
Abstract
Myocardial ischemia-reperfusion (I/R) injury is a pathological process characterized by cardiomyocyte apoptosis, which leads to cardiac dysfunction. Increasing evidence shows that abnormal expression of long noncoding RNAs (lncRNAs) plays a crucial role in cardiovascular diseases. In this study we investigated the role of lncRNAs in myocardial I/R injury. Myocardial I/R injury was induced in mice by ligating left anterior descending coronary artery for 45 min followed by reperfusion for 24 h. We showed that lncRNA KnowTID_00006395, termed lncRNA-6395 was significantly upregulated in the infarct area of mouse hearts following I/R injury as well as in H2O2-treated neonatal mouse ventricular cardiomyocytes (NMVCs). Overexpression of lncRNA-6395 led to cell apoptosis and the expression change of apoptosis-related proteins in NMVCs, whereas knockdown of lncRNA-6395 attenuated H2O2-induced cell apoptosis. LncRNA-6395 knockout mice (lncRNA-6395+/−) displayed improved cardiac function, decreased plasma LDH activity and infarct size following I/R injury. We demonstrated that lncRNA-6395 directly bound to p53, and increased the abundance of p53 protein through inhibiting ubiquitination-mediated p53 degradation and thereby facilitated p53 translocation to the nucleus. More importantly, overexpression of p53 canceled the inhibitory effects of lncRNA-6395 knockdown on cardiomyocyte apoptosis, whereas knockdown of p53 counteracted the apoptotic effects of lncRNA-6395 in cardiomyocytes. Taken together, lncRNA-6395 as an endogenous pro-apoptotic factor, regulates cardiomyocyte apoptosis and myocardial I/R injury by inhibiting degradation and promoting sub-cellular translocation of p53.
Similar content being viewed by others

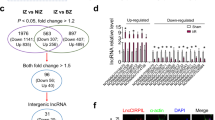
Introduction
Myocardial infarction (MI) is a leading cause of death worldwide. Effective therapy for acute MI is reperfusion of the occluded coronary artery. However, ischemic reperfusion (I/R) can cause a number of pathological changes including oxidative stress, cell apoptosis, etc, which eventually impair cardiac function. Therefore, inhibition of cell apoptosis is an attractive therapeutic strategy to reduce I/R injury and prevent heart failure after MI.
Long noncoding RNAs (lncRNAs) are a class of transcripts longer than 200 nucleotides with limited or no protein-coding potential. They participate in a wide range of biological processes such as cell proliferation, differentiation, and apoptosis [1,2,3]. Studies demonstrated that dysregulated lncRNA expression is observed in a number of diseased tissues, such as heart, kidney, liver, and so on [4,5,6]. Recently, the regulatory role of lncRNA in various physiological and pathological process has become a research hotspot. It has been reported that lncRNA OCC-1 promotes HuR protein ubiquitination degradation in colorectal cancer [7]. LncRNA ANCR attenuates the invasion and metastasis of breast cancer by promoting EZH2 phosphorylation that facilitates EZH2 degradation [8]. LncRNA ZFAS1 binds to SERCA2a protein to limit its intracellular level and inhibit its activity, and its knockdown is able to mitigate the ischemic contractile dysfunction [9].
Apoptosis-associated transcription factor p53 plays an essential role in response to diverse cellular stress stimuli [10]. It has been shown that p53 participates in the cell apoptosis process by regulating both the intrinsic mitochondrial and extrinsic death receptor pathways. The abundance, stability, and location of p53 are critical in controlling its function in cell apoptosis. LncRNAs have been demonstrated to modulate the p53 signaling network. Tripathi et al. found that lncRNA MALAT1 depletion leads to the activation of p53 and expression of its downstream target genes [11]. LncRNA Wrap53 is able to upregulate p53 expression in cancer cell [12].
