
Abstract
Nucleotide-binding oligomerization domain-like receptors (NLRs), including NLRAs, NLRBs (also known as NAIPs), NLRCs, and NLRPs, are a major subfamily of pattern recognition receptors (PRRs). Owing to a recent surge in research, NLRs have gained considerable attention due to their involvement in mediating the innate immune response and perpetuating inflammatory pathways, which is a central phenomenon in the pathogenesis of multiple diseases, including renal diseases. NLRs are expressed in different renal tissues during pathological conditions, which suggest that these receptors play roles in acute kidney injury, obstructive nephropathy, diabetic nephropathy, IgA nephropathy, lupus nephritis, crystal nephropathy, uric acid nephropathy, and renal cell carcinoma, among others. This review summarises recent progress on the functions of NLRs and their mechanisms in the pathophysiological processes of different types of renal diseases to help us better understand the role of NLRs in the kidney and provide a theoretical basis for NLR-targeted therapy for renal diseases.
Similar content being viewed by others

Introduction
Compared to the adaptive immune system, the innate immune system exerts a rapid response to pathogens or tissue injury and is initiated by the recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs) [1, 2]. DAMPs include intracellular components released during different types of cell death and molecules resulting from the enzymatic degradation of quiescent extracellular matrix (ECM) during ECM remodelling. In addition, these factors can originate in the kidney and can include crystals and uromodulin [3].
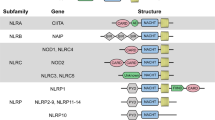
PRRs are a group of germline-encoded receptors that mediate the initial innate immune response to cellular injury and stress [4]. PRRs can be classified into five subfamilies according to their different structural domains: Toll-like receptors (TLRs), retinoic acid-inducible gene-I-like receptors, C-type lectin receptors, absent in melanoma 2 (AIM2)-like receptors, and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) [5]. NLRs are intracellular receptors located mainly in the cytoplasm, although some are also found in mitochondria. A total of 22 and 35 NLR genes have been identified in humans and mice, respectively [6, 7]. The structure of NLRs includes a C-terminal leucine-rich repeat domain that functions as a ligand sensor, a conserved nucleotide-binding (NACHT) domain that is required for nucleotide binding and oligomerization, and an N-terminal effector domain that mediates downstream protein–protein interactions, such as an acidic transactivation domain, caspase recruitment domain (CARD), pyrin domain (PYD), and baculoviral inhibitory repeat (BIR)-like domain [7, 8]. Based on their N-terminal domains, NLRs can be subdivided into four different subfamilies: NLRAs, NLRBs (also known as NAIPs), NLRCs, and NLRPs.
The NLRA subfamily, which contains an acidic transactivating domain (AA), consists of only one member: CIITA. The NLRB subfamily contains an N-terminal BIR-like domain, and BIR consists of NAIP in humans and NAIP1-7 in mice. The NLRC subfamily includes NOD1, NOD2, and NLRC3-5, in which a CARD domain is present. NLRX1, which is located in the mitochondrial matrix and is mediated by an N-terminal addressing sequence, belongs to the NLRC subfamily. The NLRP subfamily consists of 14 members named NLRP1-14, each of which contains a PYD domain. All members also contain the NACHT and LRR domains, except for NLRP10, which lacks an LRR domain. NAIPs and NLRC4 can pair with each other to form inflammasomes in animals [9, 10].
Most NLRs function in (1) antigen presentation; (2) pathogen/damage sensing; (3) inflammasome activation; and (4) inflammatory signalling inhibition (Fig. 1). However, several NLRs play definitive roles in embryonic development [11]. These findings suggest alternative functions for some NLRs in different cell types and multiple activation mechanisms with separate downstream effects for other NLRs. Although an increasing number of studies have shown links between NLRs and renal diseases, the results have been inconclusive. In this review, we summarise recent progress on the functions of NLRs and discuss whether these receptors could serve as therapeutic targets in renal diseases.

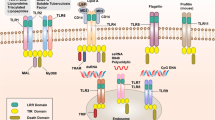
A Structure of NLRs. Based on their N-terminal domains, NLRs can be subdivided into four different subfamilies: NLRAs, NLRBs (also known as NAIPs), NLRCs and NLRPs. B Function of NLRs. Multiple NLRs serve as antigen presentation, pathogen/damage sensing, inflammasome activators, inflammatory signalling inhibitors. C The effects of NODs and NLRP3. NOD1 and NOD2 can both interact with the receptor-interacting protein 2 (Rip2) kinase and recruit the Rip2s through the CARD-CARD binding which will lead to the activation of NF-κB or MAPK pathway, resulting in the secretion of proinflammatory cytokines, such as IL-6, IL-1β and TNF-α. Additionally, NOD1 and NOD2 interact with ATG16L to mediate autophagy. NLRP3 can work in a canonical (inflammasome dependent) or a noncanonical manner (inflammasome independent). Canonical inflammasomes convert procaspase-1 into its mature form caspase-1, whereas noncanonical inflammasome activate procaspase-11.
Characteristics of NLRs in biological processes
NLRs in pathogen/damage sensing
NOD1 is expressed in tubular epithelial cells (TECs) in the kidneys of humans and mice, as well as in many other cells and organs. The expression of NOD2 can be detected in TECs and glomerular endothelial cells in humans and in podocytes, mesangial cells, and TECs in mice [12]. NOD1 can recognise γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP), which is the degradation product of gram-negative bacteria and certain gram-positive bacteria, while NOD2 can recognise muramyl dipeptide (MDP), which is the degradation product of gram-negative and gram-positive bacteria [13,14,15]. Binding with the corresponding ligand results in the rapid self-oligomerization of NOD1 or NOD2. Although the ligands identified by NOD1 and NOD2 are different, they can interact with the downstream protein receptor-interacting protein 2 (Rip2) kinase and recruit Rip2 through CARD-CARD binding, which leads to activation of the NF-κB or MAPK pathway, resulting in the secretion of proinflammatory cytokines, such as IL-6, IL-1β, and TNF-α [16]. Additionally, NOD1 and NOD2 interact with ATG16L to mediate autophagy [17]. Furthermore, NOD1 and NOD2 can participate in the recognition of DAMPs derived from cell damage [10].
Exogenously derived PAMPs and DAMPs convert NLRs from inactive monomers to activated oligomers [18]. The NLR family plays a critical role in sensing PAMPs and DAMPs in the cytosol [19]. The primary DAMPs associated with inflammatory diseases are ATP, hyaluronan, uric acid, monosodium urate (MSU) crystals, cholesterol crystals and β amyloid (Aβ) [20, 21]. Recent studies have demonstrated that the NLRP3 inflammasome can recognise pathogen- and danger-associated molecular patterns [22, 23]. Uric acid and MSU crystals directly induce insulin resistance, insulin signalling impairment, CKD and gouty arthritis by inducing NLRP3 inflammasome activation [23,24,25,26]. Extracellular ATP is released by necrotic or apoptotic cells and sensed by P2X7 receptors to activate innate immune responses, including the NLRP3 inflammasome [27, 28]. In addition, the NLRP3 inflammasome is also activated by cholesterol crystals [29, 30]. Prxs are ubiquitously expressed peroxidases that are released from cells in response to various stress conditions. These factors function as DAMPs. A recent study showed that NLRP3 inflammasome activation induced Prx2 secretion, which induced C1q-mediated classic complement pathway activation [31]. Furthermore, NOD1 and NOD2 can also participate in the recognition of DAMPs derived from cell damage [10]. The recognised bacterial peptidoglycan derivatives iE-DAP and MDP undergo conformational alterations and ATP-dependent self-oligomerization of the NACHT domain followed by downstream signalling [18, 32].
PAMPs include lipopolysaccharides (LPS), flagellin, single-stranded RNA, and unmethylated CpG DNA [33]. NOD2 variants in Crohn’s disease share a signalling defect in response to LPS [34]. NLRC4, which is activated by bacterial flagellin or the rod complex, plays a role in pathogen sensing [35,36,37]. NAIP proteins are receptors of bacterial ligands, while NLRC4 is a downstream adaptor that multimerizes with NAIPs to form an inflammasome with adaptor apoptosis-associated speck-like protein (ASC) and caspase-1, resulting in the release of IL-1β and IL-18 [11, 37].
In summary, these studies show that the NLR family contains cytosolic PRRs that play important roles in innate immune signalling.
NLRs in antigen presentation
NLRC5 has a CARD-like N-terminal domains, is predominantly expressed in cells of the myeloid and lymphoid lineages and is induced by IFN-γ, LPS, and polyinosinic:polycytidylic acid [38]. NLRC5 is not only an essential factor for MHC class I gene expression and type I interferon responses but also mediates the activation of CD8+ T cells and CD4+ T cells [39, 40]. Furthermore, NLRC5 plays a role not only in immune regulation but also in cancer immunity [41].
CIITA, which has an N-terminal CARD and acid transactivation domain, displays features similar to those of NLRC5. CIITA acts as a transcriptional coactivator and is involved in the regulation of MHC class II expression [42, 43]. Both NLRC5 and CIITA shuttle between the cytosol and the nucleus in a CrmA-dependent manner [38].
NLRs in inflammation regulation
Recent studies have demonstrated that multiple NLRs play essential roles in regulating inflammation. The role of different NLRs in regulating inflammation may be different. NLRC3, NLRC5, NLRX1 and NLRP6 may serve as negative modulators of inflammation, while NLRP3, NLRP1, NLRP2, NLRP6, and NLRP7 are able to form inflammasomes and promote the inflammatory response.
NLRs as inhibitory regulators of inflammation
A recent study showed that LPS administration increased hypothermia and enhanced macrophage infiltration and IL-6 production in NLRC3−/− mice. NF-κB, a key downstream transcription factor of TLR signalling, is activated in NLRC3−/− macrophages [44]. Mechanistically, NLRC3 inhibits NF-κB activation by decreasing the K63-linked ubiquitination of TRAF6 [45]. Thus, NLRC3 functions as a negative modulator of inflammatory pathways [46].
Similar to NLRC3, NLRC5 has been proposed to function as a negative modulator of inflammatory pathways. NLRC5 overexpression inhibits NF-κB-dependent signalling [38]. NLRP6 also inhibits NF-κB activation. NLRP6 has been shown to impede NF-κB activation during bacterial pathogen infection, and in the absence of NLRP6, bacteria are cleared more rapidly, indicating that NLRP6 may serve as a negative modulator of inflammation [47].
The lack of pyrin and caspase domains, which are essential for caspase-1 activation and recruitment, as well as the unique localisation of NLRX1 in the mitochondrial matrix, are the main causes of the specific effects of NLRX1. NLRX1 plays a significant role in suppressing the NF-κB pathway, negatively regulates antiviral immune responses, and prevents apoptosis [48]. Additionally, NLRX1 interacts with some mitochondrial proteins, such as dynamin-related protein 1, to increase mitochondrial fission and ubiquinol cytochrome c reductase core protein II, which is a subunit of complex III, to induce the generation of reactive oxygen species (ROS) [49, 50].
NLRs as positive regulators of inflammation
NLRP3 is the most well-understood NLR in kidney diseases [51]. Activation of the NLRP3 inflammasome requires two signals: the priming signal and the activation signal (Fig. 2). The first signal is related to the expression of NLRP3 and pro-IL-1β and is mediated by the NF-κB signalling pathway. IL-6 or TNF-α can also stimulate the NLRP3 inflammasome as the first signal [52]. The second signal is the activation of NLRP3 by copious amounts of priming signals, such as ROS, ATP, amyloid-beta, biglycan, crystals of MSU, hyaluronan, uric acid, and pathogens [53,54,55,56,57]. Soluble biglycan, a macromolecule in the ECM, induces the activation of NLRP3/ASC inflammasomes by signalling through TLR2/4, as well as purinergic P2X7 receptors, and activates the inflammasome in an ROS-dependent manner, releasing caspase-1 and IL-1β for the subsequent inflammatory response. A lack of ASC has been shown to reduce the secretion of IL-1β, which is related to biglycan [58]. NLRP3 ubiquitination is also involved in inflammasome activation [59]. BRCA1-BRCA2-containing complex subunit 3 (BRCC3), a deubiquitinating enzyme, can deubiquitinate the NLRP3 LRR domain and promote NLRP3 inflammasome activation [60]. Activated NLRP3 recruits ASC, which consists of two major domains (PYD and CARD), and caspase-1 to form a complex called the inflammasome. Inflammasomes induce the activation of caspase-1, resulting in the cleavage of pro-IL-1β and pro-IL-18 and inducing the release of proinflammatory cytokines, such as IL-1β and IL-18. In addition, the mature form of caspase-1 can cleave gasdermin D to further induce the form of programmed cell necrosis known as pyroptosis [61, 62].

LPS, PAMPs, DAMPs, IL-6, TNF-α could also stimulate NLRP3 inflammasome as the first signal. The second signal is that the activation of NLRP3 can be initiated by copious amount of activation signals, such as, reactive oxygen species (ROS), ATP, K+ efflux, amyloid-beta, biglycan, crystals of monosodium urate (MSU), hyaluronan, uric acid, pathogens, etc.
