
Abstract
Inflammation and apoptosis are main pathological processes that lead to the development of cardiac hypertrophy. Lupeol, a natural triterpenoid, has shown anti-inflammatory and anti-apoptotic activities as well as potential protective effects on cardiovascular diseases. In this study we investigated whether lupeol attenuated cardiac hypertrophy and fibrosis induced by pressure overload in vivo and in vitro, and explored the underlying mechanisms. Cardiac hypertrophy was induced in mice by transverse aortic constriction (TAC) surgery, and in neonatal rat cardiomyocytes (NRCMs) by stimulation with phenylephrine (PE) in vitro. We showed that administration of lupeol (50 mg ·kg-1· d-1, i.g., for 4 weeks) prevented the morphological changes and cardiac dysfunction and remodeling in TAC mice, and treatment with lupeol (50 μg/mL) significantly attenuated the hypertrophy of PE-stimulated NRCMs, and blunted the upregulated hypertrophic markers ANP, BNP, and β-MHC. Furthermore, lupeol treatment attenuated the apoptotic and inflammatory responses in the heart tissue. We revealed that lupeol attenuated the inflammatory responses including the reduction of inflammatory cytokines and inhibition of NF-κB p65 nuclear translocation, which was mediated by the TLR4-PI3K-Akt signaling. Administration of a PI3K/Akt agonist 740 Y-P reversed the protective effects of lupeol in TAC mice as well as in PE-stimulated NRCMs. Moreover, pre-treatment with a TLR4 agonist RS 09 abolished the protective effects of lupeol and restored the inhibition of PI3K-Akt-NF-κB signaling by lupeol in PE-stimulated NRCMs. Collectively, our results demonstrate that the lupeol protects against cardiac hypertrophy via anti-inflammatory mechanisms, which results from inhibiting the TLR4-PI3K-Akt-NF-κB signaling.
Similar content being viewed by others


Introduction
Cardiac hypertrophy (CH) was initially regarded as a compensatory cellular response to various physiological and pathological stimuli [1], but the compensatory mechanisms related to the structural and functional alteration of the left ventricle can become maladaptive and even results in cardiac failure under sustained and abnormal loading conditions [2]. It is usually characterized by an increase in cardiomyocyte cell volume and thus an enlargement of the heart, and caused by various stimuli including chronic pressure overload, ischemia, oxidative stress, and vascular remodeling and the inflammatory response [3]. The inflammatory response specific to the mechanisms of cardiac remodeling has been increasingly investigated [4]. In the process of cardiac inflammation, immunocytes invade the cardiac tissue and regulate the tissue responses, leading to cardiomyocyte hypertrophy and even death. Besides, the invasion of immunocytes initiates the pro-fibrotic signaling cascade. Overabundant fibrotic remodeling and collagen deposition results in ventricular wall stiffness, which causes systolic and diastolic dysfunction. Constant cardiac dysfunction would further decrease the myocardial perfusion, aggravate the inflammatory response, and eventually contribute to cardiac failure [5].
Toll-like receptor (TLR4) that belongs to the TLR family is an inflammatory protein, which plays a critical role in the innate immune system. The TLR4 activation initiates signaling cascades to stimulate severe cytokine-regulating inflammatory responses, which lead to the development of CH [6]. Previous studies have demonstrated that the activated phosphatidylinositol 3-kinase (PI3K) was also tightly involved in the inflammatory response [7], and promoted the process of cardiac remodeling through activating hypertrophy-related genes [8]. Besides, the nuclear factor-kappa B subunit 1 (NF-κB) signaling pathway was highly associated with the inflammatory response [9], and it has been reported that the NF‑κB was involved in vascular remodeling [10], apoptosis [11], and oxidative stress [12]. The activated NF-κB p65 subunits translocated to the nucleus and promoted the transcription of the targets genes, including IL-10, IL-6, and TNF-α [13], which resulted in the development of inflammation and CH. Therefore, the effective intervention of these signaling pathways would be a key therapeutic strategy in CH.
Medicinal plants recently have been reported to have therapeutic effects in cardiovascular diseases in many countries [14,15,16]. Hence, we have screened a group of medicinal plants that have potentials for cardiovascular therapy based on their anti-inflammatory and antioxidant properties. And we found that lupeol, belonging to pentacyclic triterpenoids, is an extract from vegetables like pepper and white cabbage, fruits like strawberry and mango [17], and herbal plants [18]. Pentacyclic triterpenoids, such as ursolic acid, and asiatic acid have been proven to have cardiovascular protective effects. Ursolic acid protected the streptozotocin (STZ)-induced diabetic cardiomyopathy via decreasing inflammation and oxidative stress [19]. And asiatic acid improved cardiac function through activating AMPKα signaling [20]. Besides, lupeol has a series of biological properties, including anti-inflammatory, antioxidant, and fibroblast proliferative characteristics [21]. For example, it ameliorated liver injury in mice through inhibiting the IL-1 receptor-associated kinase-mediated TLR inflammatory signaling [22]. In addition, it significantly attenuated the phosphorylation of the PI3K in vivo in a dose-dependent manner [23, 24]. It is also noteworthy that lupeol attenuated the NF-κB protein expression, exhibiting an anti-inflammatory effect [23, 24]. In cardiovascular research, previous studies also reported that lupeol protected the cardiac abnormalities from hypercholesterolemia [25] as well as improved the myocardial permeability in cyclophosphamide administered rats [23]. These findings further demonstrated that lupeol may have a potential protective effect on cardiovascular diseases. Yet whether the lupeol has therapeutic potential in the protection of the cardiac remodeling is still unknown.
Therefore, the present study aimed to investigate whether lupeol can attenuate CH and fibrosis induced by pressure overload in mice and to explore its mechanism of action.
Materials and methods
Chemicals
Lupeol (98% purity) was acquired from Shanghai Winherb Medical Technology Co. Ltd (545-47-1, Shanghai, China).
