepinephrine N-methyltransferase activity in hypothalamus and brain stem. Comp Biochem Physiol. 1983;74C:47–9.
Song S, Jiang L, Oyarzabal EA, et al. Loss of brain norepinephrine elicits neuroinflammation-mediated oxidative injury and selective Caudo-Rostral neurodegeneration. Mol Neurobiol. 2019;56:2653–69.
Caraci F, Merlo S, Drago F, Caruso G, Parenti C, Sortino MA. Rescue of noradrenergic system as a novel pharmacological strategy in the treatment of chronic pain: Focus on microglia activation. Front Pharmacol. 2019;10:1024.
Dello Russo C, Boullerne AI, Gavrilyuk V, Feinstein DL. Inhibition of microglial inflammatory responses by norepinephrine: Effects on nitric oxide and interleukin-1beta production. J Neuroinflammation. 2004;1:9.
Acknowledgements
This study is supported, in part, by the University of Kansas Medical Center Endowment Fund, and by research grants from Shenzhen Key Laboratory of Steroid Drug Discovery and Development (grant # ZDSYS20190902093417963) and the National Natural Science Foundation of China (grant # 81630096).
Author information
Authors and Affiliations
Contributions
BTZ conceived the study, participated in the research design and implementation of the study, analyzed and interpreted the data, and wrote the manuscript. HJC performed some of the experiments, analyzed the data. TXC, MJH, JHS, PL and CFL, and PW performed some of the experiments. PW participated in manuscript revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Choi, H.J., Chen, Tx., Hou, MJ. et al. Protection against glutathione depletion-associated oxidative neuronal death by neurotransmitters norepinephrine and dopamine: Protein disulfide isomerase as a mechanistic target for neuroprotection. Acta Pharmacol Sin 43, 2527–2541 (2022). https://doi.org/10.1038/s41401-022-00891-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00891-w