In previous work, we demonstrated that lncRNA KnowTID_00006395 named PCFL (pro-cardiac fibrotic lncRNA) participated in the process of ischemic myocardial fibrosis by regulating miR-378/GRB2 pathway [13]. However, in this study, we found that KnowTID_00006395 termed lncRNA-6395 was highly expressed in I/R injured heart and significantly regulated cardiomyocyte apoptosis by inhibiting ubiquitination and degradation of p53. These results provide new functional insights into p53 regulatory network in the heart and suggest that lncRNA-6395 may serve as a potential therapeutic target in the intervention of I/R injury.
Materials and methods
Mouse model of myocardial I/R injury
Male C57BL/6 mice (12 weeks old, weighing 21–25 g) were provided by the Animal Center of the Second Affiliated Hospital of Harbin Medical University. Animal care and experimental protocols were in accordance with the Institutional Animal Care and approved by the Ethics Committee of Harbin Medical University. The procedure for establishing mouse model of myocardial I/R injury was previously described [14]. In brief, C57BL/6 mice (8 weeks old, 20–30 g) were fixed on the operation board. They were then intubated and connected to a rodent ventilator (Ugo Basile, Comerio, Italy). The chest was opened and the heart was exposed. Myocardial ischemia was achieved by ligating left anterior descending coronary artery with a 7-0 silk suture for 45 min and reperfusion was conducted by releasing the ligation for 24 h. The sham-operated mice underwent the same procedures without the artery ligation. Cardiac tissues dissected from border zone (BZ) of I/R hearts were used for mRNA and protein detection.
Isolation and culture of mouse cardiomyocytes
Neonatal mouse ventricular cardiomyocytes (NMVCs) were prepared as previously described [14]. Briefly, the mouse hearts were surgically removed from 1- to 3-day-old neonatal C57BL/6 mice, cut into 1.0 mm3 pieces, washed twice in Dulbecco’s modified Eagle’s medium (DMEM). The heart tissues were digested by 0.25% trypsin, the supernatant was collected and centrifuged at 1500 r/min for 5 min at room temperature. Cell pellets were resuspended and incubated in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 µg/mL streptomycin for 2 h at 37 °C with 5% CO2 and 95% air. After fibroblasts adherence, the no-adherent and weakly attached cells, mainly cardiomyocytes, were removed to cell culture dishes and incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. After 48 h culture, cardiomyocytes that reached 70%–80% confluence were treated with H2O2 for 12 h to induce cardiomyocyte injury for the subsequent experiments.
Synthesis of siRNA and transfection
LncRNA-6395-specific siRNA (si-lncRNA-6395), p53-specific siRNA (si-p53), and the negative control (NC) were commercially synthesized by GenePharma (Shanghai, China). The target sequences of siRNA and NC were shown in Supplementary Table S1. These siRNAs were transfected into cells for lncRNA-6395 and p53 knockdown. The siRNA transfection was performed using X-treme GENE Transfection Reagent (Roche, Basle, Switzerland) according to the manufacturer’s instructions. In briefly, 80 nM of si-lncRNA-6395 or si-p53 was added to X-treme GENE Transfection Reagent medium at room temperature to form transfection complexes. The cells were incubated in serum-free DMEM containing the transfection complexes for 6 h, then the medium was replaced with 10% FBS-DMEM. At 48 h after transfection, the cardiomyocytes were collected for isolation and purification of total RNA or protein.
Construction and transfection of plasmids
Plasmids pCDNA 3.1 carrying lncRNA-6395 cDNA or p53 cDNA were constructed by Genechem (Shanghai, China). These plasmids were transfected into cells for lncRNA-6395 or p53 overexpression at a final concentration of 1 μg/mL. The plasmid DNA transfection and co-transfection (plasmid DNA and si-RNA) were performed using Lipofectamine™ 2000 Transfection Reagent (Thermo Scientific, Carlsbad, USA) according to the manufacturer’s instructions.
MTT assay
NMVCs were seeded onto 96-well plates and incubated with H2O2 at designed concentrations for 12 h. After treatment, 20 μL MTT (5 mg/mL) was added to each well of the plate. The cells were incubated for 4 h at 37 °C, MTT solution was removed, and then 150 μL dimethyl sulfoxide was added into each well and shacked for 10 min. The absorbance value of each well was measured with Eliza Reader at 490 nm.