Cell culture and treatment
To identify the anti-hypertrophic effects of lupeol in vitro, neonatal rat cardiomyocytes (NRCMs) were cultured in Dulbecco’s modified Eagle’s medium/F-12 (C11330500BT, Gibco, Grand Island, NY, USA) supplemented with 15% fetal bovine serum (FBS, 10099-141, Gibco, Grand Island, NY, USA) at 37 °C under 5% CO2 environment. To stimulate myocardial hypertrophy in vitro, cells were exposed to phenylephrine (PE) (P6126, Sigma-Aldrich, St. Louis, MO, USA), a known cardiomyocyte hypertrophy-inducing chemical, or vehicle (0.1% DMSO, D8418, Sigma-Aldrich, St. Louis, MO, USA), in the presence or absence of lupeol (50 μg/mL, dissolved in 0.1% DMSO), and incubated for 12 h.
Detection of cytotoxicity with cell-counting kit-8 assay
To detect the cytotoxicity of lupeol, the NRCMs’ cell viability was determined after the treatment with different concentrations of lupeol (0, 12.5, 25, 50, 75, and 100 μg/mL) for a specific time. Similar to the above process, different concentrations of lupeol (0, 12.5, 25, 50, and 75 μg/mL) in PE-stimulated groups also had cell viability values. The cytotoxicity and efficacy of lupeol were determined by the Enhanced Cell Counting Kit-8 (C0042, Beyotime Institute of Biotechnology, Haimen, China). And the absorbance was measured at 450 nm using a labeling apparatus, Synergy HT: Multi-Detection Microplate Reader (BioTek Instruments, Winooski, VT, USA).
Cardiomyocyte immunofluorescence analysis
Through immunofluorescence staining, the cell surface of the NRCMs was assessed. The expression and the localization of the NF-κB p65 on the NRCMs were observed. Briefly, after washing thrice with PBS, the cells were fixed with 4% formaldehyde, permeabilized with 0.2% Triton, and stained with anti-α-actinin (ab68167, Abcam, Cambridge, UK), anti-α-SMA (ab7817, Abcam, Cambridge, UK), or anti-p65 (8242, CST, Danvers, MA, USA) at a dilution of 1:100 in 1% goat serum (GTX27481, GeneTex, Sanantonio, TX, USA) at 4 °C overnight. The next day, cells were incubated with the Alexa Fluor 488 (green-fluorescent dye) secondary antibodies at a dilution of 1:200 at 37 °C for 1 h. Then the nucleus was counterstained with the DAPI (S36939, Thermo Fisher Scientific, Eugene, OR, USA) for visualization. Finally, the surface area of single cells was measured using Image-Pro Plus software (version 6.0) in a blinded manner.
Quantitative real-time RT-PCR
To measure the mRNA expression of CH and fibrosis associated markers, the total RNA from the mouse LV cardiac tissues or cells was extracted using TRIzol reagent (15596-026, Invitrogen Corporation, Waltham, MA, USA) and subsequently, the mRNA was reverse transcribed into complementary DNA (cDNA) using the cDNA Synthesis Kit (489703001, Roche, Basel, Switzerland). The Ct values of the target gene amplifications were determined using SYBR Green-1 Master Mix in a LightCycler 480 System (04896866001, Roche). The GAPDH was used as the internal control gene to normalize the target genes. The sequences of primers used in the experiments are shown in Table 1.
Animals and treatments
All animal experiments were performed, following the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH), and were permitted by the Committee on the Laboratory Animal Welfare & Ethics of Renmin Hospital of Wuhan University (IACUC Issue Numeber: WDRM20180401). Male C57BL/6 mice (23.5–27.5 g, 8–10 weeks old) were obtained from the Institute of Laboratory Animal Science, Chinese Academy of Medical Science (Beijing, China).
To acquire the model of pressure overload-induced CH, after 1 week of acclimatization, mice were randomly divided into different groups to undergo the transverse aortic constriction (TAC) surgery. After that procedure, the mice were given lupeol (Lup) (50 mg/kg) for 4 weeks, and equal volumes of vehicles were given to the mice in the control group. At the end of the study, the hearts and lungs were harvested and weighed after the mice were euthanized. The heart weight (HW)/body weight (BW) and HW/tibial length (TL) were calculated and compared between different groups.
Echocardiography and hemodynamics
To assess the cardiac function, echocardiography was performed after mice were anesthetized using 1.5% isoflurane. Then, the ventricular septum, chamber dimension, systolic and diastolic function, and ejection fraction were recorded using a MyLab 30 CV ultrasound system (Esaote S.P.A, Genoa, Italy).
Hemodynamics were monitored with a microtip catheter transducer, which was inserted into the left ventricle. Subsequently, the pressure and heart rate were continuously recorded using a pressure-volume catheter (SPR-839, Millar Instruments, Houston, TX, USA), which was connected to a PowerLab system (AD Instruments Ltd., Oxford, UK) and then stored and displayed on a private computer. Finally, the data collected were analyzed by using the PVAN data analysis software.
Cardiac morphology and histomorphometric analysis
To further assess CH and fibrosis, morphological analysis on heart sections was performed. Mouse hearts that were excised from the sacrificed mice were immediately placed in a 10% KCl to arrest in the diastolic phase. Next, the hearts were fixed in 10% formalin overnight, paraffin-embedded, and sectioned into 3–5 μm slices. Then the sections were stained with hematoxylin-eosin or picrosirius red (PSR) as well as Masson’s trichrome to assess the myocyte hypertrophy or the extracellular collagen deposition. The digital images of the stained sections were captured and then measured using a digital image analysis procedure (Image-Pro Plus, version 6.0).
Western blot
To evaluate the levels of protein expression, the Western blot was performed. The total proteins were extracted from cultured cardiomyocytes or mouse hearts using RIPA lysis buffer (G2002, Servicebio, Wuhan, China) with a protease inhibitor cocktail (cOmplete, Roche, Mannheim, Germany). Protein concentration was assessed using a BCA protein assay kit (23227, Thermo Fisher Scientific, Waltham, MA, USA) and was normalized before the Western blot. Protein samples (50 μg) were separated on a 10% SDS-PAGE and subsequently transferred to the PVDF membranes (FL00010, Millipore, Billerica, MA, USA). Next, the PVDF membranes were blocked with 5% nonfat milk for 1 h at room temperature to block the non-specific binding sites, and then incubated overnight at 4 °C with primary antibodies. TGF-β (ab66043), p-IκBα (ab133462), t-IκBα (ab7217) were purchased from Abcam (Cambridge, UK). TLR4 (sc-30002) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). p-SMAD2 (3108 S), t-SMAD2 (3103 S), Bax (2772), c-caspase3 (9661), t-caspase3 (9662 P), Bcl-2 (2870), Myd88 (4283), p-p65 (3033), t-p65 (8242), p-PI3K (4228 S), t-PI3K (4257), p-Akt (4060), t-Akt (4691), and GAPDH (2118) were purchased from Cell Signaling Technology (CST, Danvers, MA, USA). The protein expression level of the GAPDH was used as an internal standard. The next day, the protein blots were incubated with secondary antibodies for 1 h at room temperature. Finally, the blots were obtained and scanned by the Image Lab software (Bio-Rad Laboratories, Inc., Hercules, CA, USA) to assess protein expression.