NLRP3 can work in a canonical (inflammasome-dependent) or noncanonical manner (inflammasome-independent). Canonical inflammasomes convert procaspase-1 into its mature form, caspase-1, whereas noncanonical inflammasomes activate procaspase-11. Moreover, NLRP3 can promote epithelial-mesenchymal transition (EMT) in epithelial cells by enhancing TGF-β1 signalling and R-Smad activation via inflammasome-independent mechanisms [4, 63, 64]. These findings greatly increase our understanding of the activation and function of NLRP3.
In addition to NLRP3, NLRP1, NLRP2, NLRP6, and NLRP7 are also able to form inflammasomes [65,66,67]. Moreover, NLRP12 may also assemble inflammasomes [4, 6]. During Yersinia pestis infection, NLRP12 has been reported to recognise acylated lipid A, and NLRP12−/− mice are more susceptible to infection and have reduced IL-18 levels than wild-type (WT) mice. The recognition of Y. pestis expressing a stimulatory LPS by TLR4 leads to the upregulation of acute haemolytic transfusion reactions (AHTR) involving NLRP12 and proinflammatory cytokines, such as IL-18 and IL-1β. NLRP12 then recognises a ligand produced during Y. pestis infection and assembles into an inflammasome that processes IL-18 and IL-1β [68]. A recent study demonstrated that NLRP12 interacts with NLRP3 and NLRC4 to induce IL-1β maturation through caspase-1 activation [69]. Redundancy between NLRs may occur, and other NLRs may also participate in optimal responses to infection. These findings support the concept that NLRs work together to optimally protect the host.
NLRP10 has no LRR domain, which is important for the detection of PAMPs or DAMPs [70]. NLRP10 ablation markedly suppresses activation of the NF-κB signalling pathway and impairs the function of NLRP12 [71]. In this context, NLRP10 may play a regulatory role in the inflammatory response via its association with NLRP12 [72].
Role of NLRs in renal diseases
Recent studies have demonstrated that multiple NLRs play essential roles in renal diseases, including acute kidney injury (AKI), obstructive nephropathy (ON), diabetic nephropathy (DN), lupus nephritis (LN), uric acid nephropathy (UAN), and renal cell carcinoma (RCC).
NLRs in acute kidney injury
AKI is a group of life-threatening syndromes characterised by a sudden decrease in the glomerular filtration and the accumulation of waste products [73, 74]. As a risk factor for CKD and end-stage renal disease (ESRD), AKI is associated with high morbidity and mortality [40, 75, 76]. AKI is always caused by ischaemia–reperfusion injury (IRI). The IRI model is widely accepted in AKI-related research [40, 77,78,79]. Other models include sepsis, oxalate, and nephrotoxic drugs, such as cisplatin, which is a chemotherapeutic drug that is used clinically [80,81,82]. Although the mechanisms underlying AKI remain unclear, an extensive inflammatory response is regarded as a major contributor to the initiation and progression of AKI. Emerging studies indicate the importance of NLRs in the pathophysiology of AKI [73, 83,84,85] (Table 1).
NOD1 and NOD2 in acute kidney injury
NOD1 and NOD2 are the first reported NLRs found in the renal TECs of both mice and humans. Studies indicate the protective effect of NOD2 deficiency in IRI, and a more robust protective effect can be seen after the simultaneous deficiency of NOD1 and NOD2. Consistently, mice with Rip2 deficiency are also protected against IRI [12]. Although NOD1 deficiency shows less functional protection than NOD2 knockout, it has a great impact on neutrophil recruitment in vivo [86]. Additionally, progranulin, an autocrine growth factor, ameliorates renal injury and inhibits apoptosis in part by negatively regulating the NOD2-dependent pathway in mouse kidneys with AKI induced by IR [87]. In addition, ligustrazine, a type of bioactive alkaloid, can protect against AKI by inhibiting NOD2 expression and apoptosis in mice following ischaemia/reperfusion (IR) injury [88]. These findings support the therapeutic potential of NOD1 and NOD2 inhibition in AKI.
NLRC4 in acute kidney injury
NLRC4 is a member of the NOD-like receptor family that can form inflammasome complexes and lead to the subsequent activation of caspase-1 and the secretion of IL-1β and IL-18. NLRC4 expression is enhanced after IR injury, which is related to T cell immunoglobulin domain and mucin domain-containing molecule-3 (Tim-3) signalling. The administration of RMT3-23, a Tim-3 neutralising antibody, can reduce NLRC4 expression and macrophage infiltration, suggesting that Tim-3-mediated NLRC4 inflammasome activation participates in IR-induced AKI [78]. Although studies concerning the role of NLRC4 in AKI are scarce, this finding presents a novel perspective on the potential action of NLRC4 in IR-induced AKI.
NLRX1 in acute kidney injury
According to its localisation in the mitochondrial matrix and its distinct structure, NLRX1 has a unique effect on AKI. The expression of NLRX1 is decreased in the human kidney in IRI and is positively correlated with increased TEC apoptosis and oxidative stress. Furthermore, NLRX1 deletion contributes to mitochondrial dysfunction and TEC apoptosis during IRI. Mechanistically, NLRX1 can suppress oxidative phosphorylation and excessive ROS formation in IRI to regulate mitochondrial function [48]. Consistently, NLRX1 inhibits type I IFN signalling and NF-κB activation to exert its anti-inflammatory effects [89, 90]. Based on the above data, NLRX1 is regarded as a pivotal target in mediating a protective effect against renal tissue injury during IRI.
NLRC5 in acute kidney injury
Notably, the expression of NLRC5 has been shown to be markedly increased in mice suffering from IR-induced and cisplatin-induced AKI [40]. NLRC5 expression was also shown to be significantly elevated in HK-2 cells induced by IR. NLRC5 is also involved in NF-κB-mediated renal inflammation [91]. Ablation of NLRC5 inhibits oxidative stress and apoptosis in HK-2 cells by enhancing the PIK3/Akt signalling pathway [92]. NLRC5 deficiency can reduce inflammatory responses and attenuate cisplatin-induced renal injury. Mechanistically, NLRC5 knockout prevents the activation of CD4+ T cells by restoring the expression of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) [40]. A recent study showed that NLRC5 activates the inflammatory response and exacerbates TEC apoptosis by negatively regulating CEACAM1, which can act through well-known antiapoptotic signalling pathways, including extracellular signal-regulated kinase 1/2 (ERK1/2) and Akt in NRK-52E cells [40]. Moreover, NLRC5 has been recognised as a key regulator of MHC class I gene expression and may work with NLRP3 to induce inflammasomes [93, 94]. Thus, targets of NLRC5 may have novel therapeutic implications in patients suffering from AKI.
NLRP3 in acute kidney injury
NLRP3, an important member of the NOD-like receptor family, is composed of an LRR, an NOD domain, and an N-terminal effector domain [7]. In addition to its interleukin-associated and pyroptosis-inducing effects, NLRP3 contributes to immunity and tissue injury [54]. NLRP3 contributes to renal injury in a cell-specific manner. NLRP3 is expressed in leucocytes and is related to tubular apoptosis, and NLRP3 in the kidney has been reported to interfere with the tissue repair process [95]. Several studies have indicated the critical role of NLRP3 in different types of AKI (Table 2).
IR-induced acute kidney injury
Recent studies have provided evidence that the NLRP3 inflammasome plays a crucial role in IR-induced AKI. One study showed that the NLRP3 inflammasome was activated in IR-induced AKI and peaked after 3 days of reperfusion, and mitochondrial damage and cytochrome c redistribution were tested in WT IR mice [96]. The accumulation of damaged mitochondria had a positive effect on ROS overproduction, mainly in the proximal tubules, which could further activate the NLRP3 inflammasome and induce subsequent inflammation through the mROS-TXNIP-NLRP3 pathway [97, 98]. TEC pyroptosis is one of the main pathological features of renal IRI. ROS can induce NLRP3 through TEC pyroptosis in renal IR, which in turn initiates ROS production [99]. Additionally, ATP released from necrotic cells also participates in activating the NLRP3 inflammasome. Furthermore, according to this hypothesis, NALP3 may function via downstream activation of the MAPK signalling pathway during I/R injury [73]. Inhibiting the inflammatory response protects the kidneys from further damage. In NLRP3−/− mice, kidney injury and inflammation activation were attenuated, and the deletion of NLRP3 further protected the kidney from acute tubular necrosis and apoptosis [96, 100]. Moreover, another study showed that NLRP3−/− mice but not Asc−/− or capsase-1−/− mice were protected from IRI, suggesting a direct effect of NLRP3 on TECs leading to IRI, which is independent of inflammasome formation [101]. A recent study showed that in caspase-11−/− mice, IR-induced NLRP3 inflammasome activation, renal functional deterioration, and tubular morphological changes were significantly attenuated [102]. P2X7R is associated with the release of IL-1β via activation of the NLRP3 inflammasome. P2X7R deficiency protects against renal IRI and attenuates NLRP3 inflammasome formation [103].
Notably, NLRP3 has been identified as a target for ameliorating IR-induced AKI. MitoTEMPO, a mitochondrial antioxidant, could reduce the overproduction of mROS, thereby inhibiting the activation of the NLRP3 inflammasome. By using siRNA targeting TXNIP (siTXNIP), the interaction between TXNIP and the inflammasome can be inhibited, and the levels of IL-1β and IL-18 are lowered [96]. Cathelicidin-related antimicrobial peptide (CRAMP) has been shown to have a renal protective effect against I/R-induced AKI. In addition, CRAMP deficiency in mice can exacerbate inflammatory responses and apoptosis through NLRP3 inflammasome overexpression. Thus, CRAMP exerts a renoprotective effect by suppressing the activation of the NLRP3 inflammasome and further inflammation and apoptosis [104]. This evidence suggests that NLRP3 participates in IR-induced AKI through both inflammasome-dependent and inflammasome-independent mechanisms.
Cisplatin-induced acute kidney injury
In recent years, an increasing number of studies have shown that the NLRP3 inflammasome may promote the progression of cisplatin-induced AKI. The levels of NLRP3, as well as its related factors caspase-1 and IL-1β, are significantly upregulated by cisplatin [105]. The upstream mechanisms regulating NLRP3 have also been studied. Autophagy activated by cisplatin can inhibit NLRP3 expression and inflammasome assembly [105]. The upregulation of P2X7R contributes to the development of cisplatin-induced nephrotoxicity, most likely via the NLRP3 pathway. Moreover, cancer patients are commonly prescribed proton pump inhibitors (PPIs), such as lansoprazole, for gastrointestinal complications. Evidence shows that lansoprazole promotes cisplatin-induced AKI by activating NLRP3 [106].
NLRP3 may be considered a novel therapeutic target for cisplatin-induced AKI. P2X7R blockade with the antagonist A-438079 can reduce kidney injury and oxidative stress by inhibiting the activation of NLRP3 and its downstream compounds ASC and caspase-1 in C57BL/6 mice [46]. Omeprazole, a major PPI, exerts a protective effect against cisplatin-induced damage in both HK-2 cells and rat kidneys by inhibiting the TLR4/NF-κB/NLRP3 signalling pathway [107]. Treatment with the SS-31 peptide could reduce apoptosis in cisplatin-induced AKI mice and protect the kidneys from injury by inhibiting the activation of the ROS-NLRP3-IL-1β/caspase-1 pathway in mitochondria [108]. Astragaloside IV protects against cisplatin-induced kidney injury via autophagy-mediated inhibition of NLRP3 in rats [109]. Overall, although the precise mechanisms of cisplatin-induced nephrotoxicity have yet to be elucidated, these findings provide novel insight into the role of NLRP3 in the development and progression of cisplatin-induced AKI, as well as targeting NLRP3 as a therapeutic strategy for cisplatin-induced AKI.
Sepsis-induced acute kidney injury
AKI is a major complication of sepsis. The NLRP3 inflammasome plays a significant role in the occurrence and development of sepsis-induced AKI [110, 111]. The expression of NLRP3, as well as the downstream proinflammatory cytokines IL-1β and IL-18, is elevated in LPS-induced AKI. Moreover, a recent study showed that the functional difference between M1 and M2 macrophages in sepsis-induced AKI is a result of exosome-mediated pyroptosis regulated by NLRP3 [112]. By inhibiting the NLRP3 inflammasome, kidney tissues can be protected from damage [113]. An increasing number of studies have indicated that long noncoding RNAs (lncRNAs) exert crucial effects on sepsis-induced AKI. Recently, the lncRNA RMRP was shown to facilitate inflammation by activating the NLRP3 inflammasome [114]. In addition, pyroptosis, which is induced by LPS, has been reported to be involved in sepsis-induced AKI. The lncRNA plasmacytoma variant translocation 1 is involved in regulating NLRP3-mediated pyroptosis in septic AKI by targeting miR-20a-5p [115].
It is worth noting that the NLRP3 inflammasome has been identified as a target for treating sepsis-induced kidney injury. Sirtuin1 (SIRT1), a key member of the sirtuin family, suppresses activation of the NLRP3 inflammasome, and SIRT1 overexpression increases the viability of TECs, thereby protecting against sepsis-induced AKI [110]. Hyperin alleviates the inflammatory response by inhibiting the NLRP3 signalling pathway in sepsis-induced AKI [116]. Exogenous CO released after treatment with CO-releasing molecule 2 has been shown to restore the decline in renal function in sepsis-induced AKI rats in an NLRP3 inflammasome-dependent manner; however, a detailed mechanism for this has yet to be determined [117]. Mangiferin can ameliorate oxidative stress and inflammatory responses in mice with AKI induced by caecal ligation and puncture by inhibiting NLRP3 inflammasome activation and upregulating the expression of the nuclear transcription factor Nrf2 [118]. Cluster of differentiation 39, an inflammatory inhibitor, has been reported to play a protective role in renal TEC damage by reducing NLRP3 activation and partly inhibiting the overexpression of ROS [80].
