
Abstract
We previously show that L-Cysteine administration significantly suppresses hypoxia-ischemia (HI)-induced neuroinflammation in neonatal mice through releasing H2S. In this study we conducted proteomics analysis to explore the potential biomarkers or molecular therapeutic targets associated with anti-inflammatory effect of L-Cysteine in neonatal mice following HI insult. HI brain injury was induced in postnatal day 7 (P7) neonatal mice. The pups were administered L-Cysteine (5 mg/kg) at 24, 48, and 72 h post-HI. By conducting TMT-based proteomics analysis, we confirmed that osteopontin (OPN) was the most upregulated protein in ipsilateral cortex 72 h following HI insult. Moreover, OPN was expressed in CD11b+/CD45low cells and infiltrating CD11b+/CD45high cells after HI exposure. Intracerebroventricular injection of OPN antibody blocked OPN expression, significantly attenuated brain damage, reduced pro-inflammatory cytokine levels and suppressed cerebral recruitment of CD11b+/CD45high immune cells following HI insult. L-Cysteine administration reduced OPN expression in CD11b+/CD45high immune cells, concomitant with improving the behavior in Y-maze test and suppressing cerebral recruitment of CD11b+/CD45high immune cells post-HI insult. Moreover, L-Cysteine administration suppressed the Stat3 activation by inducing S-sulfhydration of Stat3. Intracerebroventricular injection of Stat3 siRNA not only decreased OPN expression, but also reversed HI brain damage. Our data demonstrate that L-Cysteine administration effectively attenuates the OPN-mediated neuroinflammation by inducing S-sulfhydration of Stat3, which contributes to its anti-inflammatory effect following HI insult in neonatal mice. Blocking OPN expression may serve as a new target for therapeutic intervention for perinatal HI brain injury.

Similar content being viewed by others


Introduction
The physiological importance of H2S has been clarified as an endogenous neuromodulator [1, 2]. H2S is produced in mammalian cells from the metabolism of L-Cysteine and homocysteine by three enzymes: cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3MST). H2S-producing enzymes are expressed in various types of tissue and cells, including brain [3]. H2S plays a significant protective role in neurodegenerative disorders [4], stroke [5], and subarachnoid hemorrhage [6]. Moreover, H2S is also involved in inflammatory response by suppressing leukocyte adherence and infiltration, and inducing neutrophils apoptosis [7]. H2S is able to decrease LPS-induced expression of pro-inflammatory cytokines in macrophage [8] and microglia [9]. Recently, we found that L-Cysteine was able to suppress microglial activation and neuroinflammation following perinatal hypoxia-ischemia (HI) brain injury by H2S releasing [10,11,12]. Although these reports demonstrated its anti-inflammatory effects, the underlying mechanisms of H2S in HI have not been fully elucidated.
Using proteomics to identify candidate proteins involved in neuronal apoptosis and neuroinflammation associated with HI insult contributes to elucidation of the potential mechanisms underlying HI brain damage. The two-dimensional differential proteome analysis of lesion cortex 2 h following HI has identified dopamine neurotransmission [13]. More recently, a 2D-DIGE-based proteome analysis of lesion hemispheres 48 h after HI has identified novel factors involved in the inflammatory response, such as Calcineurin A, Coronin-1A, as well as glial fibrillary acidic protein (GFAP) [14]. Seventy-two hours after HI insult, a time point where microglia has already activated, and with the higher level of widespread pro-inflammatory cytokine releasing within the acute injury [15, 16]. We previously found that CD16, CD86, and CD206 mRNA achieved maximal levels at 72 h after HI exposure [17]. A comprehensive proteomic profiling of proteins associated with inflammatory responses in the lesion cortex 72 h after HI insult is still lacking.
In the present study, we aimed to use TMT-based proteomic analysis to examine differences in protein expression in ipsilateral cortex of mice treated with L-Cysteine 72 h following HI. We subsequently focused on exploring the potential biomarkers or molecular therapeutic targets associated with anti-inflammatory property of L-Cysteine treatment.
Materials and methods
Reagents
L-Cysteine (as a donor of H2S) and aminooxyacetic acid (AOAA) (a nonspecific inhibitor of H2S-producing enzymes) were purchased from Sigma (Aldrich, St. Louis, MO, USA). Other reagents for the present study were listed in Supplementary Table S1.
Mouse HI model and treatment
Pregnant C57BL/6J mice (≥15 days gestation) were purchased from the Center for Experimental Animals of Shandong University and maintained under standardized environmental conditions. The study was approved by the Animal Ethics and Welfare Committee of the Shandong University. On postnatal day 7 (P7), HI brain injury models were established as mentioned before [10]. Briefly, the pups were anesthetized by inhaling 2% isoflurane, the right carotid artery of pups (male) was exposed and ligated. After 1 h, the pups were exposed to 8% oxygen at 37 °C for 1.5 h and then removed back to their cage. The total 267 pups (P7) were used in the study. The P7 pups were randomized within litters and allocated to one of eleven groups: (1) Sham + vehicle (Sham, n = 67), (2) HI + vehicle (HI, n = 67), (3) HI + L-Cysteine (n = 28), (4) HI + L-Cysteine + AOAA (n = 25), (5) HI + OPN antibody (n = 28), (6) HI + Stat3 siRNA (n = 10), (7) HI + negative control of Stat3 siRNA (Control-siRNA) (n = 10), (8) HI + OPN silencing short hair RNA lentivirus (LV-shOPN) (n = 10), (9) HI + corresponding negative control of LV-shOPN (LV-shNC) (n = 10), (10) Sham + PLX3397 (n = 6), and (11) HI + PLX3397 (n = 6). The L-Cysteine and AOAA were diluted by phosphate-buffered saline (PBS). These compounds were administered intraperitoneally (i.p.) at 24, 48, and 72 h post-HI. In the HI + L-Cysteine + AOAA group, AOAA was administered 30 min prior L-Cysteine injection. Administration route and dose of L-Cysteine (5 mg/kg) and AOAA (1 mg/kg) were determined according to our previous studies [10]. We have reported that L-Cysteine treatment increased the concentration of H2S in the ipsilateral cortex at 72 h following HI insult, this effect was reversed by AOAA pre-treatment [10].
OPN antibody, Stat3 siRNA, Control-siRNA, LV-shOPN, and LV-shNC was injected via intracerebroventricular (i.c.v.). PLX3397, a selective inhibitor of the colony-stimulating factor 1 receptor, which can completely ablate microglia in wild-type mice without inducing side effects [18]. The mice were fed with PLX3397 about 20 μL twice a day for 7 consecutive days from the first day of birth. The Sham and HI group were fed corn oil. In preliminary experiments, control IgG treatment has no effect on brain damage in HI group. Therefore, we chose to present only HI group data for comparison. The details of experimental design were contained in Supplementary Fig. S1.