TUNEL assay
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was performed to measure the number of apoptotic cells in vivo using a commercial kit (S7111, Millipore Corporation, Burlington, MA, USA). Cardiac sections were prepared and stained following the manufacturer’s protocol. The apoptotic cells had only apoptotic nuclei at the center of the cells and were captured by a digital fluorescence microscope (Olyplus DX51, Tokyo, Japan) and further analyzed using the Image-Pro Plus 6.0 software.
Immunohistochemistry
The paraffin-embedded tissue sections were incubated with the antibodies of CD45 (ab10558, Abcam, Cambridge, UK) and CD68 (ab125212, Abcam, Cambridge, UK) at 4 °C overnight and followed by the incubation with EnVisionTM + /horseradish peroxidase regent for 30 min at 37 °C. Then, these sections were stained with DAB. Sections were assessed using a fluorescence microscope (OLYMPUS DX51, Tokyo, Japan) and further analyzed with the Image-Pro Plus software (version 6.0).
Statistical analysis
All statistical analyses in this study were performed using the SPSS version 25.0 software and are presented as means ± standard deviation (SD). Differences between the multiple groups were evaluated by one-way ANOVA, followed by the post hoc Tukey test. Two groups were analyzed using an unpaired Student’s t test. All studies in vivo and in vitro were performed blind. P < 0.05 was considered statistically significant.
Results
Effects of lupeol on the survival of NRCM (Neonatal rat cardiac myocytes)
The results of lupeol cytotoxicity on primary cultured NRCMs detected by Cell Counting Kit-8 (CCK-8) assay showed that high concentrations of lupeol (100 μg/mL) resulted in a 17% reduction in cell viability. And statistical analysis revealed that no distinct difference in cell viability (P > 0.05) was observed in other groups. Based on the above results, lupeol at the concentrations of 12.5–75 μg/mL had no cytotoxicity on the NRCMs (Fig. 1a). Compared with the PBS group, the cell viability in PE-stimulated NRCMs (100 μM) was decreased by about 22%, and the treatment with 25–75 μg/mL lupeol restored the reduction (Fig. 1b). As 50 μg/mL lupeol had the most obvious protective effect on cell viability, this concentration was selected for further experiments. Besides, trials to select the appropriate time were conducted for use in subsequent experiments, and 12 h was found to cause a distinct change of cell viability.

a Cell viability was detected in NRCMs treated with different concentrations of lupeol (0, 12.5, 25, 50, 75, 100 μg/mL). b Cell viability was detected in NRCMs treated with different concentrations of lupeol as well as 100 μM PE. All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test. **P < 0.01.
Lupeol attenuated cardiomyocyte hypertrophy
To determine whether lupeol protected against cardiomyocytes hypertrophy induced by PE, the NRCMs were used as a standard experimental setting to establish an in vitro model. The cells were treated with or without PE for 12 h, and lupeol was administered at a proven safe concentration. Then the cell surface area was assessed with the α-actinin immunostaining. The results showed that lupeol caused a decrease in cardiomyocyte size (Fig. 2a, b) in the PE-stimulated NRCMs. Besides, the upregulated hypertrophic markers such as atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-major histocompatibility complex (β-MHC) (Fig. 2c) were also strikingly blunted via the lupeol treatment. These findings implied that the lupeol could attenuate the NRCMs hypertrophy induced by the PE.

a Images of NRCMs from the indicated groups stained for α-actinin (green) (n = 50, scale bar: 50 μm). b Quantification of the cell area of NRCM based on pixel size using ImageJ software. c Expression of hypertrophic gene (BNP, ANP, β-MHC) in the NRCMs with the indicated treatment. All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test. **P < 0.01.
Lupeol attenuated cardiac hypertrophy induced by pressure-overload
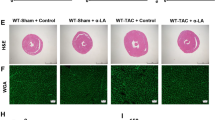
To demonstrate the effects of lupeol on CH, we next established a pressure-overload hypertrophy model via TAC surgery. After 4 weeks of the TAC surgery, CH was evaluated by echocardiography and hemodynamics parameters, the gross appearance of the heart, myocyte cross-sectional area (CSA), and the expression of hypertrophy-related genes including ANP, BNP, and β-MHC. As shown in Fig. 3a, b, compared with the control group, the TAC group showed remarkable hypertrophy, determined from the greater ventricular thickness and deteriorated cardiac function. However, the CH of mice treated with lupeol after TAC surgery was abolished, including decreased HW/BW and HW/TL ratios (Fig. 3c), the decreased gross volume of the heart (Fig. 3d), the decreased CSA (Fig. 3e, f), and decreased hypertrophic markers (Fig. 3g). Moreover, the lupeol inhibited the left ventricular dilation induced by TAC surgery, as shown by decreased LV end-diastolic dimension. Besides, the lupeol prevented the increase of interventricular septal thickness at the end-diastole and the decrease of fractional shortening (FS) compared to those TAC mice without the lupeol administration. Taken these results together, lupeol could inhibit the CH caused by continuous pressure overload.

a Echocardiographic pictures of the left ventricular chamber, and IVSd, LVEDd, fractional shortening (FS) and ejection fraction (EF) were measured (n ≥ 5 each group). b Hemodynamic parameters of all groups 4 weeks after surgery (n ≥ 5 each group). c Gravimetric analysis of heart weight (HW)/body weight (BW) and HW/tibia length (TL) (n ≥ 5 each group). d Histological analysis of whole hearts (n = 6). e Hematoxylin and eosin-stained cardiac cross-sections for the indicated groups (scale bar: 1000 and 20 μm). f Quantification analysis of the cross-sectional areas in Lup-treated mice. g The effects of lupeol on hypertrophic markers ANP, BNP and β-MHC (n = 3) mRNA expression were determined by quantitative real-time polymerase chain reaction. All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test. *P < 0.05, **P < 0.01.