At present, studies are increasingly showing that traditional Chinese medicines or monomers have therapeutic effects on AKI. Harmine, a factor in Peganum harmala L, has been shown to protect mice against LPS-induced AKI by reducing oxidative stress and inflammation, and the underlying mechanisms may correlate with the suppression of TLR4-NF-κB and deactivation of the NLRP3 inflammasome [119]. 5-O-Methyldihydroquercetin (GS1) and cilicicone B (GS2), two traditional Chinese medicines, have protective effects on mice induced by LPS. These agents can reduce the inflammatory response and oxidative stress via the MyD88/TRIF/NLRP3 pathway [120]. These findings indicate that the NLRP3 inflammasome could be a key mediator and therapeutic target in sepsis-induced AKI.
Other types of acute kidney injury
NLRP3 also participates in the development of other types of AKI. Acute calcium oxalosis can induce AKI as a result of CaOx deposition in the distal tubule, which can not only trigger local inflammation via the NLRP3/ASC/caspase-1 axis in dendritic cells but also induce necroptosis, as well as mitochondrial permeability transition-related cell necrosis. In addition, crystals can be absorbed by TECs, causing the cells to release ATP, which can induce subsequent NLRP3 inflammasome activation [121, 122].
Rhabdomyolysis-induced AKI (RIAKI), which has the characteristics and features of muscle damage and toxic contents released into the circulation, is one of the main manifestations of community-acquired AKI [123]. NLRP3 plays a vital role in the development of RIAKI during the early phase. NLRP3 inflammasomes contribute to inflammation, apoptosis, and tubular injury in the kidneys. Initial activation of the NLRP3 inflammasome is predominantly triggered in renal tubular cells. In addition, these manifestations can be ameliorated in mice that are deficient in NLRP3 or its downstream molecules, such as ASC, caspase-1, and IL-1β [124, 125]. Anisodamine, an acetylcholine receptor antagonist, exerts protective effects against RIAKI through the ER stress-mediated TXNIP-NLRP3-ASC inflammasome axis [126].
Blood transfusion may result in an AHTR, which represents a danger to patients by damaging kidney function. High concentrations of haem in AHTR could promote NLRP3 inflammasome activation in RTECs, leading to renal dysfunction (Fig. 3) [127]. These findings highlight the importance of haem-induced NLRP3 activation in RTECs in kidney function.

Blood transfusion, a common therapy in clinic, may result in AHTR occurrence after administration of a blood product. Once AHTR occurs, a mass of heme is produced. High concentrations of heme in an AHTR could promote NLRP3 inflammasome activation in RTECs leading to renal dysfunction.
The NLRP3 inflammasome also contributes to paraquat-induced AKI regulated by NF-κB and death-associated protein kinase in rats [128]. With the widespread use of contrast media in diagnosis and treatment, contrast-induced AKI (CI-AKI) has become the third most common cause of hospital-acquired renal failure [129]. The expression of NLRP3 and its adaptor ASC was increased in vitro and in vivo. A cell-based study demonstrated that the NLRP3 inflammasome was related to apoptosis in CI-AKI. Consistently, the absence of NLRP3 can lead to the inactivation of apoptosis-associated proteins and the inhibition of IL-1β and IL-18 secretion [130]. The NLRP3 inflammasome is activated to promote kidney inflammation and apoptosis, thus participating in the development of CI-AKI via the S100A8/A9-TLR4 pathway [131]. In addition, PINK1-Parkin-related mitophagy reduces TEC apoptosis and renal injury in CI-AKI by inhibiting activation of the NLRP3 inflammasome [132]. The upregulation of microRNA-30c was shown to suppress apoptosis in CI-induced AKI by targeting NLRP3 [133]. αKlotho has been shown to exert therapeutic effects on CI-AKI and reduce contrast-induced renal tubular cell pyroptosis by inhibiting NLRP3 inflammasome activation [134]. Taken together, these findings provide strong evidence for the use of NLRP3 as a therapeutic target in these types of AKI.
NLRP3 in acute kidney injury in patients with COVID-19
Recent studies have shown that AKI occurs in 37%–56% of COVID-19 patients, of which 67% develop stage 3 AKI [135, 136]. SARS-CoV-2 causes specific dysfunction of the kidney proximal tubule by binding to the angiotensin converting enzyme 2 receptor, which is highly expressed in proximal tubule cells [137]. Another mechanism through which SARS-CoV-2 can affect the kidney is the occurrence of a cytokine storm induced by viral infection that may influence the kidney both directly and indirectly by triggering sepsis, hypoxia, shock and rhabdomyolysis [138]. In SARS-CoV-2 infection, overactivation of the NLRP3 inflammasome leads to a cytokine storm [139]. Targeting NLRP3 may be a potential therapeutic strategy for AKI in patients with COVID-19.
NLRP6 in acute kidney injury
NLRP6, a poorly characterised NLR that forms atypical inflammasomes, has recently been shown to have a nephroprotective role in AKI. During nephrotoxic kidney injury, NLRP6 expression is downregulated. NLRP6 deficiency promotes the inflammatory response and exacerbates the severity of nephrotoxic AKI by suppressing ERK1/2 and p38 mitogen-activated protein kinase (MAPK) phosphorylation, as well as the expression of the nephroprotective gene Klotho [140].
NLRs in obstructive nephropathy
Progressive renal disease is associated with tubulointerstitial inflammation and fibrosis [141]. Ureteral obstruction is characterised by reduced expression of aquaporin water channels and an inflammatory response. In unilateral ureteral obstruction (UUO) nephropathy, evidence shows that the NLRP3 inflammasome is activated and plays a vital role in disease progression.
NLRP3 has a canonical effect on mice through the NLRP3-ASC-caspase-1-IL-1β axis, which is important in renal inflammation and fibrosis [51, 54]. Consistently, NLRP3-knockout mice show reduced tubular injury, alleviated mitochondrial dysfunction, and reduced tubulointerstitial inflammation and fibrosis, and these effects correlated with the inhibition of caspase-1 activity and IL-1β/IL-18 production [51, 142]. The upregulation of IL-36 in mouse TECs induced by UUO plays a significant role in the progression of renal tubulointerstitial lesions, which further induce renal inflammation and fibrosis by activating the NLRP3 inflammasome [143]. Furthermore, ROS are a hallmark of ON and have been shown to be essential binding molecules for subsequent NLRP3 inflammasome activation [144]. Apart from the canonical effects, NLRP3 also participates in the pathophysiology of UUO in an inflammasome-independent manner. NLRP3 induces the activation of TGF-β/Smad signalling and the EMT process in renal epithelial cells via an inflammasome-independent mechanism as a further explanation for the inhibition of fibrosis in NLRP3−/− mice with UUO [63]. Intriguingly, another study reported the protective effect of NLRP3 against early tubular injury. NLRP3 mRNA expression is elevated in renal TECs and myofibroblasts, prevents interstitial oedema in the kidney, and preserves renal integrity to attenuate renal injury [141]. This controversial result may be associated with the characteristics of the different stages of ON.
Notably, NLRP3 has been viewed as a target of ON treatment. A growing number of agents protect against ON by inhibiting the NLRP3 inflammasome. For example, aliskiren (an inhibitor of renin), mefunidone (a novel synthetic compound), and fluorofenidone (a low-molecular-weight pyridine agent) downregulate NLRP3 inflammasome components in ON and further inhibit the subsequent release of IL-1β [145,146,147]. Additionally, ghrelin, cyclic helix B peptide, and H2S can alleviate renal fibrosis by suppressing the NLRP3 pathway [148,149,150]. These findings support the therapeutic potential of NLRP3 inhibition in UUO.
NLRs in diabetic nephropathy
DN is characterised by both glomerular sclerosis and tubulointerstitial fibrosis and is a major cause of ESRD [151, 152]. The mechanisms underlying the development and progression of DN are complicated, and metabolic disorders, such as hyperglycaemia and inflammation, play specific roles. Studies are increasingly demonstrating that several NLRs, including NLRP3, NOD1, and NOD2, play key roles in the pathophysiological process of DN. Notably, a large amount of evidence has been reported in relation to NLRP3.
NLRP3 in diabetic nephropathy
The expression of inflammasome-related factors, such as NLRP3, IL-18, and IL-1β, is increased in patients and mouse models of DN [153]. Emerging evidence has shown that NLRP3 inflammasome activation is a central mediator of renal inflammation in DN [154]. Knockout or inhibition of NLRP3 suppresses inflammation and fibrosis to protect the kidneys from DN. Inhibition of the NLRP3 inflammasome reduces podocyte damage by suppressing lipid accumulation in DN [155]. Relative NLRP3 mRNA expression is not only an independent predictor of DN but also a potent biomarker for identifying DN in patients with T2DM [156]. However, the mechanisms underlying NLRP3 activation in DN remain unclear. Despite this, evidence indicates that multiple molecules are involved in the regulation of NLRP3 in DN.
NLRP3 and ROS in diabetic nephropathy
Recently, various theories regarding the role of NLRP3 in DN have been suggested, including potassium efflux, lysosome rupture, and ROS overproduction. The latter is widely accepted [7, 157]. High levels of mitochondrial ROS correlate with the upregulated expression of NLRP3/IL-1β in the human tubular HK-2 cells of patients with DN and db/db mice, and this effect can be reversed by treatment with the antioxidant MitoQ, indicating that the mtROS-TXNIP/NLRP3/IL-1β axis may contribute to tubular injury, while the inhibition of this axis exerts renal protective effects [158, 159]. High glucose levels induce ROS-NLRP3 inflammasome signalling activation, partly via sweet taste receptors (STRs). This response can be reversed by the STR inhibitor lactisole [160].
NLRP3 and TXNIP in diabetic nephropathy
In addition to ROS, TXNIP activates the NALP3 inflammasome in DN. TXNIP binds with the antioxidant thioredoxin (TRX) in the physiological state. In DN, TXNIP is upregulated, and high glucose levels activate the ROS/TXNIP/NLRP3 pathway, which leads to ROS-mediated dissociation of TRX and TXNIP in mesangial cells [161]. Silencing TXNIP inhibits NLRP3 inflammasome activation via the TXNIP-NLRP3 pathway [162, 163]. Thus, TXNIP may play a pivotal role in the pathogenesis of DN through the oxidative stress pathway, and TXNIP inhibition could provide a novel therapeutic strategy for DN [164]. In addition, gp91phox, a subunit of NADPH oxidase, was upregulated in DN mice, enhancing NLRP3 and TXNIP simultaneously. Furthermore, NADPH oxidase activation is driven by TXNIP, which can trigger activation of the downstream NALP3 inflammasome in podocytes [165]. Moreover, one study reported that high glucose levels induced NLRP3 inflammasome activation through the Syk/JNK pathway. JNK activation can be inhibited by the Syk inhibitor BAY61-3606 and Syk-siRNA in HG-induced HK-2 cells and rat glomerular mesangial cells, subsequently downregulating the expression of NLRP3 and mature IL-1β [153].
NLRP3 and P2X7R in diabetic nephropathy
Additionally, evidence demonstrates that a lack of P2X7R attenuates renal inflammation, fibrosis, and NLRP3 inflammasome component expression and activation induced by a high-fat diet. These findings indicate that P2X7 may participate in renal inflammation and injury by activating the NLRP3 inflammasome [166]. P2X7R is also related to DN pathogenesis, such as podocyte injury. Ophiocordyceps sinensis, an artificially developed agent, effectively downregulates the expression of P2X7R and NLRP3 to alleviate podocyte injury [167]. In DN, high glucose activates the NLRP3 inflammasome through the ATP-P2X4 signalling pathway to promote the progression of tubulointerstitial inflammation in DN. Consistently, apyrase (extracellular ATP scavenger), suramin (P2 receptor inhibitor), TNP-ATP (P2X receptor antagonist), and 5-BDBD (P2X4 inhibitor) downregulate NLRP3 expression in DN[168,169,170].
NLRP3, pyroptosis, and lncRNAs in diabetic nephropathy
In addition, pyroptosis induced by high glucose has been reported to function in DN. Pyroptosis is involved in activating ELAVL1, a protein downstream of NLRP3, and caspase-1, which promotes the release of IL-1β and IL-18. The upregulation of the lncRNA MALAT1 and the downregulation of miR23c expression in streptozotocin-induced diabetic mice and high glucose-treated HK-2 cells abrogates the inhibition of ELAVL1 and promotes downstream NLRP3 expression and ultimately pyroptosis, leading to DN development [171]. In addition, lincRNA Gm4419 knockdown ameliorated inflammation and fibrosis in mesangial cells under high glucose conditions via the NF-κB/NLRP3 pathway, suggesting that the upregulation of GM4419 contributes to inflammation and fibrosis in mesangial cells [172]. These data suggest that lncRNAs participate in the occurrence and progression of DN by targeting the NLRP3 signalling pathway.