Brain water content measurement
At 72 h post-HI, mice were anesthetized with 2% isoflurane, brain tissues were removed and divided into contralateral and ipsilateral hemispheres that was weighed (wet weight) using an analytical balance. Subsequently, the tissues were dried in an oven at 105 °C for 24 h to obtain dry weight.
Measurement of infarct size
At 72 h post-HI, the mice were deeply anesthetized with isoflurane, brain tissues were quickly removed and then frozen at −20 °C for 20 min, which was sliced 4 sections that was immersed in 1.5% 2, 3, 5-triphenyltetrazolium chloride monohydrate (TTC) solution at 37 °C for 15 min. The infarct volume was analyzed by image-analysis software (ImageJ software).
Immunofluorescent staining
At 72 h after HI injury, whole brain was removed and fixed in 4% paraformaldehyde solution for 24 h, and then embedded by paraffin. Brain tissues were cut into coronal slices (4 µm thickness) for the following experiments.
For immunofluorescent staining, brain slices were dewaxed by xylene, dexylene by anhydrous ethanol, and washed with PBS for 3 times, each 5 min. The sections were blocked with 10% donkey serum for 1 h at room temperature and incubated with primary antibody (OPN, 1:100; NeuN, 1:1000; Iba-1, 1:400; GFAP, 1:100) at 4 °C overnight. Then, tissue slices were washed twice in PBS and incubated with fluorescence secondary antibody at 37 °C for 30 min. Finally, the cell nucleus was stained with DAPI for 5 min. The microphotographs were analyzed by using Fluorescent microscopy (OLYMPUS-BX51) and the Magna Fire SP system.
Immunohistochemistry
The immunohistochemistry assay was performed as described previously [19]. In brief, anti-rabbit primary antibodies (OPN; 1:100) were chosen to stain brain slices at 4 °C overnight. The brain slides were covered with DAB and alcohol differentiation. The slides were assessed by microscopy with the above mentioned Magna Fire SP system.
Flow cytometry analysis
At 72 h after HI injury, the ipsilateral cerebral cortices were taken out and put it into Hank’s buffer, mechanically crushed with a magic pestle and passed it through a 70 μm sieve. The resultant single-cell suspension was washed twice in 0.2% bovine serum albumin-PBS and centrifuged at 400 × g for 10 min. The cells were resuspended with 40% percoll solution and 75% percoll solution was added to the lower layer to further purify the cells according to density gradient. Cells were incubated at 4 °C for 30 min with the addition of 1.5 μL antibody (CD11b, CD45). After 350 × g for 10 min, supernatant was discarded. Finally, cells were fixed, broken and incubated with antibody (OPN). Fluorescence activated Cell Sorting (FACS) analysis was performed using a flow cytometer C6 (BD Biosciences).
Intracerebroventricular (i.c.v.) treatment
For the i.c.v. treatment groups, the injection sites were identified at 2/5 of the distance from the lambda suture to each eye (the coordinates relative to bregma: 0.8 mm posterior and 1.0 mm lateral to the bregma) at a depth of about 3 mm as referenced previously [20]. Briefly, the mice were first anesthetized with 2% isoflurane and fixed on the table with tape. OPN antibody (0.61 μg/μL, 3.3 μL) (polyclonal goat anti-osteopontin, Ab11503), Stat3 siRNA (75 pmol, 0.26 μg/μL, 3.8 μL) and Control-siRNA (75 pmol, 0.26 μg/μL, 3.8 μL) were injected with 10 μL microinjector via i.c.v. injection at 24 h before HI insult [21].
In order to silence OPN, the lentivirus package was obtained from GenePharmag (Suzhou, China). P4 pups were injected with 1 × 107 TU/mL (3 μL) of LV-shOPN and LV-shNC via i.c.v. The injection rate was 1 μL/min and the needle remained at the site for about 3 min after injection.
Quantitative proteomics by LC–MS/MS assay
At 72 h after HI injury, the ipsilateral cerebral cortices were removed and digested with trypsin to obtain total protein which was isolated by dimensional gel electrophoresis. The isolated proteins were labeled with TMT reagents. The pooled peptides were separated into 15 fractions using a C18 column (Waters BEH C18 4.6 mm × 250 mm, 5 µm) on a Rigol L3000 HPLC (see Supplementary Fig. S2) by Novogene Co., Ltd (Beijing, China).
Microglial depletion drug treatment
Animals were treated with the microglial depletion drug Pexidartinib (PLX3397). PLX3397 was dissolved in 1% DMSO and then diluted with corn oil to 20 mg/kg. The mice were fed with PLX3397 about 20 μL twice a day for 7 consecutive days from the first day of birth. The sham and HI group were fed corn oil.
Reverse transcriptase quantitative real-time PCR (qRT-PCR)
The inflammatory cytokine expression levels were detected through qRT-PCR. Total RNA was extracted from ipsilateral cerebral cortices using RNA Extraction Kit according to the manufacturer’s protocol. RNA was used for cDNA synthesis that was stored in a refrigerator at −20 °C. qRT-PCR was performed using SYBR Green Mix and target primers. Amplification products were analyzed using CFX Connect Real-Time System. Primers used in qRT-PCR are shown in Table 1. Results were normalized with β-actin according to the 2−ΔΔCt method.
RT-PCR and qRT-PCR followed the same steps before cDNA amplification. RT-PCR amplification was performed using 12.5 μL Dream Taq Green PCR Master Mix (2×) and 2 μL cDNA and 2 μL primer as well as added DEPC water up to 25 μL volume. The RT-PCR conditions were 95 °C for 45 s, 55 °C for 45 s, and 72 °C for 30 s for 28 cycles. The date was analyzed by Image-Pro Plus 6.0 software.
Western blot
The ipsilateral cortex tissue removed from pups was ground in protein lysis buffer containing phenyl methane sulfonyl fluoride (PMSF), phosphatase inhibitors (PI) and RIPA buffer. The total protein concentrations were quantified using BCA Protein Assay Kits. The proteins were separated by 10% SDS–PAGE gel electrophoresis. PVDF membranes were blocked in 5% nonfat milk at room temperature for 1 h and then incubated with primary antibody (OPN, 1:1000; p-Stat3, 1:1000; Stat3: 1:1000; β-actin, 1:1000) at 4 °C for more than 12 h. After incubation with secondary antibody for 1 h. ECL kit reagents (MILLIPORE, USA) were used to develop chemiluminescent signals, which were detected using the Tanon Imaging System (Tanon-4600).