Lupeol inhibited cardiac fibrosis in vivo and in vitro
Cardiac fibrosis and CH promote each other and eventually lead to cardiac failure by decreasing pumping ability and increasing myocardial stiffness [26, 27]. Therefore, we performed PSR staining as well as Masson’s trichrome staining and quantitative analysis to further detect the role of lupeol on cardiac fibrosis. And the results showed that the mice with the TAC surgery presented remarkably increased fibrosis compared to the mice under basal conditions (Fig. 4a). However, the TAC surgery-induced fibrosis was significantly inhibited in the lupeol treatment group. Consistent with the decreased collagen volume, the mRNA expression levels of the fibrosis markers including Collagen Iα, Collagen IIIα, and CTGF were also remarkably decreased after the administration of the lupeol (Fig. 4b). Also, we conducted Western blots, and the protein levels of the TGF-β, as well as p-SMAD2, were increased in the TAC group and the opposite trend was observed in the TAC + Lup group (Fig. 4c). In addition, the effects of the lupeol on the PE-induced fibrosis in the NRCMs were also confirmed (Fig. 4d–f).

a PSR and Masson staining of cardiac fibrosis (n = 6, scale bar: 200 μm) and the percentage of fibrotic area. b, d The effects of lupeol on Col-I, Col-IIIα and CTGF mRNA expression in vivo and in vitro were assessed by quantitative real-time polymerase chain reaction (n = 3). c, e Immunoblotting analysis of TGF-β and p-SMAD2 protein expression levels in vivo and in vitro and quantification analysis of immunoblotting results, GAPDH was used as an internal control (n = 6). f Images of NRCMs from the indicated groups stained for α-SMA (green) (n = 50, scale bar: 50 μm). All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test. **P < 0.01.
Lupeol inhibited cardiomyocyte apoptosis
Cell apoptosis is a significant process in the development of pressure overload-induced CH [28]. To identify whether lupeol inhibited hypertrophy via affecting apoptosis, we next assessed the levels of the apoptotic cell through the TUNEL assay. As shown in Fig. 5a, b, the proportion of the apoptotic cells was increased in the mice with the TAC surgery, which was reduced after the lupeol treatment. Besides, the inhibitory effects of lupeol on apoptosis were further verified by the Western blot, which showed that the lupeol upregulated the level of the Bcl-2 protein, downregulated the protein levels of Bax and cleaved caspase 3 caused by the stress (Fig. 5c, d). Consistent with the alteration of protein expression, the mRNA levels also supported that the lupeol treatment inhibited the apoptotic process (Fig. 5e). Similar protein and mRNA results were obtained from the PE-treated cardiomyocyte in vitro (Fig. 5f–h). These results indicated that the lupeol could protect against the TAC surgery-induced cardiac apoptosis.

a TUNEL staining in cardiac issues (n = 6, scale bar: 50 μm). b Statistical results of TUNEL positive-cell. c Western blots showing the expression of Bcl-2, Bax and c-caspase3 in mouse heart in vivo (n = 6). d Quantitative analysis of the immunoblot data. e The mRNA expression of apoptosis markers in vivo, including Bcl-2, Bax and c-caspase 3. f The mRNA expression of apoptosis markers in vitro, including Bcl-2, Bax and c-caspase3. g Western blots showing the expression of Bax, c-caspase3 and Bcl-2 in cardiomyocytes in vitro (n = 6). h Quantitative analysis of the immunoblot data. All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test or using an unpaired Student’s t test. **P < 0.01.
Lupeol attenuated the inflammatory responses
Cardiac hypertrophy (CH) was accompanied by an inflammatory response [29], and a previous study demonstrated that the lupeol decreased the inflammatory response of wound healing in hyperglycemic rats [21]. Hence, we next investigated whether the lupeol inhibited the inflammatory response caused by the TAC surgery. We first evaluated the levels of inflammatory cytokines by the RT-PCR. Compared with the TAC surgery group, the treatment of lupeol remarkably improved the cardiac mRNA levels of inflammatory factors, including increased IL-10 levels and decreased IL-6 as well as TNF-α levels (Fig. 6a). Besides, considering that the CD45 and CD68 as indicators of inflammation, studies have shown that the TLR4 regulated the CD45+ cell infiltration [30]. Immunohistochemistry (IHC) was conducted, and the results showed that the lupeol downregulated the numbers of CD45 and CD68-positive cells in the TAC + Lup group compared to the TAC group (Fig. 6b, c). Furthermore, we conducted the WB and the results of protein expression also confirmed that the p-IκBα and NF-κB p65 levels in the TAC + lupeol group were significantly decreased but not in the single TAC group (Fig. 6d, e). Moreover, the NF-κB p65 level in the PE-stimulated NRCMs was also inhibited by lupeol (Fig. 6f, g). Further detection of the NF-κB p65 by immunofluorescent staining indicated that the p65 in unstimulated cells was mainly localized in the cytoplasm while it was translocated to the nucleus in the PE-stimulated group. This translocation could have been inhibited by the lupeol treatment (Fig. 6h), which supported that the lupeol could attenuate the inflammatory response.

a The effects of lupeol on TNF-α, IL-6 and IL-10 mRNA expression were assessed by quantitative real-time polymerase chain reaction (n = 3). b Immunohistochemical staining of CD45 and CD68 in the heart (n = 6, scale bar: 200 μm). c Quantification analysis of CD45 and CD68-positive areas. d–g Images of immunoblotting showing the expression of p-IκBα and p-p65 (d, f) in vivo and in vitro, respectively, and Quantitative analysis of the immunoblot data (e, g) (n = 6). h Images of cardiomyocytes stained for p65 (scale bar: 50 μm). All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test or using an unpaired Student’s t test. **P < 0.01.