Lactate dehydrogenase release
NMVCs were seeded onto each well of 12-well plates and incubated with H2O2 at a final concentration of 200 µM for 12 h. Lactate dehydrogenase (LDH) was measured in the cell culture supernatant. The assay was performed using the lactate dehydrogenase assay kit (Nanjing Jiancheng, Nanjing, China) according to manufacturer’s instructions. The LDH activity was measured by an enzyme-labeled instrument at 450 nm.
Western blotting
The total protein was extracted from NMVCs or heart tissues of mice with RIPA buffer containing 1% protease inhibitors, and the concentration of protein was determined by BCA assay. Protein samples (80–100 μg) were fractionated by SDS-PAGE (12% polyacrylamide gels), subsequently transferred to a Nitrocellulose Blotting Membrane (PALL, New York, USA) and blocked with 5% non-fat milk at room temperature for 2 h. The membranes were then incubated with the primary antibody overnight at 4 °C and conjugated with secondary anti-rabbit or anti-mouse (LI-COR, Lincoln, USA) polyclonal antibody at room temperature for 1 h in the dark. The relative level of protein was detected by Odyssey Infrared Imaging System (LI-COR, Lincoln, USA) and analyzed with Odyssey 3.0 software. The primary antibodies were used as follows: anti-Bcl-2 antibody (Cell Signaling Technology, MA, USA), anti-Bax antibody (Cell Signaling Technology), anti-p53 antibody (Cell Signaling Technology), mouse anti-β-actin mAb (Proteintech, Wuhan, China).
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cardiomyocytes or myocardial tissues using Trizol reagent (Invitrogen, Carlsbad, USA) according to the manufacturer’s protocol. Quality of the RNA samples was measured by NanoDrop ND-8000 (Thermo Fisher Scientific, MA, USA) to ensure the RNA/DNA ratio of 1.8–2.0. RNA was reverse transcribed to cDNA using Reverse Transcription Kit (Transgene, Beijing, China). The SYBR Green qRT-PCR assay (Roche, Basel, Switzerland) was used for relative quantification of lncRNA-6395 and p53. β-Actin was used as an internal control. The specific sequences of primers were synthesized by Invitrogen and listed in Supplementary Table S2.
Echocardiographic assessment of cardiac function
After I/R injury, transthoracic echocardiography was performed to monitor changes of the left ventricular function of mice using ultrasound machine Vevo2100 high-resolution imaging system equipped with a 10-MH2 phased-array transducer with the M-mode recordings (Visual Sonics, Toronto, Canada). Ejection fraction (EF) and left ventricular fractional shortening (FS) were measured to assess cardiac function.
TTC staining
After 24 h reperfusion, the mice were anesthetized by injection of sodium pentobarbital (40 mg/kg). The hearts were isolated and frozen at −80 °C. The frozen hearts were cut into 1–2 mm thick sections perpendicular to the long axis, and then slices were incubated with 2% TTC for 15 min at 37 °C. The images of infarct area (pale white) and non-infarct area (brick red) were taken by stereo-microscope (Zeiss, Jena, Germany), and the percentage of infarct area was calculated with Image-Pro Plus 6.0 software.
TUNEL staining
The terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining for cultured NMVCs was performed with In Situ Cell Death Detection Kit (Roche, Mannheim, Germany) according to the manufacturer’s protocol. Cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 1% Triton X-100. Nuclei were stained with DAPI. Five visual fields in each slice were randomly chosen, cells were counted by Image-Pro Plus 6.0 software. The apoptosis rate (AR) was determined as follows: AR = (the number of positive cells/the total number of counted cells) × 100%.
Annexin V-FITC/PI apoptosis assay
Annexin V-FITC/PI apoptosis assay was performed using the Annexin V-FITC apoptosis assay kit (Absin, Shanghai, China). Briefly, NMVCs were treated with 0.25% Trypsin-EDTA solution, cells were harvested and washed twice with PBS. The cells then were resuspended in 300 μL of binding buffer, and 5 μL of FITC-labeled Annexin-V and 5 μL of PI solution were added to the cell suspension. The tubes were incubated at room temperature for 15 min and subjected to flow cytometry (Beckman Coulter, Brea, USA) to assess the percentage of apoptotic cells.