BV-2 cells culture
BV-2 cell line was purchased from Pu He Company (Wu-xi, China), and BV-2 cells were cultured into 6-well plate maintaining in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 0.1 mg/mL streptomycin at 37 °C incubator with 5% CO2. The BV-2 cells were stimulated using LPS (500 ng/mL) in the absence or presence of L-Cysteine (10 μm) as well as AOAA (1 μm) to next experiment.
S-sulfhydration assay
S-sulfhydration was performed as described previously [22]. Briefly, the BV-2 cells were treated with or without LPS, L-Cysteine, AOAA for 2 h, and then were homogenized in HEN buffer (250 mM Hepes・NaOH, pH = 7.7; 1 mM EDTA; 0.1 mM Neocuproine), supplemented with PMSF, PI and NP-40 lysate, which was centrifuged at 12,000 rpm for 10 min at 4 °C. The supernatant was added to HEN buffer (containing 2.5% SDS and 20 mM methyl methanethiosulfonate) at 50 °C for 30 min. Methyl methanethiosulfonate was removed by adding acetone and the proteins were precipitated using acetone at −20 °C for 30 min. Proteins were resuspended in HENS buffer (containing 1% SDS) and 50 μL 4 mM biotin-N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propinamide (HPDP) was added for incubation at room temperature for 3 h. Biotinylated proteins were purified by streptavidin agarose beads, which were then washed with HENS buffer. The biotinylated proteins were eluted by HENS and loading buffer, and later were analyzed by Western blot.
Co-immunoprecipitation
The BV-2 cells were grown on 10-cm culture dish and stimulated with LPS for 1 h. The cells were lysed in 1 mL NP-40 buffer containing PMSF for 15 min, which was centrifuged at 12,000 rpm for 10 min at 4 °C. The cell supernatants were added anti-IgG and anti-Stat3 at 4 °C for 12 h with rotation. Then, the 30 µL protein A + G beads were added to bond with antibody. The immunoprecipitated proteins with beads were centrifuged, washed three times with NP-40 buffer and analyzed by Western blot.
Y-maze test
The Y-maze test was used to evaluate the cognitive abilities of animals as described previously at 28 d after HI insult (P35) [23]. Briefly, the test consisted of two parts: training trial and test trail. For the training trial, one arm (the novel arm) was closed. Mice were placed at the end of a pseudorandomly chosen arm (start arm) and allowed to explore the maze for 5 min. Then mice were returned to their cage for resting 30 min. In the test trail, mice were allowed to freely explore all three arms for 5 min. Tracking system SMART was used to record the mice activity. Percent of time spent in the new arm and numbers of total arm entries were analyzed.
Statistical analysis
The SPSS statistical sofware (IBM SPSS Statistics 25) was used to perform statistical analyses. All values were expressed as mean ± standard deviation. Data were carried out using one-way ANOVA, following by Bonferroni corrections for multiple post hoc comparisons of means. Statistical significance was set at P value ≤ 0.05 for all experiments unless specified separately.
Results
Differentially expressed proteins as identified using TMT quantitative proteomics following L-Cysteine treatment in HI exposure
In this study, the ipsilateral cortex proteomes of mice in Sham, HI, HI + L-Cysteine group were compared using a TMT-based quantitative proteomic analysis as summarized in Supplementary Fig. S2. The heat map (Supplementary Fig. S3a) and Volcano plots (Supplementary Fig. S3b) showed that the distributions of proteins were significantly enriched and reduced in HI and HI + L-Cysteine group. Of these proteins, 93 proteins were upregulated (fold change ≥ 1.5, and P value ≤ 0.05) (top 6: OPN, Metallothionein-2, Metallothionein-1, Galectin-3, Protein S100-A4, and Heat shock protein beta-1) (detail in Supplementary Table S2); 8 were downregulated (fold change ≤ 0.67, and P value ≤ 0.05) (top 3: Septin-7, Myelin basic protein (Fragment), and Mal, T-cell differentiation protein 2) after HI insult (Supplementary Fig. S3a) (detail in Supplementary Table S2). These different proteins were involved in oxidative stress (e.g., Heat shock protein beta-1 (HSPB1), HSPB6 and Methionine-R-sulfoxide reductase B2), immune response (e.g., OPN, CD68, Galectin-3, Galectin-1, Galectin, and CD44 antigen), complement activation (Complement C1q subcomponent subunit A, B, C, and Complement component 4B) and apoptosis (BCL2-associated athanogene 3, Protein S100-A4, Protein S100-A6).
In addition, 9 proteins were upregulated (fold change ≥ 1.5, and P value ≤ 0.05) (top 6: Pleckstrin homology domain-containing family M member 2, Septin-7, DNA polymerase delta subunit 2, HERC3, Casp9 protein and Protein tyrosine phosphatase) (detail in Supplementary Table S2); 16 were downregulated by L-Cysteine treatment (fold change ≤ 0.67, and P value ≤ 0.05) (top 6: NK-tumor recognition protein, Heat shock protein beta-1, Transcription factor BTF3, Metallothionein-2, Metallothionein-1 and Methionine-R-sulfoxide reductase B2) compared to Vehicle-treatment HI group (detail in Supplementary Table S2). These different proteins were involved in oxidative stress (e.g., HSPB1, HSPB6, and Msrb2) and immune response (e.g., OPN).
Biological interaction of differentially expressed proteins in response to L-Cysteine treatment following HI exposure
The functional annotations of the HI-changed proteins were analyzed using a Gene Ontology (GO) enrichment analysis. Supplementary Fig. S4a listed the top 6 significantly enriched GO terms (cellular component, molecular function and biological process) identified after screening with P value ≤ 0.05. In addition, GO analysis of differentially expressed proteins showed that neuropeptide Y receptor activity, protein repair, peptide-methionine (R)-S-oxide reductase activity, structural constituent of eye lens, ossification, keratin filament, and response to oxidative stress were specifically enriched in proteins decreased in HI + L-Cysteine group, whereas septin complex, DNA-directed DNA polymerase activity and cysteine-type endopeptidase activity were enriched GO terms in proteins increased in HI + L-Cysteine group (P value ≤ 0.05).
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis revealed that the different proteins in HI group were enriched in complement and coagulation cascades, lysosome, mineral absorption and apoptosis pathway (Supplementary Fig. S4b). In addition, L-Cysteine treatment regulated different proteins involved in apoptosis, signal transduction and immune system-related pathways, such as VEGF signaling pathway, MAPK signaling pathway, PI3K/Akt signaling pathway, TOLL-like receptor signaling pathway, and p53 signaling pathway (Supplementary Fig. S4b). Most importantly, the PI3K-Akt signaling pathway is a classical signaling pathway in apoptotic signal transduction, including 2 differentially expressed proteins (OPN and Casp9 protein) following L-Cysteine treatment.