Protective effects of lupeol involve the PI3K-Akt-NF-κB signaling
The results suggested that the lupeol attenuated the cardiac injury induced by the pressure-overload. Yet, the underlying molecular mechanisms by which lupeol protected against TAC-induced cardiac injuries were still unclear. Hence, we next conducted experiments to examine which signaling was responsible for the inflammatory cytokine production specific to CH. To clarify whether the PI3K-Akt pathway, which is known to be pro-inflammatory [31, 32], participated in the inflammatory response during cardiac remodeling, the phosphorylation of the PI3K and Akt were measured by the Western blot. The level of the p-PI3K and p-Akt were upregulated after the TAC surgery, and the treatment with lupeol significantly abolished the activation of the PI3K and Akt (Fig. 7a, b), which demonstrated that the PI3K-Akt signaling was indeed involved in the cardiac remodeling. Furthermore, the PI3Kα and PI3Kγ are the two isoforms of the PI3K, which play critical roles in cardiomyocyte growth and function, linking to adaptive and maladaptive CH. Hence, we specifically investigated the changes of the PI3Kγ to recapitulate the effects of the PI3K. And we found that the trend of the p-PI3Kγ protein modifications was similar to the p-PI3K (Supplementary Fig. S1). In addition, the effects of the lupeol on the PE-induced NRCMs were also noticed (Fig. 7c, d). To further detect whether the PI3K-Akt signaling regulated the CH via NF-κB p65, 740 Y-P (a PI3K/Akt agonist) was used. After pre-treatment with the 740 Y-P in the lupeol+ PE-treated NRCMs, the downregulation of mRNA levels of hypertrophic markers was restored (Fig. 7e), and the decreased protein levels of phosphorylated Akt as well as p-p65 were nullified, which completely blocked the protected effects of lupeol against hypertrophy (Fig. 7f, g). Besides, mice were intraperitoneally injected with 740 Y-P to confirm the effects of PI3K-Akt-NF-κB. As shown in the results, the improved echocardiography parameters and the expression of hypertrophy-related genes in TAC + Lup groups were abolished by 740 Y-P (Fig. 8a–f).

a Western blot showing the expression of p-PI3K and p-Akt in mouse heart. b Quantification analysis of immunoblotting results. c Western blot showing the expression of p-PI3K and p-Akt in NRCMs. d Quantification analysis of immunoblotting results. e RT-PCR was used to assess the mRNA levels of hypertrophic genes (ANP, BNP, β-MHC) after treatment with 740Y-P. f Western blots showing the the effect of 740Y-P on p-PI3K, p-Akt, and p-p65, and (g) the quantitative analysis of these blots (n = 6). All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test. *P < 0.05, **P < 0.01.

a, b Echocardiographic pictures of the left ventricular chamber, and fractional shortening (FS) and ejection fraction (EF) were measured (n ≥ 5 each group). c Gravimetric analysis of heart weight (HW)/body weight (BW) and HW/tibia length (TL) (n ≥ 5 each group). d Hematoxylin and eosin-stained cardiac cross-sections for the indicated groups (n = 6, scale bar: 1000 and 20 μm). e Quantification analysis of the cross-sectional areas. f The effect of lupeol on hypertrophic markers ANP, BNP and β-MHC (n = 3) mRNA expression were determined by quantitative real-time polymerase chain reaction. All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test. **P < 0.01.
The involvement of the TLR4 in the PI3K-Akt-NF-κB signaling
The TLR4 induced an innate immune response and played a critical role in regulating the inflammatory factors production [33, 34]. Findings of the ATP50-3 showed that it exerted neuroinflammation protective effects via modulation of the TLR4-mediated PI3K-Akt signaling pathways [35]. Hence, we next examined whether the alteration of the PI3K-Akt-NF-κB signaling could be associated with the TLR4 signaling pathway, which includes the TLR4 and Myd88. As shown in Fig. 9a, b, the lupeol blocked the increased protein levels of the TLR4 and Myd88 in the TAC surgery group. Similar protein and mRNA results were obtained from the PE-treated cardiomyocyte in vitro (Fig. 9c, d). Furthermore, the NRCMs were treated with the RS 09 (a TLR4 agonist), and the downregulated hypertrophic markers mRNA levels (Fig. 9e), as well as the inhibited PI3K-Akt-NF-κB signaling (Fig. 9f, g) were restored in the PE + Lup pretreated with the RS 09, which demonstrated that the PI3K-Akt-NF-κB was controlled by the TLR4. These results demonstrated that the TLR4-PI3K-Akt-NF-κB signaling may have been the underlying mechanism of the lupeol’s function against CH (Fig. 10).

a Images of immunoblotting showing the expression of TLR4 and Myd88 in mouse heart (n = 6). b Quantitative analysis of the immunoblot data. c Western blot showing the expression of TLR4 and Myd88 in NRCMs. d Quantification analysis of immunoblotting results. e RT-PCR was used to assess the mRNA levels of hypertrophic genes (ANP, BNP, β-MHC) after treatment with RS 09. f Western blots showing the the effect of RS 09 on PI3K-Akt-NF-κB signaling, and (g) the quantitative analysis of these blots (n = 6). All data are expressed as the mean ± SD. Statistics were performed using a one-way ANOVA and followed by a post hoc Tukey test. **P < 0.01.

The treatment with lupeol, by inhibiting the TLR4-PI3K-AKT-NF-κB signaling, alleviates inflammatory response during cardiac hypertrophy.
Discussion
In this study, we provided evidence that the lupeol inhibited CH in vivo after the TAC-induced chronic pressure overload and in vitro after the induction by PE. Besides, the lupeol attenuated cardiac fibrosis and collagen deposition. The protection of lupeol was due to the inhibition of the TLR4-PI3K-Akt-NF-κB signaling, which then inhibited the inflammatory response during CH. And these protective effects of the lupeol were nullified after administering the PI3K/Akt agonist 740 Y-P and TLR4 agonist RS 09, including the reversal of the cardiomyocyte hypertrophy as well as its downstream signaling pathway.