FISH and immunofluorescence
Fluorescence in situ hybridization (FISH) assay for lncRNA-6395 was performed according to RiboTM fluorescent in situ hybridization kit (RiboBio, Guangzhou, China). Briefly, the cultured cardiomyocytes were fixed in 4% polyoxymethylene in PBS at room temperature for 20 min, permeated with 0.5% Triton X-100 for 10 min at 4 °C. After washing with PBS solution, the cells were prehybridized at 37 °C for 1 h and then incubated with lncRNA-6395 specific probe (RiboBio, Guangzhou, China) at 37 °C overnight. Then cells were washed and blocked by 3% BSA, subsequently incubated with p53 antibody (Cell Signaling Technology, MA, USA) overnight, then cells were washed and conjugated with secondary antibody. The nuclei were stained by DAPI. Cells were observed with a fluorescence microscope (Nikon 80i, Tokyo, Japan).
LncRNA-pull down assay
LncRNA-6395 and its antisense RNAs were labeled with biotin RNA labeling mix (Roche, Basel, Switzerland) and transcribed with T7 RNA polymerase (Roche, Basel, Switzerland). The RNA was purified with G50 Sephadex Quick Spin columns (Roche, Mannheim, Germany). The protein samples from the whole heart (200–250 μg/sample) were incubated with purified biotinylated RNA (15–20 μg/sample) at 4 °C overnight, RNA-protein complexes were isolated by streptavidin agarose beads, the beads were then washed 5 times. The proteins pulled down was analyzed by Western blot.
Statistical analysis
Data are presented as the mean ± SEM. One-way ANOVA followed by Tukey post hoc analysis was used for multiple comparisons by GraphPad Prism software 8.0, and unpaired Student’s t-test was performed for comparison of two groups. A value of P < 0.05 was considered statistically significant difference.
Results
LncRNA-6395 is upregulated in I/R injured hearts and H2O2-treated NMVCs
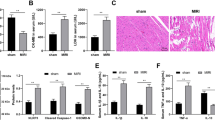
The I/R injury model was successfully established in mice with 45 min ischemia and 24 h reperfusion. Left ventricular EF and FS were significantly decreased in the I/R mice (Fig. 1a–c). Myocardial infarct size was measured by TTC staining assay in sham and I/R group (Fig. 1d, e). Quantitative real-time RT-PCR (qRT-PCR) was performed to detect the expression of lncRNA-6395. The expression of lncRNA-6395 was markedly increased in the infarct and border region compared to the remote region of the I/R hearts (Fig. 1f). In addition, H2O2, a commonly used inducer of apoptosis [15] also induced lncRNA-6395 expression in a dose-dependent manner in cultured NMVCs after treatment with 100, 200, and 300 μM H2O2 (Fig. 1g). H2O2 200 µM induced a significant moderate increase in lncRNA-6395 expression and was applied in the subsequent experiments. These data suggested that lncRNA-6395 is likely involved in I/R injury.

a Representative echocardiography images of mouse hearts in Sham and I/R groups. b, c Quantitative analysis of left ventricular ejection fraction (EF) and fractional shortening (FS) in Sham and I/R groups. n = 6, **P < 0.01 vs Sham. d Representative images of TTC staining in Sham and I/R groups. Red indicates the non-infarct area and border area, white indicates the infarct area. e Infarct size measurement by TTC staining in sham and I/R groups. n = 8, **P < 0.01 vs Sham. f Expression of lncRNA-6395 detected by qRT-PCR in different regions of I/R hearts. n = 5, **P < 0.01 vs Remote. g LncRNA-6395 expression in NMVCs treated with H2O2 100, 200, and 300 µM. n = 5, **P < 0.01 vs Control. Data are expressed by mean ± SEM.