Cellular localization of OPN protein in the ipsilateral hemisphere following HI
As above TMT-based quantitative proteomic analysis showed, OPN was the most enriched protein in ipsilateral cortex following HI insult, raised our interest as it appeared in close association with the presence of microglia/macrophages and also involved in the pathogenesis of immune-mediated diseases [24]. Next, we concentrated on the role of OPN in the induction of neuroinflammation after HI exposure. In HI mice, OPN protein expression was largely increased in the ipsilateral cortex (Fig. 1b, b2), weak or negligible in the contralateral hemisphere (Fig. 1b, b1) and in the Sham group (Fig. 1a). OPN colocalized with activated Iba1+ microglia/macrophages in the lesion core, exhibiting amoeboid morphological characteristics. Moreover, a few granular OPN accumulation was detected in the lesion core and scattered among the amoeboid microglia/macrophages (Fig. 2a). A few OPN colocalized with the neuronal marker NeuN in the cortex region of the ipsilateral hemisphere at 72 h after HI injury (Fig. 2b), none colocalized with astrocytes (GFAP+) (Supplementary Fig. S5a). Depletion of microglial/macrophage with PLX3397 caused a significant reduction in the expression levels of OPN, IL-1β, and TNF-α following HI insult (Supplementary Fig. S5b).

a Representative photographs of OPN staining within the brain of Sham mice. Scale bar = 1 mm. b Representative photographs of OPN staining within the brain of HI mice taken at 72 h following insult. Scale bar = 1 mm. b1 Magnified views of boxed regions in (b) shows OPN staining within contralateral cortex. Scale bar = 50 μm. b2 Magnified views of boxed regions in (b) shows OPN staining within ipsilateral cortex. Scale bar = 50 μm.

a Representative photographs of double immunofluorescent staining of Iba-1 (green), OPN (red) within the ipsilateral cortex at 72 h following HI insult. a1 Magnified views of boxed regions (undamaged areas) in a shows Iba-1+/OPN+ staining. a2 Magnified views of boxed regions (in the peri-infarct areas) in (a) shows Iba-1+/OPN+ staining. a3 Magnified views of boxed regions (the infarct areas) in (a) shows Iba-1+/OPN+ staining. b Representative photographs of double immunofluorescent staining of NeuN (green), OPN (red) within the ipsilateral cortex at 72 h following HI insult. b1 Magnified views of boxed regions (undamaged areas) in (b) shows NeuN+/OPN+ staining. b2 Magnified views of boxed regions (in the peri-infarct areas) in (b) shows NeuN+/OPN+ staining. b3 Magnified views of boxed regions (the infarct areas) in (b) shows NeuN+/OPN+ staining. Yellow arrowhead: NeuN+/OPN+ cells in the infarct areas. Scale bar = 50 μm.
OPN produced by CD11b+/CD45low cells and infiltrating CD11b+/CD45high cells
FACS analysis revealed that HI remarkably increased the percentage of CD11b+/OPN+ cells in the ipsilateral cortex at 72 h post-HI ([F(3,12) = 226.247, P < 0.001]; post hoc P < 0.001) (Fig. 3d). Next, we examined the subcellular localization of OPN in infiltrating monocytes/macrophages (CD11b+/OPN+/CD45high cells) or resident microglia (CD11b+/OPN+/CD45low cells). The results showed that HI expousure led to 8.52-fold increase in number of infiltrating CD11b+/OPN+/CD45high cells in contrast to Sham group ([F(3,12) = 136.644, P < 0.001]; post hoc P < 0.001) (Fig. 3e). However, OPN-expressing microglia indicated a significant 0.56-fold decrease in HI group as compared to Sham group ([F(3,12) = 115.926, P < 0.001]; post hoc P < 0.001) (Fig. 3f).

a Immune cell populations were quantified in single-cell suspensions isolated from ipsilateral cortex 72 h following HI insult. Samples were progressively gated based first on size (FSC) and granularity (SSC). b CD11b+/OPN+ cells (red rectangle) populations in the ipsilateral cortex following HI were evident. c CD11b+/OPN+ /CD45high cells (blackish green rectangle) and CD11b+/OPN+/CD45low cells (purplish red rectangle) populations in the ipsilateral cortex following HI were evident. d Bar graphs show quantitative analysis of CD11b+/OPN+ cells population. e Bar graphs show quantitative analysis of CD11b+/OPN+/CD45high cells population. f Bar graphs show quantitative analysis of CD11b+/OPN+/CD45low cells population. g Bar graphs show the number of CD11b+ cells in each group. h Bar graphs show the number of CD11b+/OPN+ cells in each group. In all panels, n = 4 pools per group, three brains per pool. Values represent the mean ± SD, ***P < 0.001 according to ANOVA with Bonferroni correction.
We also found HI exposure increased the percentage of CD11b+/CD45high cells ([F(3,8) = 1130.483, P < 0.001]; post hoc P < 0.001), and reduced the percentage CD11b+/CD45low cells in the injured ipsilateral cortex ([F(3,8) = 2867.575, P < 0.001]; post hoc P < 0.001) (Supplementary Fig. S6). L-Cysteine treatment remarkably suppressed cerebral recruitment of the percentage of CD11b+/CD45high cells (post hoc P < 0.001) (Supplementary Fig. S6), confirming our previous observations [11]. Additionally, L-Cysteine treatment decreased HI-induced the percentage of CD11b+/OPN+ cells (post hoc P < 0.001) (Fig. 3d) and the percentage of CD11b+/OPN+/CD45high cells (post hoc P < 0.001) (Fig. 3e), and increased the percentage of OPN-expressing microglia (post hoc P < 0.001) (Fig. 3f). Moreover, L-Cysteine treatment increased the absolute number of CD11b+ cells ([F(3,12) = 5823.432, P < 0.001]; post hoc P < 0.001) (Fig. 3g) and the absolute number of CD11b+/OPN+ cells ([F(3,12) = 68.238, P < 0.001]; post hoc P < 0.01) (Fig. 3h). AOAA reversed the effect of L-Cysteine on OPN+ cells change following HI damage (Fig. 3).
Blocking OPN expression suppressed HI-induced brain damage
To investigate the function of OPN on HI-induced brain damage, mice were treated with OPN antibody (injected i.c.v.) to block OPN expression. The inhibition of OPN expression was verified by RT-PCR and Western blot (Fig. 4a, b). The results showed that OPN antibody decreased HI-induced CD11b+/OPN+ cells ([F(2,9) = 158.156, P < 0.001]; post hoc P < 0.001) with FACS analysis (Fig. 4c). OPN antibody administration significantly attenuated HI-induced edema ([F(2,9) = 12.411, P < 0.01]; post hoc P < 0.01) (Fig. 4d). Moreover, HI resulted in 60.0% ± 18.8% infarction of the ipsilateral hemisphere as measured 72 h following HI, which was significantly reduced to 26.4% ± 5.6% in OPN antibody groups ([F(2,9) = 27.023, P < 0.001]; post hoc P < 0.01) (Fig. 4e). At the same time, knockdown of OPN expression with LV-shOPN significantly attenuated HI-induced edema ([F(3,8) = 13.113, P < 0.01]; post hoc P < 0.05) and infarction ([F(3,8) = 169.468, P < 0.001]; post hoc P < 0.001) (Supplementary Fig. S8a, b).