Cardiac hypertrophy (CH) is characterized by the increase of cardiomyocyte volume and accumulation of collagen as well as the extracellular matrix. The inflammatory cells promote the development of structural remodeling by secreting inflammatory mediators and factors. The secretion and accumulation of the TNF-α can inhibit the Ca2+ pump in cardiomyocytes and have a negative ionotropic effect on cardiomyocytes. The TNF-α can also activate multiple apoptotic pathways and promote the apoptosis of cardiomyocytes. The secretion of the IL-6 can promote inflammation and damage, stimulate intracellular signaling pathways such as the JAK/STAT and MAPK, and accelerate pathological structural remodeling. On the other hand, these inflammatory factors further promote the recruitment of inflammatory cells and secrete more inflammatory factors. They also act on myocardial cells, interstitial fibroblasts, and endothelial cells to accelerate myocardial cell apoptosis and myocardial interstitial fibrosis. Previous studies showed that the lupeol attenuated the inflammatory responses, which were associated with CH. In this study, we demonstrated that the treatment with lupeol remarkably improved the cardiac mRNA levels of inflammatory factors, including IL-10, IL-6, and TNF-α. The IHC results also showed that the lupeol reduced the numbers of CD45 and CD68-positive cells in the TAC + Lup group compared to the TAC group. However, the concrete effects and mechanisms of the lupeol’s action on the inflammatory response of cardiac remodeling have not yet been studied comprehensively. Mounting evidence has demonstrated that the TLR4 and the PI3K-Akt-NF-κB signaling played critical roles in the process of CH [8, 36]. And pharmacological interventions that targeted these molecules attenuated the cardiac remodeling [37, 38].
The NF-κB, as an important transcription factor, plays a crucial role in the primary stage of immune responses as well as the expression of proinflammatory cytokines during the process of CH [39]. The inhibition of the NF-κB suppressed the expression of proinflammatory cytokines and then attenuated the cardiac remodeling [40]. It was reported that the lupeol alleviated the inflammatory response by inhibiting the NF-κB pathway in macrophages [41]. In this research, the phosphorylation of the IκBα, as well as NF-κB was increased in the model of CH, and the lupeol decreased these upregulations in the TAC + lupeol group. Then, we aimed to know which signaling was responsible for the activation and translocation of the NF-κB, considering the PI3K-Akt and NF-κB signaling pathways were closely related [35, 42, 43]. The primary activation of the Akt signaling supported the growth and development of the heart, however, continuous Akt signaling activation eventually contributed to CH [44]. Previous research from our lab and others have demonstrated that the activated PI3K-Akt signaling participated in the process of pressure overload-induced CH partly through controlling the general protein translational machinery. Our team also found that inhibiting the expression of the Akt reduced CH and blunted the myofibroblast activation and transformation in vitro. The upregulation of the expression of the Akt abolished the protective effects of leflunomide on CH [45, 46]. Besides, studies also have found that the lupeol inhibited the inflammation by blunting the phosphorylation of the PI3K-Akt and the expression of the NF-κB [24]. Mohammad et al. demonstrated that the administration of the lupeol inhibited the inflammation in skin tumorigenesis in CD-1 mice via the inhibition of activated PI3K and Akt as well as the phosphorylation of the NF-κB [23]. Consistent with these previous results, our research found that in the model of hypertrophy induced by pressure overload in vivo or PE stimulation in vitro, the PI3K and Akt were activated, however, the treatment with lupeol nullified these effects. The PI3Kα and PI3Kγ are the two isoforms of the PI3K, which play critical roles in cardiomyocyte growth and function, linking to adaptive and maladaptive CH. Hence, we next investigated the changes of the PI3Kγ to further elucidate the effects of the PI3K and we found that the trend of the p-PI3Kγ modifications was similar to the p-PI3K. To further verify that the lupeol can inhibit the NF-κB expression through the PI3K signaling pathway, we used the PI3K agonist, 740 Y-P, which could have bound to the GST fusion proteins containing the N- and C-terminal SH2 domains of the p85 [47]. The results proved that the PI3K agonists can reverse the cardioprotective effects and the inhibition of PI3K by lupeol. Therefore, lupeol can protect against CH by inhibiting the PI3K activation.
Furthermore, we aimed to clarify how the PI3K-Akt-NF-κB signaling was activated. The TLR4 signaling pathway that increases myocardial damage and inflammation had a considerable role in the development of cardiac remodeling [48]. A previous study found that the TLR4 signaling was involved in the pressure overload-induced myocardial inflammation, and the TLR4 degradation restored the deteriorated phenotype [49]. In the TLR4 signaling pathway, as a critical activator, the MyD88-dependent signaling regulated the NF-κB signaling [50]. In addition, a study has shown that the lupeol downregulated the TLR inflammatory signaling, and then attenuated the GaIN/LPS-induced liver injury. In the present study, the expression of the TLR4 and the phosphorylation of the IκBα, as well as NF-κB were increased in the model of CH, and lupeol decreased these upregulations in the TAC + Lup group. Next, a TLR4 agonist, RS 09 was used to verify its downstream signaling pathways. The results showed that the RS 09 restored the downregulated mRNA levels of hypertrophic markers and the inhibited PI3K-Akt-NF-κB pathway in the PE + Lup group, showing that lupeol exerted cardioprotective effects completely via the TLR4 signaling. Herein, our study provided experimental evidence that the lupeol might have the potential in the treatment of CH.
In conclusion, this current study showed that the lupeol protected against CH induced by pressure overload via inhibiting the TLR4-PI3K-Akt-NF-κB signaling-induced inflammatory response. These results provided evidence for the application of lupeol in the potential treatment of CH. In the future, the structural basis of the TLR4 inhibition and modulation by the lupeol, as well as the way of lupeol enters into cells could be further studied.
References
Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407.
Lei H, Hu J, Sun K, Xu D. The role and molecular mechanism of epigenetics in cardiac hypertrophy. Heart Fail Rev. 2021;26:1505–14.
Camici PG, Tschöpe C, Di Carli MF, Rimoldi O, Van Linthout S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc Res. 2020;116:806–16.
Tschöpe C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. 2021;18:169–93.
Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, et al. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. 2019;114:19.
Xiao Z, Kong B, Yang H, Dai C, Fang J, Qin TY, et al. Key player in cardiac hypertrophy, emphasizing the role of toll-like receptor 4. Front Cardiovasc Med. 2020;7:579036.
Dai W, Tang T, Dai Z, Shi D, Mo L, Zhang Y. Probing the mechanism of hepatotoxicity of hexabromocyclododecanes through toxicological network analysis. Environ Sci Technol. 2020;54:15235–45.
Wang X, Chen L, Zhao X, Xiao LL, Yi ST, Kong YW, et al. A cathelicidin-related antimicrobial peptide suppresses cardiac hypertrophy induced by pressure overload by regulating IGFR1/PI3K/Akt and TLR9/AMPKα. Cell Death Dis. 2020;11:96.
Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5:209.
Lin K, Luo W, Yan J, Shen SY, Shen QR, Wang J, et al. TLR2 regulates angiotensin II-induced vascular remodeling and EndMT through NF-κB signaling. Aging (Albany NY). 2020;13:2553–74.
Lee TL, Lai TC, Lin SR, Lin SW, Chen YC, Pu CM, et al. Conditioned medium from adipose-derived stem cells attenuates ischemia/reperfusion-induced cardiac injury through the microRNA-221/222/PUMA/ETS-1 pathway. Theranostics. 2021;11:3131–49.
Surai PF, Kochish II, Kidd MT. Redox homeostasis in Poultry: Regulatory roles of NF-κB. Antioxid (Basel). 2021;10:186.
Chen Y, Yang B, Stanton C, Ross RP, Zhao JX, Zhang H, et al. Bifidobacterium pseudocatenulatum ameliorates DSS-induced colitis by maintaining intestinal mechanical barrier, blocking proinflammatory cytokines, inhibiting TLR4/NF-κB signaling, and altering gut microbiota. J Agric Food Chem. 2021;69:1496–512.
Perez-Cornago A, Crowe FL, Appleby PN, Bradbury KE, Wood AM, Jakobsen MU, et al. Plant foods, dietary fibre and risk of ischaemic heart disease in the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort. Int J Epidemiol. 2021;50:212–22.
Zou LX, Chen C, Yan X, Lin QY, Fang J, Li PB, et al. Resveratrol attenuates pressure overload-induced cardiac fibrosis and diastolic dysfunction via PTEN/Akt/Smad2/3 and NF-κB signaling pathways. Mol Nutr Food Res. 2019;63:e1900418.
Zhang X, Zhu JX, Ma ZG, Wu HM, Xu SC, Song P, et al. Rosmarinic acid alleviates cardiomyocyte apoptosis via cardiac fibroblast in doxorubicin-induced cardiotoxicity. Int J Biol Sci. 2019;15:556–67.
Liu K, Zhang X, Xie L, Deng M, Chen HJ, Song JW, et al. Lupeol and its derivatives as anticancer and anti-inflammatory agents: molecular mechanisms and therapeutic efficacy. Pharmacol Res. 2021;164:105373.
Tsai FS, Lin LW, Wu CR. Lupeol and its role in chronic diseases. Adv Exp Med Biol. 2016;929:145–75.
Wang XT, Gong Y, Zhou B, Yang JJ, Cheng Y, Zhao JG, et al. Ursolic acid ameliorates oxidative stress, inflammation and fibrosis in diabetic cardiomyopathy rats. Biomed Pharmacother. 2018;97:1461–7.
Ma ZG, Dai J, Wei WY, Zhang WB, Xu SC, Liao HH, et al. Asiatic acid protects against cardiac hypertrophy through activating AMPKα signalling pathway. Int J Biol Sci. 2016;12:861–71.
Beserra FP, Vieira AJ, Gushiken LFS, Souza EOD, Hussni MF, Hussni CA, et al. Lupeol, a dietary triterpene, enhances wound healing in streptozotocin-induced hyperglycemic rats with modulatory effects on inflammation, oxidative stress, and angiogenesis. Oxid Med Cell Longev. 2019;2019:3182627.
Kim SJ, Cho HI, Kim SJ, Kim JS, Kwak JH, Lee DU, et al. Protective effects of lupeol against D-galactosamine and lipopolysaccharide-induced fulminant hepatic failure in mice. J Nat Prod. 2014;77:2383–8.
Saleem M, Afaq F, Adhami VM, Mukhtar H. Lupeol modulates NF-kappaB and PI3K/Akt pathways and inhibits skin cancer in CD-1 mice. Oncogene. 2004;23:5203–14.
Pereira Beserra F, Xue M, Maia GLA, Leite Rozza A, Helena Pellizzon C, Jackson CJ. Lupeol, a pentacyclic triterpene, promotes migration, wound closure, and contractile effect in vitro: possible involvement of PI3K/Akt and p38/ERK/MAPK pathways. Molecules. 2018;23:2819.
Sudhahar V, Kumar SA, Sudharsan PT, Varalakshmi P. Protective effect of lupeol and its ester on cardiac abnormalities in experimental hypercholesterolemia. Vasc Pharmacol. 2007;46:412–8.
Bacmeister L, Schwarzl M, Warnke S, Stoffers B, Blankenberg S, Westermann D, et al. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol. 2019;114:19.
Burke RM, Burgos Villar KN, Small EM. Fibroblast contributions to ischemic cardiac remodeling. Cell Signal. 2021;77:109824.
Martinelli I, Timotin A, Moreno-Corchado P, Marsal D, Kramar S, Loy H, et al. Galanin promotes autophagy and alleviates apoptosis in the hypertrophied heart through FoxO1 pathway. Redox Biol. 2021;40:101866.
Kumari S, Katare PB, Elancheran R, Nizami HL, Paramesha B, Arava S, et al. Musa balbisiana fruit rich in polyphenols attenuates isoproterenol-induced cardiac hypertrophy in rats via inhibition of inflammation and oxidative stress. Oxid Med Cell Longev. 2020;2020:7147498.
Deng S, Hu Y, Zhou J, Wang YF, Wang YG, Li SC, et al. TLR4 mediates alveolar bone resorption in experimental peri-implantitis through regulation of CD45+ cell infiltration, RANKL/OPG ratio, and inflammatory cytokine production. J Periodontol. 2020;91:671–82.
Wang D, Jin M, Zhao X, Zhao TY, Lin W, He ZL, et al. FGF1ΔHBS ameliorates chronic kidney disease via PI3K/Akt mediated suppression of oxidative stress and inflammation. Cell Death Dis. 2019;10:464.
Liu Y, Tong C, Tang Y, Liu Y, Shi XY, Shi L, et al. Tanshinone IIA alleviates blast-induced inflammation, oxidative stress and apoptosis in mice partly by inhibiting the PI3K/Akt/FoxO1 signaling pathway. Free Radic Biol Med. 2020;152:52–60.