NLRP3 and IL-22 in diabetic nephropathy
IL-22 has anti-inflammatory effects on DN, markedly downregulates the expression of NLRP3, and further suppresses IL-1β maturation. Wang et al. showed that IL-22 gene therapy not only alleviates systemic syndromes, such as hyperglycaemia, but also inhibits renal fibrosis, the accumulation of ECM, and the proliferation of mesangial cells by suppressing the NLRP3/caspase-1/IL-1β pathway [173]. The impact of glycation end products (AGEs) on cytokines remains controversial. Advanced AGEs can promote mitochondrial ROS production and activate inflammasomes to release IL-1β in podocytes via RAGE-mediated signalling [174]. Other studies have suggested that AGEs fail to induce IL-1β release in bone marrow-derived macrophages and THP-1 cells, inhibiting NLRP3 inflammasome activation [175].
NLRP3 as a target for diabetic nephropathy treatment
An increasing number of studies have inhibited the activation of NLRP3 in the treatment of DN. Generally, targeting NLRP3 leads to the suppression of multiple signalling pathways, such as oxidative phosphorylation, and the excessive formation of ROS and NF-κB activation. For example, liquiritigenin, thrombomodulin domain 1, and tetrahydrostilbene glucoside can reduce the inflammatory response in DN mice by inhibiting the NF-κB signalling pathway, suppressing NLRP3 inflammasome activation, and enhancing Nrf2 antioxidant activity [176,177,178]. Ginsenoside compound K, minocycline, and apocynin exert protective effects against DN by inhibiting oxidative stress and the ROS/NLRP3 and NF-κB/p38 signalling pathways and stabilising Nrf2 [179,180,181].
In addition, several Chinese herbs or monomers have been shown to exert protective effects against DN by suppressing the NLRP3 signalling pathway. For example, Huangkui capsule can reduce renal tubular EMT, which is the major cause of interstitial fibrosis in DN, most likely by inhibiting NLRP3 inflammasome activation and the TLR4/NF-κB signalling pathway [182]. Piperine, luteolin, and naringin inhibit the inflammatory response in rats by suppressing NF-κB and NLRP3 activation, thus reducing podocyte apoptosis [168, 183,184,185]. Salidroside, a component of Rhodiola rosea, downregulates the level of ROS and ECM overproduction in mesangial cells treated with high glucose in rats through the TXNIP-NLRP3 inflammasome pathway [163].
These findings underscore the complex mechanisms of NLRP3 in the development of DN and suggest NLRP3 as a potential therapeutic target in DN (Table 2).
NOD1 and NOD2 in diabetic nephropathy
NOD2 is expressed in glomerular mesangial cells, endothelial cells, podocytes, TECs, and inflammatory cells [12]. It was recently discovered that NOD2 receptors also function in DN. In vitro, NOD2 receptors have been reported to induce inflammation and insulin resistance [186]. NOD2 overexpression is positively related to the severity of renal injury in biopsy specimens from patients with DN. Recent studies indicate that glomerular endothelial cells (GEnCs) play a pivotal role in restricting the development of proteinuria and further renal dysfunction [187]. NOD2 activation can promote the endothelial-to-mesenchymal transition (EndMT) in GEnCs via the MEK/ERK signalling pathway. NOD2 knockdown drastically diminished high glucose-induced EndMT [188]. NOD2 deficiency can attenuate the high glucose-induced downregulation of nephrin, which is vital for maintaining podocyte integrity [186]. Furthermore, NOD1 receptors contribute to the pathogenesis and development of DN via the NOD1-RICK-NF-κB inflammatory signalling pathway, which plays a critical role in the progression of insulin resistance [189]. Consistently, NOD1 is involved in metabolic inflammation and insulin resistance induced by obesity, and NOD1 deficiency protects against high-fat diet-induced insulin resistance [190, 191]. Based on these findings, therapies based on NOD1 may provide a therapeutic benefits.
NLRs in lupus nephritis
The occurrence and development of LN, a serious complication of systemic lupus erythematosus, is thought to be facilitated by inflammation. Evidence shows that the NLRP3 inflammasome in podocytes participates in LN development, contributing to cell injury and proteinuria [192,193,194]. Furthermore, a positive relationship between NLRP3 activation and disease activity index has recently been demonstrated [195]. The NLRP3 inflammasome promotes the production of inflammatory cytokines, such as IL-1β and IL-18, which are increased in the renal tissues of MRL/lpr mice. Indeed, the upstream signalling pathways that regulate NLRP3 expression in LN have been extensively studied. The NLRP3 inflammasome is activated in podocytes via the RIP3-dependent pathway, and the inhibition of RIP3 slows disease progression. Studies have reported that glycogen synthase kinase 3β and P2X7R activation aggravate the development and progression of LN by activating the NLRP3/IL-1β pathway [196,197,198]. These data indicate that the NLRP3 inflammasome is involved in the pathophysiology of LN.
The overexpression of A20 (also known as tumour necrosis factor alpha-induced protein 3) has been reported to inhibit immune complex deposition and the inflammatory response in LN induced by pristane by blunting the NF-κB/NLRP3 pathway in mice [199]. A recent study showed that FcγRI silencing attenuated LN injury by suppressing NF-κB-regulated NLRP3 inflammasome activation and reducing NLRP3 inflammasome-associated inflammatory cytokines in the kidneys of MRL/lpr mice [200]. Blockade of the AMPK pathway further inhibited the activation of NLRP3, exacerbating pyroptosis in TECs and LN progression [201].
To date, NLRP3 has been viewed as a target for alleviating LN severity. For example, the anaesthetic isoflurane reduces renal IC deposition, as well as the level of BUN, proteinuria, and macrophage infiltration, via NLRP3 activation and inflammasome formation [202]. Xenon, an inert anaesthetic gas, decreases neutrophil infiltration in the glomerulus and thus protects the kidneys from injury by inhibiting activation of the NF-κB/NLRP3 inflammasome [203]. In addition, tris dipalladium has been shown to alleviate tubulointerstitial inflammation and restore renal function by suppressing MAPK (ERK, JNK)-mediated NLRP3 activation, as well as the autophagy/NLRP3 pathway [204]. Procyanidin B2, icariin, and Bay11-7082 significantly inhibited LN development in MRL/lpr mice by suppressing NLRP3 inflammasome activation [205, 206]. Thus, NLRP3 inhibitors are important therapeutic tools that should be further developed to prevent the progression of LN.
NLRs in other kidney diseases
NLRs in IgA nephropathy
IgA nephropathy (IgAN), which is characterised by IgA1 immune complex deposition in the glomerular mesangium, is one of the most common types of primary glomerulonephritis with chronic, progressive characteristics, and 20%–40% of patients progress to ESRD within 20 years [207, 208]. NLRP3 expression was significantly increased in a mouse model and in patients with IgAN. Mechanistically, IgA immune complexes contribute to mitochondrial dysfunction and the overproduction of mROS by activating the NLRP3 inflammasome in macrophages. NLRP3 deficiency protects against renal injury in IgAN [209]. In addition, IgA1 could induce NLRP3 expression in podocytes and macrophage transdifferentiation in podocytes, resulting in a subsequent inflammatory response and renal fibrosis in IgAN [210].
NLRP3 is regarded as a potential therapeutic target for alleviating the progression of IgAN. For example, osthole and antroquinonol, which are isolated from traditional Chinese medicines, play protective roles in IgAN development by inhibiting ROS production and NLRP3 inflammasome activation [211, 212]. Resveratrol alleviates sclerosis and inflammation in glomeruli by inhibiting mitochondrial damage and NLRP3 inflammasome activation [213]. Compound K, a metabolite of intestinal bacteria, serves as a therapeutic agent for IgAN by enhancing autophagy-mediated NLRP3 inflammasome inhibition [214].
According to recent studies, NLRP3 is predominantly expressed in primary renal tubular cells (PRTCs). NLRP3 expression is transiently induced in PRTCs by TGF-β1 but decreases over time as cells lose their epithelial phenotype in vitro. Surprisingly, lower levels of NLRP3 mRNA expression have been associated with a worse prognosis in IgAN patients. This outcome may be due to the loss of the tubular epithelial phenotype and cell death associated with decreased NLRP3 mRNA and protein levels in the tubules. The loss of NLRP3 in advancing IgAN seems plausible in progressive tubular atrophy and renal fibrosis, although the functional role of NLRP3 in IgAN pathogenesis has yet to be fully elucidated [215].
NLRs in diet-induced nephropathy
Chronic kidney disease can be initiated by related metabolic syndromes. NLRX1, a member of the NLR family that is localised specifically in mitochondria and lacks the necessary domain to form inflammasomes, can induce ROS generation and affect NF-κB- and JNK-dependent signalling [49]. Diet-induced renal dysfunction can be prevented by NLRX1 deletion in mice. The absence of NLRX1 prevents high plasma creatinine compared with that in Western diet (WD)-fed WT mice and prevents hyperuricaemia after WD feeding compared to control feeding. Tubular PLIN2 surrounding neutral lipid droplets in NLRX1 KO kidneys was reduced compared to WT kidneys. It has been suggested that the prevention of WD-associated nephropathy in NLRX1 deletion plays a protective role in WD-associated nephropathy by inhibiting renal lipid accumulation and toxicity [216]. In addition, the presence of NLRP3 is pivotal for the development of WD-induced nephropathy, most likely via macrophage infiltration and cholesterol accumulation, which could be reversed by NLRP3-knockout [217].
NLRs in crystal nephropathy
Apart from cytotoxicity, cystine crystals, which accumulate in the lysosome, can induce IL-1β secretion via the NLRP3 inflammasome, indicating a novel pathway for the pathogenesis of nephropathic cystinosis [218]. Crystals can also induce inflammation by activating NLRP3 and inducing the subsequent secretion of IL-1β in dendritic cells. The TNF receptor pathway, as well as RIPK3/MLKL, have been reported to activate the NLRP3 inflammasome, eliciting a subsequent renal inflammatory response. Furthermore, certain cytokines can induce necrosis in renal tubules [219]. CaOx crystals can trigger innate immunity via the release of IL-1β through the NLRP3/ASC/caspase-1 axis in mononuclear phagocytes [122]. In addition, uromodulin can be deposited in the tubule to form nanoparticles, which could function as an endogenous danger signal to activate the NLRP3 inflammasome and its downstream molecule ASC. These reactions should take place when crystals are taken up into lysosomes of monocytes and dendritic cells, leading to cathepsin leakage, oxidative stress, and potassium efflux [220].
NLRs in HIV-associated nephropathy
Podocyte dysfunction plays a vital role in the development of HIV-associated nephropathy (HIVAN) [221]. HIV can mediate podocyte pyroptosis and damage T lymphocytes by promoting NLRP3 inflammasome formation and the activation of the downstream molecule caspase-1. The underlying mechanism may be related to the generation of ROS and K+ efflux, since this effect can be partly attenuated by Tempol (an antioxidant) and glyburide (potassium efflux inhibitor) [222]. Further study of this mechanism will help to improve our understanding of HIV and provide a potential new strategy for HIVAN in the future.
NLRs in uric acid nephropathy
UAN is characterised by the overproduction of uric acid, which results in hyperuricaemia. Urate crystal deposition-mediated activation of the NLRP3 inflammasome and IL-1β release play critical roles in the development and progression of UAN [223, 224]. The lncRNA ANRIL can participate in the pathogenesis of UAN via the miR-122-5p/BRCC3 pathway to further activate the NLRP3 inflammasome [225]. Weicao capsule, a Chinese herbal, can protect against kidney injury by promoting autophagy and inhibiting NLRP3-induced inflammation [226]. G31P, an antagonist of CXCR1/CXCR2, has been shown to alleviate the inflammatory response and inhibit inflammatory progression in UAN by inhibiting NLRP3 activation [227]. These findings suggest that NLRP3 inhibition may be a treatment for UAN.
NLRs in obstructive sleep apnoea-associated CKD
Chronic intermittent hypoxia, which is a major feature of the pathogenesis of obstructive sleep apnoea (OSA), can lead to oxygen deficiency in the kidney, accelerating local ROS generation, and this accumulation can further trigger NLRP3 activation and the subsequent release of the inflammatory cytokine IL-1β. Furthermore, it has been demonstrated that there may be positive feedback between miR-155 and NLRP3. Consistently, the overexpression of miR-155 can enhance the activation of NLRP3 by inhibiting the FOXO3a gene [228]. This finding provides novel insights into NLRP3 blockade as an OSA-related CKD treatment.
NLRs in renal cell carcinoma
RCC has a high rate of incidence and mortality, ranking second in the uropoietic system [229]. In 75% of cases, clear-cell renal cell carcinoma (ccRCC) is the most commonly diagnosed histological type among RCCs and is related to dysfunction of the von Hippel Lindau gene [230, 231]. Evidence is increasingly demonstrating that NLRs play important roles in RCC.
NLRP3 in renal cell carcinoma
The role of NLRP3 in the progression and development of RCC remains controversial. NLRP3 expression is elevated in human RCC tissues [232]. Consistently, NLRP3, caspase-1, and IL-1β expression was increased in ccRCC in a pilot study [233]. Notably, NLRP3 is considered to be a potential target for RCC treatment. Resveratrol suppresses tumour cell proliferation, migration, and invasion by inhibiting NLRP3 expression [232].