a OPN antibody was injected (i.c.v.) at 24 h before HI insult. Three days following HI, the levels of OPN mRNA were determined with RT-PCR in each group. n = 4/group. b Three days following HI, the levels of OPN protein were determined with Western blot in each group. Values were normalized to β-actin. n = 5/group. c CD11b+/OPN+ cells populations (purplish red rectangle) in the ipsilateral cortex following HI were analyzed with FACS. Bar graphs show quantitative analysis of CD11b+/OPN+ cells population. In all panels, n = 4 pools per group, three brains per pool. d Representative brain images as obtained from each group described above at 72 h following HI. Brain water content was determined. n = 4/group. e Representative samples were stained with TTC. Quantified infarct volume is indicated by the white area. n = 4/group. Values represent the mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001 according to ANOVA with Bonferroni correction.
Blocking OPN expression suppressed HI-induced CD11b+/CD45high infiltrating cells
The blocking expression of OPN partially reduced the HI-recruited percentage of CD11b+/CD45high immune cells ([F(2,9) = 1158.456, P < 0.001]; post hoc P < 0.001) and reversed HI-reduced the percentage of CD11b+/CD45low cells ([F(2,9) = 297.269, P < 0.001]; post hoc P < 0.01) (Fig. 5a). At the same time, a significantly decreased absolute number of CD11b+/CD45high immune cells ([F(2,9) = 74.857, P < 0.001]; post hoc P < 0.001) and an increased absolute number of CD11b+/CD45low cells ([F(2,9) = 344.714, P < 0.001]; post hoc P < 0.001) were observed following administration with OPN antibody in HI mice, associated with suppressed pro-inflammatory cytokines levels 72 h following HI: CD16 ([F(2,15) = 24.214, P < 0.001]; post hoc P < 0.001), CD11b ([F(2,15) = 29.042, P < 0.001]; post hoc P < 0.001), CD86 ([F(2,15) = 26.788, P < 0.001]; post hoc P < 0.01), IL-1β ([F(2,15) = 730.297, P < 0.001]; post hoc P < 0.001), TNF-α ([F(2,15) = 9.527, P < 0.01]; post hoc P < 0.01), IL-6 ([F(2,9) = 8.087, P < 0.05]; post hoc P < 0.05) (Fig. 5b). However, we found that the blocking expression of OPN upregulated IFN-γ mRNA level ([F(2,9) = 5.980, P < 0.05]; post hoc P < 0.05) (Fig. 5b).

a Gating through CD11b+/CD45low and CD11b+/CD45high identified microglia and infiltrating immune cells. Activated macrophages in the upper right quadrant (deep-green rectangle) (CD11b+/CD45high). Microglia in the lower left quadrant (red rectangle) (CD11b+/CD45low). In all panels, n = 4 pools per group, three brains per pool. b Three days following HI, the levels of cytokines expression were determined with qRT-PCR in each group. n = 4–6/group. Values represent the mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001 according to ANOVA with Bonferroni correction.
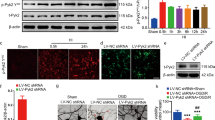
Stat3 silencing inhibited OPN expression and brain damage following HI
Stat3 activation is essential for transcriptional activation of OPN [25]. Next, we aimed to confirm whether the Stat3 pathway was involved in OPN activities following HI insult. Following Stat3-specific siRNA administration, Stat3 expression was successfully decreased ([F(3,12) = 9.283, P < 0.01]; post hoc P < 0.05) (Fig. 6a). Stat3 silencing resulted in a significant decrease in OPN expression ([F(3,12) = 13.476, P < 0.001]; post hoc P < 0.05) (Fig. 6a) in the ipsilateral hemisphere and attenuated HI-induced edema ([F(3,8) = 13.845, P < 0.01]; post hoc P < 0.05) and infarction ([F(3,8) = 109.033, P < 0.001]; post hoc P < 0.01) (Fig. 6d, e). Moreover, knockdown of OPN expression with LV-shOPN attenuated HI-induced p-Stat3 ([F(3,12) = 22.351, P < 0.001]; post hoc P < 0.001) (Fig. 6b), but no effect on Stat3 level at 72 h following HI insult. In addition, we conducted immunoprecipitation with Stat3 antibody using cell lysates from BV-2 cells to address whether OPN could bind to Stat3. As shown in Fig. 6c, endogenous OPN and Stat3 formed a complex in BV-2 cells, indicating that there was a direct or indirect interaction between OPN and Stat3.

a The levels of OPN and Stat3 at 72 h post-HI were determined with Western blot in each group following Stat3 siRNA administration. Values were normalized to β-actin. n = 4/group. b The levels of OPN and p-Stat3 at 72 h post-HI were determined with Western blot in each group following LV-shOPN administration. Values were normalized to β-actin. n = 4/group. c Co-immunoprecipitation experiments were conducted on BV-2 cell lysates with anti-IgG and anti-Stat3, and then the precipitation was subsequently analyzed by Western blot with anti-Stat3, anti-OPN. n = 5/group. d Representative brain images as obtained from each group at 72 h following HI. Brain water content was determined. n = 3/group. e Representative samples were stained with TTC. n = 3/group. Values represent the mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001 according to ANOVA with Bonferroni correction.
L-Cysteine treatment downregulated OPN expression through inducing S-sulfhydration of Stat3
Next, we studied the role of H2S on Stat3/OPN pathway following HI insult. We found that L-Cysteine administration remarkably reversed HI-induced OPN expression ([F(3,16) = 27.277, P < 0.001]; post hoc P < 0.001) and p-Stat3 ([F(3,12) = 20.968, P < 0.001]; post hoc P < 0.01). AOAA treatment reversed the effect of L-Cysteine on OPN expression (post hoc P < 0.05) (Fig. 7a) and p-Stat3 (post hoc P < 0.05) (Fig. 7b). Moreover, L-Cysteine administration remarkably reversed LPS-induced OPN expression ([F(3,16) = 10.913, P < 0.001]; post hoc P < 0.05) (Fig. 7c) and p-Stat3 expression ([F(3,12) = 26.352, P < 0.001]; post hoc P < 0.01) (Fig. 7d) in BV-2 cells. We further confirm the inhibitory effect of H2S on OPN expression and Stat3 phosphorylation in macrophages. We performed the experiment in the RAW 264.7 macrophage cell line and obtained similar results. The results showed that L-Cysteine suppressed LPS-induced OPN expression ([F(3,12) = 9.885, P < 0.01]; post hoc P < 0.05) and phosphorylation of Stat3 ([F(3,12) = 17.485, P < 0.001]; post hoc P < 0.01) in macrophage in vitro (Supplementary Fig. S9a, b). Uncropped Western blots for all replicates were shown in Supplementary Fig. S10.