Li B, Leung JCK, Chan LYY, Yiu WH, Tang SCW. A global perspective on the crosstalk between saturated fatty acids and toll-like receptor 4 in the etiology of inflammation and insulin resistance. Prog Lipid Res. 2020;77:101020.
Iannucci A, Caneparo V, Raviola S, Debernardi I, Colangelo D, Miggiano R, et al. Toll-like receptor 4-mediated inflammation triggered by extracellular IFI16 is enhanced by lipopolysaccharide binding. PLoS Pathog. 2020;16:e1008811.
Zhong J, Qiu X, Yu Q, Chen H, Yan C. A novel polysaccharide from acorus tatarinowii protects against LPS-induced neuroinflammation and neurotoxicity by inhibiting TLR4-mediated MyD88/NF-κB and PI3K/Akt signaling pathways. Int J Biol Macromol. 2020;163:464–75.
Wen J, Shen J, Zhou Y, Zhao X, Dai Z, Jin Y. Pyrroloquinoline quinone attenuates isoproterenol hydrochloride‑induced cardiac hypertrophy in AC16 cells by inhibiting the NF‑κB signaling pathway. Int J Mol Med. 2020;45:873–85.
Prakoso D, De Blasio MJ, Tate M, Kiriazis H, Donner DG, Qian HW, et al. Gene therapy targeting cardiac phosphoinositide 3-kinase (p110α) attenuates cardiac remodeling in type 2 diabetes. Am J Physiol Heart Circ Physiol. 2020;318:H840–H852.
Mian MOR, He Y, Bertagnolli M, Mai-Vo TA, Fernandes RO, Boudreau F, et al. TLR (toll-like receptor) 4 antagonism prevents left ventricular hypertrophy and dysfunction caused by neonatal hyperoxia exposure in rats. Hypertension. 2019;74:843–53.
Sun J, Niu C, Ye W, An N, Chen G, Huang XZ, et al. FGF13 is a novel regulator of NF-κB and potentiates pathological cardiac hypertrophy. iScience. 2020;23:101627.
Zhou H, Li N, Yuan Y, Jin YG, Wu QQ, Yan L, et al. Leukocyte immunoglobulin-like receptor B4 protects against cardiac hypertrophy via SHP-2-dependent inhibition of the NF-κB pathway. J Mol Med (Berl). 2020;98:691–705.
Buakaew W, Pankla Sranujit R, Noysang C, Thongsri Y, Potup P, Nuengchamnong N, et al. Phytochemical constituents of citrus hystrix DC. leaves attenuate inflammation via NF-κB signaling and NLRP3 inflammasome activity in macrophages. Biomolecules. 2021;11:105.
Yang L, Hu X, Mo YY. Acidosis promotes tumorigenesis by activating Akt/NF-κB signaling. Cancer Metastasis Rev. 2019;38:179–88.
He L, Pan Y, Yu J, Wang B, Dai G, Ying X. Decursin alleviates the aggravation of osteoarthritis via inhibiting PI3K-Akt and NF-κB signal pathway. Int Immunopharmacol. 2021;97:107657.
Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114:368–78.
Ma ZG, Yuan YP, Zhang X, Xu SC, Wang SS, Tang QZ. Piperine attenuates pathological cardiac fibrosis via PPAR-γ/Akt pathways. EBioMedicine. 2017;18:179–87.
Wang HB, Huang SH, Xu M, Yang J, Yang J, Liu MX, et al. Galangin ameliorates cardiac remodeling via the MEK1/2-ERK1/2 and PI3K-Akt pathways. J Cell Physiol. 2019;234:15654–67.
Derossi D, Williams EJ, Green PJ, Dunican DJ, Doherty P. Stimulation of mitogenesis by a cell-permeable PI3-kinase binding peptide. Biochem Biophys Res Commun. 1998;251:148–52.
Liu T, Zhang M, Niu H, Jing Liu J, Ma RL, Wang YI, et al. Astragalus polysaccharide from astragalus melittin ameliorates inflammation via suppressing the activation of TLR-4/NF-κB p65 signal pathway and protects mice from CVB3-induced virus myocarditis. Int J Biol Macromol. 2019;126:179–86.
Lu X, He Y, Tang C, Wang XY, Que LL, Zhu GQ, et al. Triad3A attenuates pathological cardiac hypertrophy involving the augmentation of ubiquitination-mediated degradation of TLR4 and TLR9. Basic Res Cardiol. 2020;115:19.
Rathore M, Girard C, Ohanna M, Tichet M, Jouira RB, Garcia E, et al. Cancer cell-derived long pentraxin 3 (PTX3) promotes melanoma migration through a toll-like receptor 4 (TLR4)/NF-κB signaling pathway. Oncogene. 2019;38:5873–89.
Acknowledgements
This study was supported by the Key Project from the National Natural Science Foundation (82000229), National Key R&D Program of China (2018YFC1311300), Development Center for Medical Science and Technology National Health and Family Planning Commission of the People’s Republic of China (The prevention and control project of the cardiovascular disease, 2016ZX-008-01), and Science and Technology Planning Projects of Wuhan (2018061005132295).
Author information
Authors and Affiliations
Contributions
DL, MX, and QZT designed research; DL, YYG, XFC, HLQ, SC, XFZ, and QZ performed the experiments; DL and YYG analyzed data; DL and MX wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, D., Guo, Yy., Cen, Xf. et al. Lupeol protects against cardiac hypertrophy via TLR4-PI3K-Akt-NF-κB pathways. Acta Pharmacol Sin 43, 1989–2002 (2022). https://doi.org/10.1038/s41401-021-00820-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00820-3
Keywords
This article is cited by
-
Diagnostic and prognostic potential of long non-coding RNA NORAD in patients with acute deep vein thrombosis and its role in endothelial cell function
Thrombosis Journal (2024)
-
D-allose Inhibits TLR4/PI3K/AKT Signaling to Attenuate Neuroinflammation and Neuronal Apoptosis by Inhibiting Gal-3 Following Ischemic Stroke
Biological Procedures Online (2023)
-
Chronic β-adrenergic stress contributes to cardiomyopathy in rodents with collagen-induced arthritis
Acta Pharmacologica Sinica (2023)
-
Agathis dammara Extract and its Monomer Araucarone Attenuate Abdominal Aortic Aneurysm in Mice
Cardiovascular Drugs and Therapy (2023)