Interestingly, contradictory results have been reported by other groups. The expression of NLRP3 in ccRCC cancer tissues is dramatically inhibited, and NLRP3 may be a factor for suppressing tumour growth in RCC [234]. Pyroptosis mediated by caspase-1 is involved in inhibiting the formation and development of tumours. The inhibition of BRD4 has an antitumour effect on RCC via NF-κB-NLRP3-caspase-1-mediated pyroptosis [235]. Liver X receptors (LXRα), which can regulate ccRCC metastasis by suppressing the NLRP3 inflammasome, could be a biomarker for diagnosing and evaluating the prognosis of ccRCC [234]. These findings clearly demonstrate that NLRP3 plays an antitumour role in RCC.
NOD1 and NOD2 in renal cell carcinoma
NOD1 and NOD2 have been found to play roles in cancer. Sequencing data revealed that the NOD2 gene has a close relationship with the prognosis of kidney cancer patients, indicating the potential of NOD2 as a biomarker for the survival of kidney cancer. However, the existing association between NOD2 and kidney cancer requires further investigation [236]. The different levels of NOD1 and NOD2 expression in specific cell types point towards diverse pathways associated with carcinogenesis [237].
NLRC5 in renal cell carcinoma
An analysis of human clinical data revealed that NLRC5 can promote the proliferation, migration, and invasion of tumour cells via the Wnt/β-catenin signalling pathway. Moreover, evidence has demonstrated that NLRC5 depletion suppresses the proliferation, migration, and invasion of ccRCC cells. In contrast, NLRC5 overexpression may promote these behaviours [238]. These findings indicate that NLRC5 is a potential therapeutic target for ccRCC.
NLR pathway-targeted pharmacological agents
NLR inflammasomes, which are potent contributors to the activation of inflammatory cytokines, have been confirmed to play general roles in the pathogenesis and progression of kidney diseases. Considering the contribution of inflammasomes in the progression of renal diseases, inflammasome inhibitors seem to have a promising future in the treatment and prevention of renal diseases. An increasing number of recent studies have verified the efficacy of NLRP3 inhibitors on multiple renal diseases. For example, the diarylsulfonylurea-based compound CP-456,773, also known as CRID3 and MCC950, is a selective NLRP3 inhibitor [239]. This compound markedly reduces IL-1β and IL-18 and strongly attenuates renal fibrosis, attenuating crystal-induced kidney fibrosis and acute allograft damage in rat kidney transplants [240, 241]. MCC950, which blocks the ATPase domain of NLRP3, attenuates apoptosis in CI-AKI by upregulating HIF1A and BNIP3-mediated mitophagy [242, 243]. Increased levels of APOL1 risk variants are an important trigger for APOL1-associated kidney disease development [244].
Glyburide is a sulfonylurea drug that is widely used for the treatment of type 2 diabetes. This drug has a specific inhibitory effect on the NLRP3 inflammasome [245, 246]. However, the doses of glyburide used in vivo to inhibit NLRP3 are very high, which results in hypoglycaemia, so it is limited to treating T2D only [247]. JC124, a novel small molecule, has been confirmed to ameliorate the hypoglycaemic effect of glyburide and exerts protective effects against various inflammatory diseases [248].
Colchicine has been verified to inhibit multiple inflammasome pathways and is used to treat gout, coronary artery disease and other inflammatory and fibrotic diseases. However, a high dose of colchicine is required to inhibit the NLRP3 inflammasome, and the use of this treatment in patients with renal insufficiency may be limited by the risk of colchicine toxicity because of the increased half-life of the drug [249, 250].
To date, immunomodulatory strategies targeting inflammasomes have focused mainly on the downstream effectors IL-1 and caspase-1. For example, anakinra (recombinant IL-1Ra) targets IL-1 and is effective in treating gout flares in patients with advanced CKD and diabetes [251, 252].
In summary, small molecule inhibitors targeting NLRP3 and other inflammasome components have been considered potential therapeutic agents through findings in experimental models of kidney diseases and in the clinic [5]. Nevertheless, future studies are needed to determine the exact potential of these NLR inhibitors and may focus on the development of specific and novel NLR inflammasome inhibitors with improved pharmacokinetic properties that are more cost-effective than currently available treatments.
Conclusion
Due to the diverse functions of NLRs, understanding their activation and regulation presents new opportunities to modulate histological damage, the inflammatory response, and apoptosis in renal diseases. The exploration of abundant NLRs has expanded our understanding of their roles in kidney diseases. NLRs are differentially expressed in a variety of renal tissues, and upstream (Table 3) and downstream (Table 4) signalling pathways of NLRs are involved in renal disease. These findings suggest that NLRs may exert important physiological and pathological effects. Increasingly, evidence suggests that targeting NLRs is a new strategy for the treatment of numerous kidney diseases, including AKI, ON, DN, RCC, IgAN, UAN, LN, and crystal nephropathy. In recent years, research on the functions of NLRs in renal diseases has increased exponentially. However, several challenges remain to be addressed. For example, while NLRP3 is a well-studied NLR in many renal diseases, the interplay between NLRP3 and other NLRs remains a topic of uncertainty. Further investigation is also needed to elucidate the inhibitory effects of certain NLRs, such as NLRX1 and NLRP6. In addition, the function of NLRC5 in renal diseases remains controversial. Studying the different roles of NLRs in renal diseases will not only increase our understanding of the pathological processes of the kidney but also provide further evidence that targeting NLRs is a viable strategy for the treatment of renal diseases.
Data availability
The data used to support the findings of this study are included within the article.
References
Leemans JC, Kors L, Anders HJ, Florquin S. Pattern recognition receptors and the inflammasome in kidney disease. Nat Rev Nephrol. 2014;10:398–414.
Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, De Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36:464–76.
Anders HJ, Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol. 2014;25:1387–400.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22.
Komada T, Muruve DA. The role of inflammasomes in kidney disease. Nat Rev Nephrol. 2019;15:501–20.
Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22:1007–18.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–7.
Meunier E, Broz P. Evolutionary convergence and divergence in NLR function and structure. Trends Immunol. 2017;38:744–57.
Keestra-Gounder AM, Tsolis RM. NOD1 and NOD2: beyond peptidoglycan sensing. Trends Immunol. 2017;38:758–67.
Lupfer C, Kanneganti TD. Unsolved mysteries in NLR biology. Front Immunol. 2013;4:285.
Shigeoka AA, Kambo A, Mathison JC, King AJ, Hall WF, Da Silva Correia J, et al. Nod1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury. J Immunol. 2010;184:2297–304.
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72.
Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jéhanno M, Viala J, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300:1584–7.
Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4:702–7.
Cfranchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106–28.
Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62.
Askari N, Correa RG, Zhai D, Reed JC. Expression, purification, and characterization of recombinant NOD1 (NLRC1): a NLR family member. J Biotechnol. 2012;157:75–81.
Inohara C, Mcdonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–83.
Kang JW, Kim SJ, Cho HI, Lee SM. DAMPs activating innate immune responses in sepsis. Ageing Res Rev. 2015;24:54–65.
Wiersinga WJ, Leopold SJ, Cranendonk DR, Van Der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5:36–44.
Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–86.
Wan X, Xu C, Lin Y, Lu C, Li D, Sang J, et al. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J Hepatol. 2016;64:925–32.
Denoble AE, Huffman KM, Stabler TV, Kelly SJ, Hershfield MS, Mcdaniel GE, et al. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc Natl Acad Sci USA. 2011;108:2088–93.
Cui D, Liu S, Tang M, Lu Y, Zhao M, Mao R, et al. Phloretin ameliorates hyperuricemia-induced chronic renal dysfunction through inhibiting NLRP3 inflammasome and uric acid reabsorption. Phytomedicine. 2020;66:153111.
Wu J, Luo Y, Jiang Q, Li S, Huang W, Xiang L, et al. Coptisine from Coptis chinensis blocks NLRP3 inflammasome activation by inhibiting caspase-1. Pharmacol Res. 2019;147:104348.
Banoth B, Cassel SL. Mitochondria in innate immune signaling. Transl Res. 2018;202:52–68.
Zeng CY, Li CG, Shu JX, Xu LH, Ouyang DY, Mai FY, et al. ATP induces caspase-3/gasdermin E-mediated pyroptosis in NLRP3 pathway-blocked murine macrophages. Apoptosis. 2019;24:703–17.
Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66:1037–46.
Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. 2018;122:1722–40.
Park CH, Lee HS, Kwak MS, Shin JS. Inflammasome-dependent peroxiredoxin 2 secretion induces the classical complement pathway activation. Immune Netw. 2021;21:e36.
Maharana J, Sahoo BR, Bej A, Jena I, Parida A, Sahoo JR, et al. Structural models of zebrafish (Danio rerio) NOD1 and NOD2 NACHT domains suggest differential ATP binding orientations: insights from computational modeling, docking and molecular dynamics simulations. PLoS One. 2015;10:e0121415.
Wu M, Guo L, Zhu KC, Guo HY, Liu B, Jiang SG, et al. Genomic structure and molecular characterization of Toll-like receptors 1 and 2 from golden pompano Trachinotus ovatus (Linnaeus, 1758) and their expression response to three types of pathogen-associated molecular patterns. Dev Comp Immunol. 2018;86:34–40.
Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003;124:140–6.
Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol. 2006;7:569–75.
Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA. 2010;107:3076–80.
Tenthorey JL, Chavez RA, Thompson TW, Deets KA, Vance RE, Rauch I. NLRC4 inflammasome activation is NLRP3- and phosphorylation-independent during infection and does not protect from melanoma. J Exp Med. 2020;217:e20191736.
Benko S, Magalhaes JG, Philpott DJ, Girardin SE. NLRC5 limits the activation of inflammatory pathways. J Immunol. 2010;185:1681–91.
Downs I, Vijayan S, Sidiq T, Kobayashi KS. CITA/NLRC5: a critical transcriptional regulator of MHC class I gene expression. Biofactors. 2016;42:349–57.
Li Q, Wang Z, Zhang Y, Zhu J, Li L, Wang X, et al. NLRC5 deficiency protects against acute kidney injury in mice by mediating carcinoembryonic antigen-related cell adhesion molecule 1 signaling. Kidney Int. 2018;94:551–66.
Triantafilou K. Enigmatic inflammasomes. Immunology. 2021;162:249–51.
Feng F, Cheng Q, Li B, Liu C, Wang H, Li B, et al. Establishment and characterization of 38 novel patient-derived primary cancer cell lines using multi-region sampling revealing intra-tumor heterogeneity of gallbladder carcinoma. Hum Cell. 2021;34:918–31.
Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265:106–9.
Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol. 2012;13:823–31.
Uchimura T, Oyama Y, Deng M, Guo H, Wilson JE, Rampanelli E, et al. The innate immune sensor NLRC3 acts as a rheostat that fine-tunes T cell responses in infection and autoimmunity. Immunity. 2018;49:1049–61.
Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity. 2014;40:329–41.
Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. 2012;488:389–93.
Stokman G, Kors L, Bakker PJ, Rampanelli E, Claessen N, Teske GJD, et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J Exp Med. 2017;214:2405–20.
Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci. 2009;122:3161–8.
Imbeault E, Mahvelati TM, Braun R, Gris P, Gris D. Nlrx1 regulates neuronal cell death. Mol Brain. 2014;7:90.
Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010;21:1732–44.
Ratajczak MZ, Bujko K, Cymer M, Thapa A, Adamiak M, Ratajczak J, et al. The Nlrp3 inflammasome as a “rising star” in studies of normal and malignant hematopoiesis. Leukemia. 2020;34:1512–23.
Agostini L, Martinon F, Burns K, Mcdermott MF, Hawkins PN, Tschoppj. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–25.
Lorenz G, Darisipudi MN, Anders HJ. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol Dial Transpl. 2014;29:41–8.
Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA. 2009;106:20388–93.
Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, et al. ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis. 2012;3:e403.
Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, et al. NLRP3/cryopyrin Is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem. 2009;284:12762–71.
Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;284:24035–48.
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287:36617–22.
Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–8.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Wang W, Wang X, Chun J, Vilaysane A, Clark S, French G, et al. Inflammasome-independent NLRP3 augments TGF-β signaling in kidney epithelium. J Immunol. 2013;190:1239–49.
Anders HJ, Lech M. NOD-like and Toll-like receptors or inflammasomes contribute to kidney disease in a canonical and a non-canonical manner. Kidney Int. 2013;84:225–8.
Fan JJ, Gao B, Song AQ, Zhu YJ, Zhou J, Li WZ, et al. Spinal cord NLRP1 inflammasome contributes to dry skin induced chronic itch in mice. J Neuroinflammation. 2020;17:122.
Ghimire L, Paudel S, Jin L, Jeyaseelan S. The NLRP6 inflammasome in health and disease. Mucosal Immunol. 2020;13:388–98.
Zhang Q, Sun Y, He Z, Xu Y, Li X, Ding J, et al. Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain Behav Immun. 2020;S0889-1591:30075–1.
Vladimer GI, Weng D, Paquette SW, Vanaja SK, Rathinam VA, Aune MH, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37:96–107.
Chen H, Deng Y, Gan X, Li Y, Huang W, Lu L, et al. NLRP12 collaborates with NLRP3 and NLRC4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol Neurodegener. 2020;15:26.
Su MY, Kuo CI, Chang CF, Chang CI. Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS One. 2013;8:e67843.
Li ZG, Shui SF, Han XW, Yan L. NLRP10 ablation protects against ischemia/reperfusion-associated brain injury by suppression of neuroinflammation. Exp Cell Res. 2020;389:111912.