Overexpression of lncRNA-6395 decreases cell viability and aggravates cell apoptosis
We then investigated whether the upregulation of lncRNA-6395 is a contributor to cardiac apoptosis or is merely a consequence. As depicted in Fig. 2a, lncRNA-6395 overexpressing plasmid was transiently transfected into the cultured NMVCs, which resulted in threefold increase in lncRNA-6395 level compared to empty vector transfection. Forced expression of lncRNA-6395 prominently reduced cell viability (Fig. 2b) and further aggravated 200 µM H2O2-induced cell viability (Supplementary Fig. S1a), and elevated LDH activities of NMVCs with or without H2O2 stimulation (Fig. 2c and Supplementary Fig. S1b). In addition, forced overexpression of lncRNA-6395 reduced the protein level of Bcl-2 (Fig. 2d) and enhanced Bax expression (Fig. 2e). Flow cytometry (Fig. 2f) and TUNEL (Fig. 2g) assay showed that lncRNA-6395 overexpression resulted in a significant increase in cell apoptosis rates compared with Vector group.

a Level of lncRNA-6395 detected by qRT-PCR assay in NMVCs transfected with lncRNA-6395 plasmid and empty vector served as a negative control. n = 3, **P < 0.01 vs Vector. b, c Cell viability detected by MTT assay and LDH activities in NMVCs transfected with lncRNA-6395 plasmid. n = 4, *P < 0.05 vs Vector. d, e Bcl-2 and Bax expression detected by Western blot assay in NMVCs with lncRNA-6395 overexpression. β-Actin served as a loading control. n = 5, **P < 0.01 vs Vector. f, g Flow cytometry and TUNEL assay for apoptosis detection in NMVCs with different treatments. n = 4, **P < 0.01 vs Vector. Scale bar, 20 μm. Data are expressed by mean ± SEM.
Knockdown of lncRNA-6395 promotes cell viability and reduces cell apoptosis
Upregulation of lncRNA-6395 induced by H2O2 was significantly inhibited by transfection of lncRNA-6395 siRNA (si-6395) (Fig. 3a). In 200 µM H2O2-treated cardiomyocytes, lncRNA-6395 knockdown resulted in increase of cell viability (Fig. 3b) and Bcl-2 expression (Fig. 3c), and decrease of LDH activities (Fig. 3d) and Bax expression (Fig. 3e). Consistently, flow cytometry and TUNEL staining assays showed that knockdown of lncRNA-6395 suppressed cell apoptosis induced by H2O2 (Fig. 3f, g). However, silencing of lncRNA-6395 produced no effects on cell viability (Supplementary Fig. S2a) and LDH activities (Supplementary Fig. S2b) in NMVCs without H2O2 treatment. These data indicated that lncRNA-6395 knockdown promotes NMVC viability and reduces cell apoptosis induced by H2O2.

a Level of lncRNA-6395 in NMVCs with H2O2 treatment and lncRNA-6395 siRNA transfection. n = 3, **P < 0.01 vs Control, ##P < 0.01 vs H2O2. b Cell viability detected by MTT assay. c Expression of Bcl-2 in NMVCs by Western blot analysis. β-Actin served as a loading control. d Activities of LDH in different groups of NMVCs. e Expression of Bax in NMVCs by Western blot analysis. β-Actin served as a loading control. f, g Flow cytometry and TUNEL staining assay in NMVCs with different treatments. n = 4, **P < 0.01 vs Control, ##P < 0.01 vs H2O2. Scale bar, 20 μm. Data are expressed by mean ± SEM.