a The levels of OPN at 72 h post-HI were determined with Western blot in each group following L-Cysteine or AOAA administration. Values were normalized to β-actin. n = 5/group. b The levels of p-Stat3 and Stat3 at 72 h post-HI were determined with Western blot in each group following L-Cysteine or AOAA administration. n = 4/group. c The levels of OPN in BV-2 cells were determined with Western blot in each group at 1 h following LPS (500 ng/mL), L-Cysteine or AOAA administration. n = 5/group. d The levels of p-Stat3 and Stat3 in BV-2 cells were determined with Western blot in each group at 2 h following LPS (500 ng/mL), L-Cysteine or AOAA administration. n = 4/group. e S-sulfhydration of Stat3 under L-Cysteine treatment in the presence or absence of 500 ng/mL LPS. n = 4/group. Values represent the mean ± SD, *P < 0.05; **P < 0.01; ***P < 0.001 according to ANOVA with Bonferroni correction.
Protein sulfhydration at cysteine sites is an important regulatory mode of H2S signaling [26]. S-sulfhydration assay revealed that L-Cysteine induced S-sulfhydration in Stat3 following LPS stimulation ([F(3,12) = 8.828, P < 0.01]; post hoc P < 0.05). AOAA reversed the effect of L-Cysteine on S-sulfhydration in Stat3 (post hoc P < 0.05) (Fig. 7e).
L-Cysteine treatment improved neurobehavioral impairment following HI
In the Y-maze test, HI mice spent less time in the novel arm ([F(3,28) = 11.055, P < 0.001]; post hoc P < 0.001) and had reduced ratio entry ([F(3,28) = 7.873, P < 0.01]; post hoc P < 0.05) than sham animals, indicating that HI was able to impair spatial learning and memory at 28 d after insult (Supplementary Fig. S7b). L-Cysteine treatment increased time spent in the new arm (post hoc P < 0.05) and enhanced ratio entry (post hoc P < 0.01) compared with the vehicle-treated HI group (Supplementary Fig. S7b). AOAA treatment reversed the effects of L-Cysteine on spatial learning and memory function.
Discussion
In the present study, we demonstrated that L-Cysteine treatment modified proteome in the ipsilateral cortex involved in oxidative stress and immune response, and effectively attenuated brain injury and improved cognitive function in HI mice, which was consistent with the results of our previous study [10, 11]. Moreover, L-Cysteine treatment was able to suppress cerebral recruitment of mononuclear inflammatory cells by reducing OPN expression in microglia/macrophage via inducing S-sulfhydration of Stat3.
Previous two-dimensional differential proteome analysis has identified dopamine neurotransmission in lesion cortex 2 h following HI [13]. A 2D-DIGE-based proteome analysis has identified novel factors involved in the inflammatory response, such as Calcineurin A, Coronin-1A, as well as GFAP in lesion hemisphere 48 h after HI [14]. In the present study, we used TMT proteomic method to investigate the proteins expression in ipsilateral cortex 72 h following HI. The results showed that 93 proteins were upregulated and 8 proteins were downregulated following HI. As expected, these proteins were involved in oxidative stress, inflammation response, complement activation and apoptosis.
More importantly, we found that OPN was the most highly expressed protein in ipsilateral cortex following HI insult by proteomics analysis. OPN, also called secreted phosphoprotein 1, is secreted by activated immune cells, including macrophages, microglia, T cells, dendritic cells and involved in the pathogenesis of immune-mediated diseases [24]. The basal OPN level in adult brains is low, but CNS injury upregulates OPN expression in active microglia/macrophage [27, 28] and astrocyte [29]. Studies also found that OPN was strongly upregulated in the brain after HI in neonatal mice and rats [30, 31]. High levels of plasma OPN correlated with severe brain injury in neonatal mice [32]. In line with previous study, we found that punctate OPN-immunoreactive profiles were mainly located within amoeboid-like brain microglia/macrophage. Further FACS analysis of CD11b myelomonocytic cells in brain showed HI-induced enrichment of OPN+ monocyte subpopulations in the CD45high cells (infiltrating cells), while the number of CD11b+/OPN+/CD45low cells was decreased. Additionally, HI-induced microglia apoptosis resulted in a decrease in the number of CD11b+/CD45low cells, responsible for the reduction in the number of CD11b+/OPN+/CD45low cells. The cell location and function of OPN in microglia/macrophage had inconsistent results in a variety of injury models. For example, OPN deficiency in macrophages increased the pro-inflammatory cytokine production in the basal state and in response to LPS [33]. OPN puncta was synthesized and released by amoeboid-like brain macrophages, and became closely associated with, or completely engulfed by reactive astrocytes in lesioned striatum at 28 d following mitochondrial toxin 3-nitropropionic acid exposure [29]. However, we found that the majority of OPN was colocalized with amoeboid-like microglia/ macrophages in the lesion core. Depletion of microglial/macrophage caused a significant reduction in HI-induced OPN expression. Blocking OPN expression attenuated brain damage, reduced pro-inflammatory cytokine levels post-HI insult. Recently study also found that anti-OPN or OPN knockdown inhibited retinal microglia activation and pro-inflammatory cytokines expression [34]. There are several reasons why inconsistencies reported in the role of OPN in microglia/macrophage, such as different brain maturation at the time of injury, injury context and spatiotemporal model.
Although studies have shown that OPN may contribute to repair-promoting processes in the HI in neonatal mice [35], recent OPN peptide treatment did not show neuroprotective effect in the injured neonatal brain [36]. OPN can trigger inflammation by upregulating IL-13 receptors, CXCR3, and CD40 ligand [37]. Studies also suggest that OPN acts as a pro-inflammatory role in animal model of colitis [38]. In this study, we found that inhibiting OPN expression was able to reduce cerebral recruitment of CD11b+/CD45high immune cells, neuroinflammation and brain damages following HI. We speculated that HI exposure activated microglia, recruited CD11b+/CD45high immune cells, and led to up-regulation of OPN expression, which could further recruit CD11b+/CD45high immune cells infiltrating to CNS and boost the detrimental effects on HI injury. We also found that L-Cysteine treatment suppressed cerebral recruitment of CD11b+/CD45high immune cells, concomitant with reducing OPN expression in CD11b+/CD45high cells following HI insult. Taken together, intervention of vicious circle between OPN and CD11b+/CD45high immune cells contributes to L-Cysteine neuroprotective effect on HI brain damage.