Singh DP, Kaur G, Bagam P, Pinkston R, Batra S. Membrane microdomains regulate NLRP10- and NLRP12-dependent signalling in A549 cells challenged with cigarette smoke extract. Arch Toxicol. 2018;92:1767–83.
Hao JL, Li YF, Li RS. A novel mechanism of NALP3 inducing ischemia reperfusion injury by activating MAPK pathway in acute renal failure. Med Hypotheses. 2013;80:463–5.
Hoste EAJ, Kellum JA, Selby NM, Zarbock A, Palevsky PM, Bagshaw SM, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol. 2018;14:607–25.
Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371:58–66.
Gao L, Zhong X, Jin J, Li J, Meng XM. Potential targeted therapy and diagnosis based on novel insight into growth factors, receptors, and downstream effectors in acute kidney injury and acute kidney injury-chronic kidney disease progression. Signal Transduct Target Ther. 2020;5:9.
Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–21.
Guo Y, Zhang J, Lai X, Chen M, Guo Y. Tim-3 exacerbates kidney ischaemia/reperfusion injury through the TLR-4/NF-κB signalling pathway and an NLR-C4 inflammasome activation. Clin Exp Immunol. 2018;193:113–29.
Wang JN, Yang Q, Yang C, Cai YT, Xing T, Gao L, et al. Smad3 promotes AKI sensitivity in diabetic mice via interaction with p53 and induction of NOX4-dependent ROS production. Redox Biol. 2020;32:101479.
Yang M, Lu L, Kang Z, Ma T, Wang Y. Overexpressed CD39 mitigates sepsis‑induced kidney epithelial cell injury via suppressing the activation of NLR family pyrin domain containing 3. Int J Mol Med. 2019;44:1707–18.
Wang JN, Liu MM, Wang F, Wei B, Yang Q, Cai YT, et al. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin Sci. 2019;133:1609–27.
Liu X-Q, Jin J, Li Z, Jiang L, Dong Y-H, Cai Y-T, et al. Rutaecarpine derivative Cpd-6c alleviates acute kidney injury by targeting PDE4B, a key enzyme mediating inflammation in cisplatin nephropathy. Biochem Pharmacol. 2020;180:114132.
Turner MD, Chaudhry A, Nedjai B. Tumour necrosis factor receptor trafficking dysfunction opens the TRAPS door to pro-inflammatory cytokine secretion. Bioscience. 2011;32:105–12.
Mulay SR, Honarpisheh MM, Foresto-Neto O, Shi C, Desai J, Zhao ZB, et al. Mitochondria permeability transition versus necroptosis in oxalate-induced AKI. J Am Soc Nephrol. 2019;30:1857–69.
Rabb H, Griffin MD, Mckay DB, Swaminathan S, Pickkers P, Rosner MH, et al. Inflammation in AKI: current understanding, key questions, and knowledge gaps. J Am Soc Nephrol. 2016;27:371–9.
Masumoto J, Yang K, Varambally S, Hasegawa M, Tomlins SA, Qiu S, et al. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med. 2006;203:203–13.
Zhou M, Tang W, Fu Y, Xu X, Wang Z, Lu Y, et al. Progranulin protects against renal ischemia/reperfusion injury in mice. Kidney Int. 2015;87:918–29.
Jiang G, Xin R, Yuan W, Zhang L, Meng X, Sun W, et al. Ligustrazine ameliorates acute kidney injury through downregulation of NOD2‑mediated inflammation. Int J Mol Med. 2020;45:731–42.
Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–7.
Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity. 2011;34:843–53.
Zhang P, Yu C, Yu J, Li Z, Lan H-Y, Qin Z. Arid2-IR promotes NF-κB-mediated renal inflammation by targeting NLRC5 transcription. Cell Mol Life Sci. 2021;78:2387–404.
Han F, Gao Y, Ding CG, Xia XX, Wang YX, Xue WJ, et al. Knockdown of NLRC5 attenuates renal I/R injury in vitro through the activation of PI3K/Akt signaling pathway. Biomed Pharmacother. 2018;103:222–7.
Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc Natl Acad Sci USA. 2010;107:13794–9.
Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF. Enhanced TLR-induced NF-κB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res. 2012;22:822–35.
Bakker PJ, Butter LM, Claessen N, Teske GJ, Sutterwala FS, Florquin S, et al. A tissue-specific role for Nlrp3 in tubular epithelial repair after renal ischemia/reperfusion. Am J Pathol. 2014;184:2013–22.
Wen Y, Liu YR, Tang TT, Pan MM, Xu SC, Ma KL, et al. mROS-TXNIP axis activates NLRP3 inflammasome to mediate renal injury during ischemic AKI. Int J Biochem Cell Biol. 2018;98:43–53.
Hall AM, Rhodes GJ, Sandoval RM, Corridon PR, Molitoris BA. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013;83:72–83.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5.
Wang R, Zhao H, Zhang Y, Zhu H, Su Q, Qi H, et al. Identification of MicroRNA-92a-3p as an essential regulator of tubular epithelial cell pyroptosis by targeting Nrf1 via HO-1. Front Genet. 2021;11:616947.
Kim HJ, Lee DW, Ravichandran K, O Keys D, Akcay A, Nguyen Q, et al. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther. 2013;346:465–72.
Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, et al. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol. 2010;185:6277–85.
Yin F, Zheng P-Q, Zhao L-Q, Wang Y-Z, Miao N-J, Zhou Z-L, et al. Caspase-11 promotes NLRP3 inflammasome activation via the cleavage of pannexin1 in acute kidney disease. Acta Pharmacol Sin. 2021;43:86–95.
Qian Y, Qian C, Xie K, Fan Q, Yan Y, Lu R, et al. P2X7 receptor signaling promotes inflammation in renal parenchymal cells suffering from ischemia-reperfusion injury. Cell Death Dis. 2021;12:132.
Pan LL, Liang W, Ren Z, Li C, Chen Y, Niu W, et al. Cathelicidin-related antimicrobial peptide protects against ischaemia reperfusion-induced acute kidney injury in mice. Br J Pharmacol. 2020;177:2726–42.
Qu X, Gao H, Tao L, Zhang Y, Zhai J, Song Y, et al. Autophagy inhibition-enhanced assembly of the NLRP3 inflammasome is associated with cisplatin-induced acute injury to the liver and kidneys in rats. J Biochem Mol Toxicol. 2018;33:e22208.
Ye L, Pang W, Huang Y, Wu H, Huang X, Liu J, et al. Lansoprazole promotes cisplatin-induced acute kidney injury via enhancing tubular necroptosis. J Cell Mol Med. 2021;25:2703–13.
Gao H, Wang X, Qu X, Zhai J, Tao L, Zhang Y, et al. Omeprazole attenuates cisplatin-induced kidney injury through suppression of the TLR4/NF-κB/NLRP3 signaling pathway. Toxicology. 2020;440:152487.
Yang SK, Han YC, He JR, Yang M, Zhang W, Zhan M, et al. Mitochondria targeted peptide SS-31 prevent on cisplatin-induced acute kidney injury via regulating mitochondrial ROS-NLRP3 pathway. Biomed Pharmacother. 2020;130:110521.
Qu X, Gao H, Tao L, Zhang Y, Zhai J, Sun J, et al. Astragaloside IV protects against cisplatin-induced liver and kidney injury via autophagy-mediated inhibition of NLRP3 in rats. J Toxicol Sci. 2019;44:167–75.
Gao Q, Zhu H. The overexpression of Sirtuin1 (SIRT1) alleviated lipopolysaccharide (LPS)-induced acute kidney injury (AKI) via inhibiting the activation of nucleotide-binding oligomerization domain-like receptors (NLR) family pyrin domain containing 3 (NLRP3) inflammasome. Med Sci Monit. 2019;25:2718–26.
Gonçalves GM, Zamboni DS, Câmara NO. The role of innate immunity in septic acute kidney injuries. Shock. 2010;34:22–6.
Juan C-X, Mao Y, Cao Q, Chen Y, Zhou L-B, Li S, et al. Exosome-mediated pyroptosis of miR-93-TXNIP-NLRP3 leads to functional difference between M1 and M2 macrophages in sepsis-induced acute kidney injury. J Cell Mol Med. 2021;25:4786–99.
Deng H, Chen F, Wang Y, Jiang H, Dong Z, Yuan B, et al. The role of activated NLRP3 inflammatory body in acute kidney injury in rats caused by sepsis and NLRP3-TXNIP signaling pathway. Saudi J Biol Sci. 2020;27:1251–9.
Zhang X, Huang Z, Wang Y, Wang T, Li J, Xi P. Long non-coding RNA RMRP contributes to sepsis-induced acute kidney injury. Yonsei Med J. 2021;62:262–73.
Zheng Y, Li Z, Yin M, Gong X. Heme oxygenase‑1 improves the survival of ischemic skin flaps (Review). Mol Med Rep. 2021;23:1.
Chunzhi G, Zunfeng L, Chengwei Q, Xiangmei B, Jingui Y. Hyperin protects against LPS-induced acute kidney injury by inhibiting TLR4 and NLRP3 signaling pathways. Oncotarget. 2016;7:82602–8.
Wang P, Huang J, Li Y, Chang R, Wu H, Lin J, et al. Exogenous carbon monoxide decreases sepsis-induced acute kidney injury and inhibits NLRP3 inflammasome activation in rats. Int J Mol Sci. 2015;16:20595–608.
He L, Peng X, Zhu J, Chen X, Liu H, Tang C, et al. Mangiferin attenuate sepsis-induced acute kidney injury via antioxidant and anti-inflammatory effects. Am J Nephrol. 2014;40:441–50.
Niu X, Yao Q, Li W, Zang L, Li W, Zhao J, et al. Harmine mitigates LPS-induced acute kidney injury through inhibition of the TLR4-NF-κB/NLRP3 inflammasome signalling pathway in mice. Eur J Pharmacol. 2019;849:160–9.
Zeng M, Qi M, Wang Y, Xu R, Wu Y, Li M, et al. 5-O-methyldihydroquercetin and cilicicone B isolated from Spina Gleditsiae ameliorate lipopolysaccharide-induced acute kidney injury in mice by inhibiting inflammation and oxidative stress via the TLR4/MyD88/TRIF/NLRP3 signaling pathway. Int Immunopharmacol. 2020;80:106194.
Mulay SR, Desai J, Kumar SV, Eberhard JN, Thomasova D, Romoli S, et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat Commun. 2016;7:10274.
Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest. 2013;123:236–46.
Chatzizisis YS, Misirli G, Hatzitolios AI, Giannoglou GD. The syndrome of rhabdomyolysis: complications and treatment. Eur J Intern Med. 2008;19:568–74.
Komada T, Usui F, Kawashima A, Kimura H, Karasawa T, Inoue Y, et al. Role of NLRP3 inflammasomes for rhabdomyolysis-induced acute kidney injury. Sci Rep. 2015;5:10901.
Song SJ, Kim SM, Lee SH, Moon JY, Hwang HS, Kim JS, et al. Was ameliorated in NLRP3 KO mice via alleviation of mitochondrial lipid peroxidation in renal tubular cells. Int J Mol Sci. 2020;21:8564.
Yuan X, Zheng Y, Chen C, Wang C. Anisodamine inhibits endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation in rhabdomyolysis-induced acute kidney injury. Apoptosis. 2017;22:1524–31.
Liu Z, Chen Y, Niu B, Yin D, Feng F, Gu S, et al. NLRP3 inflammasome of renal tubular epithelial cells induces kidney injury in acute hemolytic transfusion reactions. Clin Transl Med. 2021;11:e373.
Liu Z, Wang X, Wang Y, Zhao M. NLRP3 inflammasome activation regulated by NF-κB and DAPK contributed to paraquat-induced acute kidney injury. Immunol Res. 2017;65:687–98.
Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. Am J Kidney Dis. 2002;39:930–6.
Shen J, Wang L, Jiang N, Mou S, Zhang M, Gu L, et al. NLRP3 inflammasome mediates contrast media-induced acute kidney injury by regulating cell apoptosis. Sci Rep. 2016;6:34682.
Tan X, Zheng X, Huang Z, Lin J, Xie C, Yan L. Involvement of S100A8/A9-TLR4-NLRP3 inflammasome pathway in contrast-induced acute kidney injury. Cell Physiol Biochem. 2017;43:209–22.
Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26:101254.
Xu J, Ma L, Ping F. MicroRNA-30c attenuates contrast-induced acute kidney injury by suppressing NLRP3 inflammasome. Int Immunopharmacol. 2020;87:106457.
Zhu X, Li S, Lin Q, Shao X, Wu J, Zhang W, et al. αKlotho protein has therapeutic activity in contrast-induced acute kidney injury by limiting NLRP3 inflammasome-mediated pyroptosis and promoting autophagy. Pharmacol Res. 2021;167:105531.
Glowacka M, Lipka S, Mlynarska E, Franczyk B, Rysz J. Acute kidney injury in COVID-19. Int J Mol Sci. 2021;22:8081.
Costa RLD, Soria TC, Salles EF, Gerecht AV, Corvisier MF, Menezes MAM, et al. Acute kidney injury in patients with Covid-19 in a Brazilian ICU: incidence, predictors and in-hospital mortality. J Bras Nefrol. 2021;43:349–58.
Werion A, Belkhir L, Perrot M, Schmit G, Aydin S, Chen Z, et al. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 2020;98:1296–307.