P53 is a downstream target of lncRNA-6395
To understand the molecular mechanism by which lncRNA-6395 promotes NMVC apoptosis, prediction of lncRNA-6395 targets was performed by RNA-protein interaction prediction (http://pridb.gdcb.iastate.edu/RPISeq/). The prediction score obtained by RF algorithm was 0.85, and the score obtained by SMV algorithm was 0.97 suggested that p53 was a potential interacting protein with lncRNA-6395. Fluorescent in situ hybridization with a specific probe for lncRNA-6395 and p53 respectively showed that both lncRNA-6395 and p53 were mainly expressed in the cytoplasm of NMVCs (Fig. 4a). Furthermore, RNA pull-down assay showed that lncRNA-6395 successfully pulled down p53, while the antisense sequence of it failed to do so (Fig. 4b), which confirmed the interaction between lncRNA-6395 and p53. We next examined the influence of lncRNA-6395 on p53 expression in NMVCs. Transfection of lncRNA-6395 plasmids dramatically promoted p53 protein level (Fig. 4c), while p53 mRNA level was not changed obviously (Fig. 4d). However, knockdown of lncRNA-6395 resulted in the reduction of p53 protein in NMVCs treated with H2O2 (Fig. 4e), but not mRNA level of p53 (Fig. 4f). We further investigated the mechanism by which lncRNA-6395 regulates the protein level of p53. Ubiquitin-proteasome degradation is the main metabolic pathway of endogenous protein. Knockdown of lncRNA-6395 resulted in a decrease in p53 protein, while ubiquitin-proteasome inhibitor MG132 (5 μmol/L) counteracted the downregulation of p53 (Fig. 4g). In addition, overexpression of lncRNA-6395 reduced, whereas knockdown of lncRNA-6395 increased the ubiquitination of p53 (Supplementary Fig. S3). Therefore, we speculated that lncRNA-6395 participated in the ubiquitination-mediated degradation of p53.

a Representative images for the localization of lncRNA-6395 and p53 in NMVCs, lncRNA-6395 stained in red, p53 stained in green. Scale bar, 200 μm. b Blotting of p53 pulled down by lncRNA-6395 sense, antisense, n = 3. c, d Expression of p53 in NMVCs transfected with lncRNA-6395 plasmids by Western blot and qRT-PCR assay. n = 3-5, **P < 0.01 vs Vector. e, f Expression of p53 in NMVCs transfected with lncRNA-6395 siRNA by Western blot and qRT-PCR assay. n = 4, **P < 0.01 vs Control, ##P < 0.01 vs H2O2. g Expression of p53 by Western blot assay in NMVCs treated with ubiquitin-proteasome inhibitors MG132. n = 5, **P < 0.01 vs si-6395 + H2O2. h, i Representative images of lncRNA-6395 and p53 immunofluorescence staining. n = 5, Scale bar, 50 μm. j Expression of p53 by Western blot assay in NMVCs transfected with p53 overexpressing plasmid. β-Actin served as a loading control. k Expression of lncRNA-6395 by qRT-PCR assay in NMVCs transfected with p53 overexpressing plasmid. n = 4, **P < 0.01 vs Vector. l Expression of p53 by Western blot assay in NMVCs transfected with p53 siRNA. β-Actin served as a loading control. m Expression of lncRNA-6395 by qRT-PCR assay in NMVCs transfected with p53 siRNA. n = 4, **P < 0.01 vs NC. Data are expressed by mean ± SEM.
In addition, we found that lncRNA-6395 overexpression led to the nuclear translocation of p53, while knockdown of lncRNA-6395 inhibited the nuclear accumulation of p53 induced by H2O2, which may partly explain the detrimental effects of lncRNA-6395 on NMVCs (Fig. 4h, i). We then evaluated whether p53 can influence the expression of lncRNA-6395. P53 overexpression plasmids transfection significantly increased the protein and mRNA level of p53 (Fig. 4j and Supplementary Fig. S4a). Overexpression of p53 in NMVCs did not alter the expression of lncRNA-6395 (Fig. 4k). As expected, knockdown of p53 failed to change the expression of lncRNA-6395 (Fig. 4l, m and Supplementary Fig. S4b).
LncRNA-6395 regulates NMVC apoptosis through p53
Given the effects of lncRNA-6395 on p53 protein stability and localization as shown above, we next evaluated whether lncRNA-6395 regulates cell viability and apoptosis via acting on p53. Knockdown of lncRNA-6395 effectively abolished H2O2-induced reduction of cell viability and LDH activities. However, the effects of lncRNA-6395 were significantly restored by overexpression of p53 (Fig. 5a, b). Similar results were obtained for apoptosis-related protein expression. LncRNA-6395 knockdown reduced the protein level of p53 and Bax, and increased Bcl-2 expression. However, these effects were significantly reversed by p53 overexpression (Fig. 5c, d, e). Furthermore, flow cytometry and TUNEL assay revealed that enforced expression of p53 canceled the inhibitory effects of lncRNA-6395 knockdown on cell apoptosis (Fig. 5f, g).