Stat3 pathway is a critical immunological signaling cascade and plays a vital role in regulating microglial activation and inflammatory response [39]. Several studies have demonstrated that Stat3 activation promotes neuroinflammation in ischemia stroke [39, 40]. Stat3 was strongly upregulated following HI insult in the neonatal mice, associated with tissue repair [40]. Inhibition of Stat3 activation attenuated brain damage following HI injury [39]. Consistent with these findings, we found that Stat3 was activated in microglia in vitro and in vivo, and the effect was reversed by L-Cysteine administration. Stat3 silencing produced a similar effect with L-Cysteine administration after HI exposure in neonatal mice.
Stat3 plays a direct role in the transcriptional regulation of OPN gene [33]. We found that Stat3 silencing suppressed HI-induced OPN expression. And knockdown of OPN expression also attenuated HI-induced p-Stat3 level. Further, co-IP demonstrated a direct protein-protein interaction between OPN and Stat3. Taken together, we speculated that OPN play a role in neuroinflammation through interacting with Stat3 in microglia. Stat3 could be phosphorylated on a tyrosine residue by receptor-associated tyrosine kinases or nonreceptor tyrosine kinases. Tyrosine-phosphorylated Stat3 forms dimers and translocates to the nucleus, and induces transcription of target genes. Stat3 dimerization can be mediated by interchain disulfide bridging involving Cys259 [41, 42]. Studies indicate that protein S-sulfhydration, termed as the addition of one sulfhydryl to the cysteine residue of target proteins and the subsequent generation of a persulfide group, is a major mechanism by which H2S elicits cellular signaling [26]. For example, H2S induced S-sulfhydration of NF-κB at Cys38, thereby restraining NF-κB phosphorylation and NF-κB transactivation-mediated inflammation induced by oxidized low-density lipoprotein [43]. H2S induced S-sulfhydration of Stat3, which impaired the dimer formation of Stat3 and resulted in the anti-calcific effects in aortic smooth muscle cells treated by high glucose [25]. In this study, we found that the sulfhydrated Stat3 was decreased in LPS-induced BV-2 cells, whereas it was significantly increased after L-Cysteine treatment. Moreover, L-Cysteine administration suppressed LPS-induced phosphorylated Stat3 and reduced CD11b+/CD45high infiltrating cells. Based on these data, we speculated that L-Cysteine treatment induced S-sulfhydration of Stat3, thereby restraining dimerization and nuclear translocation of Stat3, resulted in reducing OPN expression in microglia/macrophage, which suppressed phosphorylated Stat3 expression following HI insult in neonatal mice. However, whether L-Cysteine directly inhibited the phosphorylation of STAT3 was unknown in the current study.
Our study had certain limitations. First, under inflammatory stimulation, activated glial cells lead to a local increase in OPN expression. In this study, we investigated protein expression differences of lesion cortex with TMT proteomic method, including different nerve cells. Further study on proteomic profiling of microglia from HI models could address the OPN or immune-mediated protein change. Second, L-Cysteine treatment upregulated 9 proteins and downregulated 16 proteins, compared to HI group as above demonstrated. We only focus on the L-Cysteine treatment effect on OPN expression after HI. Third, in a previous study, OPN mRNA increased as early as 12 h, and lasted for 5 d after HI [44]. Our study did not describe the time course change of OPN protein expression after HI and L-Cysteine treatment.
Our data indicated that proteomic analysis was able to evaluate the neuroprotective effects of multiple-target of L-Cysteine treatment. L-Cysteine effectively attenuated OPN-mediated neuroinflammation by inducing S-sulfhydration of Stat3, which contributes to its protective effect following HI in neonatal mice. Blocking OPN expression may potentially provide a new target for therapeutic intervention for perinatal HI brain injury.
References
Szabo C, Papapetropoulos A. International union of basic and clinical pharmacology. CII: pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol Rev. 2017;69:497–564.
Paul BD, Snyder SH. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem Pharmacol. 2018;149:101–9.
Kimura H. Hydrogen sulfide and polysulfides as biological mediators. Molecules. 2014;19:16146–57.
Xuan A, Long D, Li J, Ji W, Zhang M, Hong L, et al. Hydrogen sulfide attenuates spatial memory impairment and hippocampal neuroinflammation in beta-amyloid rat model of Alzheimer’s disease. J Neuroinflammation. 2012;9:202.
Chan SJ, Wong PT. Hydrogen sulfide in stroke: protective or deleterious? Neurochem Int. 2017;105:1–10.
Cui Y, Duan X, Li H, Dang B, Yin J, Wang Y, et al. Hydrogen sulfide ameliorates early brain injury following subarachnoid hemorrhage in rats. Mol Neurobiol. 2016;53:3646–57.
Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–20.
Whiteman M, Li L, Rose P, Tan CH, Parkinson DB, Moore PK. The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid Redox Signal. 2010;12:1147–54.
Zhou Y, Wu Z, Cao X, Ding L, Wen Z, Bian JS. HNO suppresses LPS-induced inflammation in BV-2 microglial cells via inhibition of NF-kappaB and p38 MAPK pathways. Pharmacol Res. 2016;111:885–95.
Liu S, Xin D, Wang L, Zhang T, Bai X, Li T, et al. Therapeutic effects of L-Cysteine in newborn mice subjected to hypoxia-ischemia brain injury via the CBS/H2S system: role of oxidative stress and endoplasmic reticulum stress. Redox Biol. 2017;13:528–40.
Xin D, Chu X, Bai X, Ma W, Yuan H, Qiu J, et al. L-Cysteine suppresses hypoxia-ischemia injury in neonatal mice by reducing glial activation, promoting autophagic flux and mediating synaptic modification via H2S formation. Brain Behavior Immun. 2018;73:222–34.
Shrivastava K, Llovera G, Recasens M, Chertoff M, Gimenez-Llort L, Gonzalez B, et al. Temporal expression of cytokines and signal transducer and activator of transcription factor 3 activation after neonatal hypoxia/ischemia in mice. Dev Neurosci. 2013;35:212–25.
Hu X, Rea HC, Wiktorowicz JE, Perez-Polo JR. Proteomic analysis of hypoxia/ischemia-induced alteration of cortical development and dopamine neurotransmission in neonatal rat. J Proteome Res. 2006;5:2396–404.
Rosenkranz K, May C, Meier C, Marcus K. Proteomic analysis of alterations induced by perinatal hypoxic-ischemic brain injury. J Proteome Res. 2012;11:5794–803.
Kaur C, Sivakumar V, Zou Z, Ling EA. Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct Funct. 2014;219:151–70.