Buonaguro FM, Ascierto PA, Morse GD, Buonaguro L, Puzanov I, Tornesello ML, et al. Covid-19: Time for a paradigm change. Rev Med Virol. 2020;30:e2134.
Ratajczak MZ, Kucia M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia. 2020;34:1726–9.
Valiño-Rivas L, Cuarental L, Nuñez G, Sanz AB, Ortiz A, Sanchez-Niño MD. Loss of NLRP6 expression increases the severity of acute kidney injury. Nephrol Dial Transplant. 2019;35:587–98.
Pulskens WP, Butter LM, Teske GJ, Claessen N, Dessing MC, Flavell Ra Sutterwala FS, et al. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS One. 2014;9:e85775.
Guo H, Bi X, Zhou P, Zhu S, Wei D. NLRP3 Deficiency attenuates renal fibrosis and ameliorates mitochondrial dysfunction in a mouse unilateral ureteral obstruction model of chronic kidney disease. Mediators Inflamm. 2017;2017:8316560.
Chi HH, Hua KF, Lin YC, Chu CL, Hsieh CY, Hsu YJ, et al. IL-36 signaling facilitates activation of the NLRP3 inflammasome and IL-23/IL-17 axis in renal inflammation and fibrosis. J Am Soc Nephrol. 2017;28:2022–37.
Wu M, Li R, Hou Y, Song S, Han W, Chen N, et al. Thioredoxin-interacting protein deficiency ameliorates kidney inflammation and fibrosis in mice with unilateral ureteral obstruction. Lab Invest. 2018;98:1211–24.
Wang W, Luo R, Lin Y, Wang F, Zheng P, Levi MY, et al. Aliskiren restores renal AQP2 expression during unilateral ureteral obstruction by inhibiting the inflammasome. Am J Physiol Ren Physiol. 2015;308:F910–22.
Zhang J, Zheng L, Yuan X, Liu C, Yuan Q, Xie F, et al. Mefunidone ameliorates renal inflammation and tubulointerstitial fibrosis via suppression of IKKβ phosphorylation. Int J Biochem Cell Biol. 2016;80:109–18.
Zheng L, Zhang J, Yuan X, Tang J, Qiu S, Peng Z, et al. Fluorofenidone attenuates interleukin-1β production by interacting with NLRP3 inflammasome in unilateral ureteral obstruction. Nephrology. 2018;23:573–84.
Ling L, Yang M, Ding W, Yong G. Ghrelin attenuates UUO-induced renal fibrosis via attenuation of Nlrp3 inflammasome and endoplasmic reticulum stress. Am J Transl Res. 2019;11:131–41.
Zhou Y, Zhu X, Wang X, Peng Y, Du J, Yin H, et al. H2S alleviates renal injury and fibrosis in response to unilateral ureteral obstruction by regulating macrophage infiltration via inhibition of NLRP3 signaling. Exp Cell Res. 2020;387:111779.
Qi R, Zhang W, Zheng L, Xu M, Rong R, Zhu T, et al. Cyclic helix B peptide ameliorates renal tubulointerstitial fibrosis induced by unilateral ureter obstruction via inhibiting NLRP3 pathway. Ann Transl Med. 2020;8:167.
Qian Y, Feldman E, Pennathur S, Kretzler M, Brosius FC. From fibrosis to sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes. 2008;57:1439–45.
Si X, Li P, Zhang Y, Zhang Y, Lv W, Qi D. Renoprotective effects of olmesartan medoxomil on diabetic nephropathy in streptozotocin-induced diabetes in rats. Biomed Rep. 2014;2:24–8.
Qiao Y, Tian X, Men L, Li S, Chen Y, Xue M, et al. Spleen tyrosine kinase promotes NLR family pyrin domain containing 3 inflammasome‑mediated IL-1β secretion via c‑Jun N‑terminal kinase activation and cell apoptosis during diabetic nephropathy. Mol Med Rep. 2018;18:1995–2008.
Ding H, Li J, Li Y, Yang M, Nie S, Zhou M, et al. MicroRNA-10 negatively regulates inflammation in diabetic kidney via targeting activation of the NLRP3 inflammasom. Mol Ther. 2021;S1525-0016:00141–6.
Wu M, Yang Z, Zhang C, Shi Y, Han W, Song S, et al. Inhibition of NLRP3 inflammasome ameliorates podocyte damage by suppressing lipid accumulation in diabetic nephropathy. Metabolism. 2021;118:154748.
El-Horany HE, Abd-Ellatif RN, Watany M, Hafez YM, Okda HI. NLRP3 expression and urinary HSP72 in relation to biomarkers of inflammation and oxidative stress in diabetic nephropathy patients. IUBMB Life. 2017;69:623–30.
Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–5.
Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018;16:32–46.
Li N, Zhao T, Cao Y, Zhang H, Peng L, Wang Y, et al. Tangshen formula attenuates diabetic kidney injury by imparting anti-pyroptotic effects via the TXNIP-NLRP3-GSDMD axis. Front Pharmacol. 2021;11:623489.
Zhou L, Huang W, Xu Y, Gao C, Zhang T, Guo M, et al. Sweet taste receptors mediated ROS-NLRP3 inflammasome signaling activation: implications for diabetic nephropathy. J Diabetes Res. 2018;2018:7078214.
Feng H, Gu J, Gou F, Huang W, Gao C, Chen G, et al. High glucose and lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway in mesangial cells. J Diabetes Res. 2016;2016:6973175.
Gu C, Liu S, Wang H, Dou H. Role of the thioredoxin interacting protein in diabetic nephropathy and the mechanism of regulating NOD‑like receptor protein 3 inflammatory corpuscle. Int J Mol Med. 2019;43:2440–50.
Wang S, Zhao X, Yang S, Chen B, Shi J. Salidroside alleviates high glucose-induced oxidative stress and extracellular matrix accumulation in rat glomerular mesangial cells by the TXNIP-NLRP3 inflammasome pathway. Chem Biol Interact. 2017;278:48–53.
Tan CY, Weier Q, Zhang Y, Cox AJ, Kelly DJ, Langham RG. Thioredoxin-interacting protein: a potential therapeutic target for treatment of progressive fibrosis in diabetic nephropathy. Nephron. 2015;129:109–27.
Gao P, He FF, Tang H, Lei CT, Chen S, Meng XF, et al. NADPH oxidase-induced NALP3 inflammasome activation is driven by thioredoxin-interacting protein which contributes to podocyte injury in hyperglycemia. J Diabetes Res. 2015;2015:504761.
Solini A, Menini S, Rossi C, Ricci C, Santini E, Blasetti Fantauzzi C, et al. The purinergic 2X7 receptor participates in renal inflammation and injury induced by high-fat diet: possible role of NLRP3 inflammasome activation. J Pathol. 2013;231:342–53.
Wang C, Hou XX, Rui HL, Li LJ, Zhao J, Yang M, et al. Artificially cultivated ophiocordyceps sinensis alleviates diabetic nephropathy and its podocyte injury via inhibiting P2X7R expression and NLRP3 inflammasome activation. J Diabetes Res. 2018;2018:1390418.
Chen F, Wei G, Xu J, Ma X, Wang Q. Naringin ameliorates the high glucose-induced rat mesangial cell inflammatory reaction by modulating the NLRP3 Inflammasome. BMC Complement Alter Med. 2018;18:192.
Fu Y, Wu N, Zhao D. Function of NLRP3 in the pathogenesis and development of diabetic nephropathy. Med Sci Monit. 2017;23:3878–84.
Chen K, Zhang J, Zhang W, Zhang J, Yang J, Li K, et al. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int J Biochem Cell Biol. 2013;45:932–43.
Li X, Zeng L, Cao C, Lu C, Lian W, Han J, et al. Long noncoding RNA MALAT1 regulates renal tubular epithelial pyroptosis by modulated miR-23c targeting of ELAVL1 in diabetic nephropathy. Exp Cell Res. 2017;350:327–35.
Yi H, Peng R, Zhang LY, Sun Y, Peng HM, Liu HD, et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017;8:e2583.
Wang S, Li Y, Fan J, Zhang X, Luan J, Bian Q, et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017;8:e2937.
Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol. 2009;20:742–52.
Son S, Hwang I, Han SH, Shin JS, Shin OS, Jw YU. Advanced glycation end products impair NLRP3 inflammasome-mediated innate immune responses in macrophages. J Biol Chem. 2017;292:20437–48.
Yang SM, Ka SM, Wu HL, Yeh YC, Kuo CH, Hua KF, et al. Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice via anti-NF-κB/NLRP3 inflammasome-mediated inflammation, enhancement of NRF2 antioxidant activity and inhibition of apoptosis. Diabetologia. 2014;57:424–34.
Zhu X, Shi J, Li H. Liquiritigenin attenuates high glucose-induced mesangial matrix accumulation, oxidative stress, and inflammation by suppression of the NF-κB and NLRP3 inflammasome pathways. Biomed Pharmacother. 2018;106:976–82.
Li J, Wang B, Zhou G, Yan X, Zhang Y. Tetrahydroxy stilbene glucoside alleviates high glucose-induced MPC5 podocytes injury through suppression of NLRP3 inflammasome. Am J Med Sci. 2018;355:588–96.
Song W, Wei L, Du Y, Wang Y, Jiang S. Protective effect of ginsenoside metabolite compound K against diabetic nephropathy by inhibiting NLRP3 inflammasome activation and NF-kappaB/p38 signaling pathway in high-fat diet/streptozotocin-induced diabetic mice. Int Immunopharmacol. 2018;63:227–38.
Xin R, Sun X, Wang Z, Yuan W, Jiang W, Wang L, et al. Apocynin inhibited NLRP3/XIAP signalling to alleviate renal fibrotic injury in rat diabetic nephropathy. Biomed Pharmacother. 2018;106:1325–31.
Shahzad K, Bock F, Al-Dabet MM, Gadi I, Nazir S, Wang H, et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci Rep. 2016;6:34228.
Han W, Ma Q, Liu Y, Wu W, Tu Y, Huang L, et al. Huangkui capsule alleviates renal tubular epithelial-mesenchymal transition in diabetic nephropathy via inhibiting NLRP3 inflammasome activation and TLR4/NF-κB signaling. Phytomedicine. 2019;57:203–14.
Samra YA, Said HS, Elsherbiny NM, Liou GI, El-Shishtawy MM, Eissa LA. Cepharanthine and piperine ameliorate diabetic nephropathy in rats: role of NF-κB and NLRP3 inflammasome. Life Sci. 2016;157:187–99.
Miean KH, Mohamed S. Flavonoid (myricetin, quercetin, kaempferol, luteolin, and apigenin) content of edible tropical plants. J Agric Food Chem. 2001;49:3106–12.
Yu Q, Zhang M, Qian L, Wen D, Wu G. Luteolin attenuates high glucose-induced podocyte injury via suppressing NLRP3 inflammasome pathway. Life Sci. 2019;225:1–7.
Du P, Fan B, Han H, Zhen J, Shang J, Wang X, et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int. 2013;84:265–76.
Haraldsson B, Nyström J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–87.
Shang J, Zhang Y, Jiang Y, Li Z, Duan Y, Wang L, et al. NOD2 promotes endothelial-to-mesenchymal transition of glomerular endothelial cells via MEK/ERK signaling pathway in diabetic nephropathy. Biochem Biophys Res Commun. 2017;484:435–41.
Huang W, Gou F, Long Y, Li Y, Feng H, Zhang Q, et al. High glucose and lipopolysaccharide activate NOD1- RICK-NF-κB inflammatory signaling in mesangial cells. Exp Clin Endocrinol Diabetes. 2016;124:512–7.
Chan KL, Tam TH, Boroumand P, Prescott D, Costford SR, Escalante NK, et al. Circulating NOD1 activators and hematopoietic NOD1 contribute to metabolic inflammation and insulin resistance. Cell Rep. 2017;18:2415–26.
Rivers SL, Klip A, Adria G. NOD1: an interface between innate immunity and insulin resistance. Endocrinology. 2019;160:1021–30.
Carney EF. Lupus nephritis: role of NLRP3 inflammasomes in podocyte injury. Nat Rev Nephrol. 2017;13:444.
Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, et al. Podocyte activation of NLRP3 inflammasomes contributes to the development of proteinuria in lupus nephritis. Arthritis Rheumatol. 2017;69:1636–46.
Dario U. Lupus nephritis: NLRP3 inflammasome ignites podocyte dysfunction. Nat Rev Rheumatol. 2017;13:451.
Huang T, Yin H, Ning W, Wang X, Chen C, Lin W, et al. Expression of inflammasomes NLRP1, NLRP3 and AIM2 in different pathologic classification of lupus nephritis. Clin Exp Rheumatol. 2020;38:680–90.
Zhao J, Wang H, Dai C, Wang H, Zhang H, Huang Y, et al. P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/caspase 1 pathway. Arthritis Rheumatol. 2013;65:3176–85.
Zhao J, Wang H, Huang Y, Zhang H, Wang S, Gaskin F, et al. Lupus nephritis: glycogen synthase kinase 3β promotion of renal damage through activation of the NLRP3 inflammasome in lupus-prone mice. Arthritis Rheumatol. 2015;67:1036–44.
Guo C, Fu R, Zhou M, Wang S, Huang Y, Hu H, et al. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J Autoimmun. 2019;103:102286.
Li M, Shi X, Qian T, Li J, Tian Z, Ni B, et al. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-κB and NLRP3 inflammasome activation in macrophages of mice. Int J Clin Exp Med. 2015;8:17430–40.
Zhang H, Liu L, Li L. Lentivirus-mediated knockdown of FcγRI (CD64) attenuated lupus nephritis via inhibition of NF-κB regulating NLRP3 inflammasome activation in MRL/lpr mice. J Pharm Sci. 2018;137:342–9.
Peng X, Yang T, Liu G, Liu H, Peng Y, Liyu H. Piperine ameliorated lupus nephritis by targeting AMPK-mediated activation of NLRP3 inflammasome. Int Immunopharmacol. 2018;65:448–57.
Yuan Y, Zhangsuo L. Isoflurane attenuates murine lupus nephritis by inhibiting NLRP3 inflammasome activation. Int J Clin Exp Med. 2015;8:17730–8.
Yang SR, Hua KF, Chu LJ, Hwu YK, Yang SM, Wu CY, et al. Xenon blunts NF-κB/NLRP3 inflammasome activation and improves acute onset of accelerated and severe lupus nephritis in mice. Kidney Int. 2020;98:378–90.
Wu CY, Hua KF, Chu CL, Yang SR, Arbiser JL, Yang SS, et al. Tris DBA ameliorates accelerated and severe lupus nephritis in mice by activating regulatory T cells and autophagy and inhibiting the NLRP3 inflammasome. J Immunol. 2020;204:1448–61.
Su B, Ye H, You X, Ni H, Chen X, Linlin L. Icariin alleviates murine lupus nephritis via inhibiting NF-κB activation pathway and NLRP3 inflammasome. Life Sci. 2018;208:26–32.
He J, Sun M, Sujian T. Procyanidin B2 prevents lupus nephritis development in mice by inhibiting NLRP3 inflammasome activation. Innate Immun. 2018;24:307–15.
D’amico G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol. 2004;24:179–96.
Mestecky J, Raska M, Julian BA, Gharavi AG, Renfrow MB, Moldoveanu Z, et al. IgA nephropathy: molecular mechanisms of the disease. Annu Rev Pathol. 2013;8:217–40.
Tsai YL, Hua KF, Chen A, Wei CW, Chen WS, Wu CY, et al. NLRP3 inflammasome: pathogenic role and potential therapeutic target for IgA nephropathy. Sci Rep. 2017;7:41123.
Peng W, Pei G-Q, Tang Y, Tan L, Wei Q. IgA1 deposition may induce NLRP3 expression and macrophage transdifferentiation of podocyte in IgA nephropathy. J Transl Med. 2019;17:406.
Hua KF, Yang SM, Kao TY, Chang JM, Chen HL, Tsai YJ, et al. Osthole mitigates progressive IgA nephropathy by inhibiting reactive oxygen species generation and NF-κB/NLRP3 pathway. PLoS One. 2013;8:e77794.
Yang SM, Ka SM, Hua KF, Wu TH, Chuang YP, Lin YW, et al. Antroquinonol mitigates an accelerated and progressive IgA nephropathy model in mice by activating the Nrf2 pathway and inhibiting T cells and NLRP3 inflammasome. Free Radic Biol Med. 2013;61:285–97.
Chang YP, Ka SM, Hsu WH, Chen A, Chao LK, Lin CC, et al. Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J Cell Physiol. 2015;230:1567–79.
Wu CY, Hua KF, Hsu WH, Suzuki Y, Chu LJ, Lee YC, et al. IgA nephropathy benefits from compound K treatment by inhibiting NF-κB/NLRP3 inflammasome and enhancing autophagy and SIRT1. J Immunol. 2020;205:202–12.
Chun J, Chung H, Wang X, Barry R, Taheri ZM, Platnich JM, et al. NLRP3 localizes to the tubular epithelium in human kidney and correlates with outcome in IgA nephropathy. Sci Rep. 2016;6:24667.
Kors L, Rampanelli E, Stokman G, Butter LM, Held NM, Claessen N, et al. Deletion of NLRX1 increases fatty acid metabolism and prevents diet-induced hepatic steatosis and metabolic syndrome. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1883–95.
Bakker PJ, Butter LM, Kors L, Teske GJ, Aten J, Sutterwala FS, et al. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int. 2014;85:1112–22.
Prencipe G, Caiello I, Cherqui S, Whisenant T, Petrini S, Emma F, et al. Inflammasome activation by cystine crystals: implications for the pathogenesis of cystinosis. J Am Soc Nephrol. 2014;25:1163–9.
Mulay SR, Anders HJ. Crystal nephropathies: mechanisms of crystal-induced kidney injury. Nat Rev Nephrol. 2017;13:226–40.
Darisipudi MN, Thomasova D, Mulay SR, Brech D, Noessner E, Liapis H, et al. Uromodulin triggers IL-1β-dependent innate immunity via the NLRP3 inflammasome. J Am Soc Nephrol. 2012;23:1783–9.
Husain M, Meggs LG, Vashistha H, Simoes S, Griffiths KO, Kumar D, et al. Inhibition of p66ShcA longevity gene rescues podocytes from HIV-1-induced oxidative stress and apoptosis. J Biol Chem. 2009;284:16648–58.
Haque S, Lan X, Wen H, Lederman R, Chawla A, Attia M, et al. HIV promotes NLRP3 inflammasome complex activation in murine HIV-associated nephropathy. Am J Pathol. 2016;186:347–58.
Zhu W, Pang M, Dong L, Huang X, Wang S, Zhou L. Anti-inflammatory and immunomodulatory effects of iridoid glycosides from Paederia scandens (LOUR.) MERRILL (Rubiaceae) on uric acid nephropathy rats. Life Sci. 2012;91:369–76.
Wu M, Ma Y, Chen X, Liang N, Qu S, Haibing C. Hyperuricemia causes kidney damage by promoting autophagy and NLRP3-mediated inflammation in rats with urate oxidase deficiency. Dis Model Mech. 2021;14:dmm048041.
Hu J, Wu H, Wang D, Yang Z, Dong J. LncRNA ANRIL promotes NLRP3 inflammasome activation in uric acid nephropathy through miR-122-5p/BRCC3 axis. Biochimie. 2019;157:102–10.
Hu J, Wu H, Wang D, Yang Z, Zhuang L, Yang N, et al. Weicao capsule ameliorates renal injury through increasing autophagy and NLRP3 degradation in UAN rats. Int J Biochem Cell Biol. 2018;96:1–8.
Ye Y, Zhang Y, Wang B, Walana W, Wei J, Gordon JR, et al. CXCR1/CXCR2 antagonist G31P inhibits nephritis in a mouse model of uric acid nephropathy. Biomed Pharmacother. 2018;107:1142–50.
Wu X, Chang SC, Jin J, Gu W, Shanqun L. NLRP3 inflammasome mediates chronic intermittent hypoxia-induced renal injury implication of the microRNA-155/FOXO3a signaling pathway. J Cell Physiol. 2018;233:9404–15.
Turajlic S, Larkin J, Swanton C. SnapShot: renal cell carcinoma. Cell. 2015;163:1556–e1.
Cohen HT, Mcgovern FJ. Renal-cell carcinoma. N Engl J Med. 2005;353:2477–90.
Gossage L, Eisen T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nat Rev Clin Oncol. 2010;7:277–88.
Tian X, Zhang S, Zhang Q, Kang L, Ma C, Feng L, et al. Resveratrol inhibits tumor progression by down-regulation of NLRP3 in renal cell carcinoma. J Nutr Biochem. 2020;85:108489.
Kumar A, Nallabelli N, Sharma U, Kumari N, Singh SK, Kakkar N, et al. In vitro evidence of NLRP3 inflammasome regulation by histone demethylase LSD2 in renal cancer: a pilot study. Mol Biol Rep. 2020;47:7273–6.
Wang K, Xu T, Ruan H, Xiao H, Liu J, Song Z, et al. LXRalpha promotes cell metastasis by regulating the NLRP3 inflammasome in renal cell carcinoma. Cell Death Dis. 2019;10:159.
Tan YF, Wang M, Chen ZY, Wang L, Xiu-Heng L. Inhibition of BRD4 prevents proliferation and epithelial-mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell Death Dis. 2020;11:239.
Xu D, Zhang S, Zhang S, Liu H, Li P, Yu L, et al. NOD2 maybe a biomarker for the survival of kidney cancer patients. Oncotarget. 2017;6:101489–99.
Mey L, Jung M, Roos F, Blaheta R, Hegele A, Kinscherf R, et al. NOD1 and NOD2 of the innate immune system is differently expressed in human clear cell renal cell carcinoma, corresponding healthy renal tissue, its vasculature and primary isolated renal tubular epithelial cells. J Cancer Res Clin Oncol. 2019;145:1405–16.
Wang Q, Ding H, He Y, Li X, Cheng Y, Xu Q, et al. NLRC5 mediates cell proliferation, migration, and invasion by regulating the Wnt/beta-catenin signalling pathway in clear cell renal cell carcinoma. Cancer Lett. 2019;444:9–19.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
Ludwig-Portugall I, Bartok E, Dhana E, Evers BD, Primiano MJ, Hall JP, et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016;90:525–39.
Zou XF, Gu JH, Duan JH, Hu ZD, Cui ZL. The NLRP3 inhibitor Mcc950 attenuates acute allograft damage in rat kidney transplants. Transpl Immunol. 2020;61:101293.
Lin Q, Li S, Jiang N, Jin H, Shao X, Zhu X, et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy. 2021;17:2975–90.
Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15:556–9.
Wu J, Raman A, Coffey NJ, Sheng X, Wahba J, Seasock MJ, et al. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J Clin Invest. 2021;131:e136329.
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187:61–70.
Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front Immunol. 2019;10:2538.
Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606.
Fulp J, He L, Toldo S, Jiang Y, Boice A, Guo C, et al. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J Med Chem. 2018;61:5412–23.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41.
Leung YY, Yao Hui LL, Kraus VB. Colchicine-Update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum. 2015;45:341–50.
Huang J, Yang Y, Hu R, Chen L. Anti-interleukin-1 therapy has mild hypoglycaemic effect in type 2 diabetes. Diabetes Obes Metab. 2018;20:1024–8.
Loustau C, Rosine N, Forien M, Ottaviani S, Juge PA, Liote F, et al. Effectiveness and safety of anakinra in gout patients with stage 4-5 chronic kidney disease or kidney transplantation: a multicentre, retrospective study. Jt Bone Spine. 2018;85:755–60.
Zhang Y, Yuan F, Cao X, Zhai Z, Ganghuang, Du X, et al. P2X7 receptor blockade protects against cisplatin-induced nephrotoxicity in mice by decreasing the activities of inflammasome components, oxidative stress and caspase-3. Toxicol Appl Pharmacol. 2014;281:1–10.
Levy M, Shapiro H, Thaiss CA, Eran E. NLRP6: a multifaceted innate immune sensor. Trends Immunol. 2017;38:248–60.
Wang W, Faubel S, Ljubanovic D, Mitra A, Falk SA, Kim J, et al. Endotoxemic acute renal failure is attenuated in caspase-1-deficient mice. Am J Physiol Ren Physiol. 2005;288:F997–1004.
Mcmartin K. Are calcium oxalate crystals involved in the mechanism of acute renal failure in ethylene glycol poisoning? Clin Toxicol. 2009;47:859–69.
Kim SM, Lee SH, Kim YG, Kim SY, Seo JW, Choi YW, et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am J Physiol Ren Physiol. 2015;308:F993–1003.
Shahzad K, Bock F, Al-Dabet MM, Gadi I, Kohli S, Nazir S, et al. Caspase-1, but not caspase-3, promotes diabetic nephropathy. J Am Soc Nephrol. 2016;27:2270–5.
Qiu YY, Tang LQ. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol Res. 2016;114:251–64.
Wu M, Han W, Song S, Du Y, Liu C, Chen N, et al. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol. 2018;478:115–25.
Liu Y, Xu Z, Ma F, Jia Y, Wang G. Knockdown of TLR4 attenuates high glucose-induced podocyte injury via the NALP3/ASC/Caspase-1 signaling pathway. Biomed Pharmacother. 2018;107:1393–401.
Sun Z, Ma Y, Chen F, Wang S, Chen B, Shi J. Artesunate ameliorates high glucose-induced rat glomerular mesangial cell injury by suppressing the TLR4/NF-kappaB/NLRP3 inflammasome pathway. Chem Biol Interact. 2018;293:11–9.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81970584 and 82100727). Promotion plan of basic and clinical cooperative research in Anhui Medical University (No. 2019xkjT014; No. 2020xkjT016). The Open Fund of Inflammation and Immune Mediated Diseases Laboratory of Anhui Province (No. IMMDL202002). Provincial Natural Science Foundation (No.1908085QH377).
Author information
Authors and Affiliations
Contributions
XMM provided direction and guidance throughout the preparation of this manuscript. JJ, TJZ, and GLR wrote and edited the manuscript. LC collected and prepared the related papers. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Jin, J., Zhou, Tj., Ren, Gl. et al. Novel insights into NOD-like receptors in renal diseases. Acta Pharmacol Sin 43, 2789–2806 (2022). https://doi.org/10.1038/s41401-022-00886-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00886-7
Keywords
This article is cited by
-
Pyroptosis in renal inflammation and fibrosis: current knowledge and clinical significance
Cell Death & Disease (2023)