McRae A, Gilland E, Bona E, Hagberg H. Microglia activation after neonatal hypoxic-ischemia. Brain Res Dev Brain Res. 1995;84:245–52.
Chu X, Liu D, Li T, Ke H, Xin D, Wang S, et al. Hydrogen sulfide-modified extracellular vesicles from mesenchymal stem cells for treatment of hypoxic-ischemic brain injury. J Control Release. 2020;328:13–27.
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–97.
Chu X, Cao L, Yu Z, Xin D, Li T, Ma W, et al. Hydrogen-rich saline promotes microglia M2 polarization and complement-mediated synapse loss to restore behavioral deficits following hypoxia-ischemic in neonatal mice via AMPK activation. J Neuroinflammation. 2019;16:104.
Kim JY, Grunke SD, Levites Y, Golde TE, Jankowsky JL. Intracerebroventricular viral injection of the neonatal mouse brain for persistent and widespread neuronal transduction. J Vis Exp. 2014;91:51863.
Knox-Concepcion KR, Figueroa JD, Hartman RE, Li Y, Zhang L. Repression of the glucocorticoid receptor increases hypoxic-ischemic brain injury in the male neonatal rat. Int J Mol Sci. 2019;20:3493.
Modis K, Ju Y, Ahmad A, Untereiner AA, Altaany Z, Wu L, et al. S-sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol Res. 2016;113:116–24.
Yau JL, McNair KM, Noble J, Brownstein D, Hibberd C, Morton N, et al. Enhanced hippocampal long-term potentiation and spatial learning in aged 11beta-hydroxysteroid dehydrogenase type 1 knock-out mice. J Neurosci. 2007;27:10487–96.
Rittling SR, Singh R. Osteopontin in immune-mediated diseases. J Dent Res. 2015;94:1638–45.
Zhou YB, Zhou H, Li L, Kang Y, Cao X, Wu ZY, et al. Hydrogen sulfide prevents elastin loss and attenuates calcification induced by high glucose in smooth muscle cells through suppression of Stat3/cathepsin S signaling pathway. Int J Mol Sci. 2019;20:4202.
Zhang D, Du J, Tang C, Huang Y, Jin H. H2S-induced sulfhydration: biological function and detection methodology. Front Pharmacol. 2017;8:608.
Gliem M, Krammes K, Liaw L, van Rooijen N, Hartung HP, Jander S. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia. 2015;63:2198–207.
Ellison JA, Velier JJ, Spera P, Jonak ZL, Wang X, Barone FC, et al. Osteopontin and its integrin receptor alpha(v)beta3 are upregulated during formation of the glial scar after focal stroke. Stroke. 1998;29:1698–706.
Riew TR, Kim S, Jin X, Kim HL, Lee JH, Lee MY. Osteopontin and its spatiotemporal relationship with glial cells in the striatum of rats treated with mitochondrial toxin 3-nitropropionic acid: possible involvement in phagocytosis. J Neuroinflammation. 2019;16:99.
Algra SO, Groeneveld KM, Schadenberg AW, Haas F, Evens FC, Meerding J, et al. Cerebral ischemia initiates an immediate innate immune response in neonates during cardiac surgery. J Neuroinflammation. 2013;10:24.
Smith PLP, Mottahedin A, Svedin P, Mohn CJ, Hagberg H, Ek J, et al. Peripheral myeloid cells contribute to brain injury in male neonatal mice. J Neuroinflammation. 2018;15:301.
Li Y, Dammer EB, Zhang-Brotzge X, Chen S, Duong DM, Seyfried NT, et al. Osteopontin is a blood biomarker for microglial activation and brain injury in experimental hypoxic-ischemic encephalopathy. eNeuro. 2017;4:0253.
Patouraux S, Rousseau D, Rubio A, Bonnafous S, Lavallard VJ, Lauron J, et al. Osteopontin deficiency aggravates hepatic injury induced by ischemia-reperfusion in mice. Cell Death Dis. 2014;5:e1208.
Yu H, Zhong H, Li N, Chen K, Chen J, Sun J, et al. Osteopontin activates retinal microglia causing retinal ganglion cells loss via p38 MAPK signaling pathway in glaucoma. FASEB J. 2021;35:e21405.
Chen W, Ma Q, Suzuki H, Hartman R, Tang J, Zhang JH. Osteopontin reduced hypoxia-ischemia neonatal brain injury by suppression of apoptosis in a rat pup model. Stroke. 2011;42:764–9.
Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, et al. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–805.
Nazmi A, Albertsson AM, Rocha-Ferreira E, Zhang X, Vontell R, Zelco A, et al. Lymphocytes contribute to the pathophysiology of neonatal brain injury. Front Neurol. 2018;9:159.
Kourepini E, Aggelakopoulou M, Alissafi T, Paschalidis N, Simoes DC, Panoutsakopoulou V. Osteopontin expression by CD103- dendritic cells drives intestinal inflammation. Proc Natl Acad Sci USA. 2014;111:E856–65.
Wang XL, Qiao CM, Liu JO, Li CY. Inhibition of the SOCS1-JAK2-STAT3 signaling pathway confers neuroprotection in rats with ischemic stroke. Cell Physiol Biochem. 2017;44:85–98.
McGuckin CP, Jurga M, Miller AM, Sarnowska A, Wiedner M, Boyle NT, et al. Ischemic brain injury: a consortium analysis of key factors involved in mesenchymal stem cell-mediated inflammatory reduction. Arch Biochem Biophys. 2013;534:88–97.
Li L, Shaw PE. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem Biophys Res Commun. 2004;322:1005–11.
Ahmad R, Raina D, Meyer C, Kufe D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)–>signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008;68:2920–6.
Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu M, et al. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor kappaB (NF-kappaB) pathway. J Biol Chem. 2014;289:9741–53.
van Velthoven CT, Heijnen CJ, van Bel F, Kavelaars A. Osteopontin enhances endogenous repair after neonatal hypoxic-ischemic brain injury. Stroke. 2011;42:2294–301.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos. 82072535, 81873768, and 81671213 to ZW, 81772436 to SY) and the Major Scientific and Technological Innovation Project in Shandong Province (2019JZZY011112 to SY).
Author information
Authors and Affiliations
Contributions
ZW designed study and analysed the data; DXL wrote and revised the paper; TTL did the laboratory work and analysed the data; DQX performed proteomic analysis; XLC, HFK, and YJZ performed behavioral testing; SWY revised the paper and analysed the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, Tt., Xin, Dq., Ke, Hf. et al. L-Cysteine attenuates osteopontin-mediated neuroinflammation following hypoxia-ischemia insult in neonatal mice by inducing S-sulfhydration of Stat3. Acta Pharmacol Sin 43, 1658–1669 (2022). https://doi.org/10.1038/s41401-021-00794-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00794-2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}