a, b Cell viability detected by MTT assay and activities of LDH in different groups of NMVCs. n = 3, *P < 0.05, **P < 0.01 vs Control, #P < 0.05 vs H2O2, &P < 0.05 vs si-6395 + H2O2. c–e Expression of p53, Bcl2, Bax in NMVCs by Western blot analysis. β-Actin served as a loading control. n = 3-4, **P < 0.01 vs Control, ##P < 0.01 vs H2O2, &P < 0.05, &&P < 0.01 vs si-6395 + H2O2. f, g Flow cytometry and TUNEL staining assay in NMVCs with different treatments. n = 4, **P < 0.01 vs Control, ##P < 0.01 vs H2O2, &&P < 0.01 vs si-6395 + H2O2. Scale bar, 20 μm. Data are expressed by mean ± SEM.
Knockdown of p53 attenuates the pro-apoptotic effects of lncRNA-6395
We employed siRNA for p53 to investigate lncRNA-6395 effects on cell viability and apoptosis, LDH activities, and apoptosis-related proteins. As expected, knockdown of p53 attenuated the effects of lncRNA-6395 overexpression on NMVCs. Knockdown of p53 recovered the reduced cell viability and enhanced LDH activities by lncRNA-6395 overexpression (Fig. 6a, b). Silencing p53 canceled inhibitory effect of lncRNA-6395 on Bcl-2, abolished increased p53 and Bax expressions (Fig. 6c–e), and blocked cell apoptosis induced by lncRNA-6395 by flow cytometry or TUNEL assay (Fig. 6f, g).

a, b Cell viability detected by MTT assay and activities of LDH in different groups of NMVCs. n = 3, **P < 0.01 vs Control, #P < 0.05, ##P < 0.01 vs lncRNA-6395. c–e Expression of Bcl-2, p53, Bax in NMVCs by Western blot analysis. β-Actin served as a loading control. n = 4, **P < 0.01 vs Control, #P < 0.05, ##P < 0.01 vs lncRNA-6395. f, g Flow cytometry and TUNEL staining assay in NMVCs with different treatments. n = 4, **P < 0.01 vs Control, ##P < 0.01 vs lncRNA-6395. Scale bar, 20 μm. Data are expressed by mean ± SEM.
Knockdown of lncRNA-6395 rescues cardiac I/R injury in mice
To further understand the role of lncRNA-6395 in vivo, we evaluated the effects of lncRNA-6395 on myocardial I/R injury in lncRNA-6395 heterozygous knockout mice (lncRNA-6395+/−). As shown in Fig. 7a, the level of lncRNA-6395 in lncRNA-6395+/− was decreased than that of WT controls. Compared to WT mice, lncRNA-6395+/− mice demonstrated improved heart function after I/R injury, as reflected by increased EF and FS (Fig. 7b–d), minimized infarct size (Fig. 7e, f), increased apoptosis-related protein Bcl-2 (Fig. 7g), and decreased plasma LDH activities (Fig. 7h), Bax (Fig. 7i) and p53 protein (Fig. 7j). Consistent with the in vitro experiment, there was no difference in p53 mRNA level between WT and lncRNA-6395+/− mice after I/R injury (Fig. 7k). These data indicated that knockdown of lncRNA-6395 in vivo has a protective effect against I/R injury.

a Expression of lncRNA-6395 detected by qRT-PCR in WT and lncRNA-6395+/− mice. n = 4, **P < 0.01 vs WT. b Representative images of echocardiography in different groups of mice. c, d Left ventricular ejection fraction (EF) and fractional shortening (FS) in lncRNA-6395+/− mice subjected to I/R injury. n = 8, **P < 0.01 vs WT, ##P < 0.01 vs I/R. e Representative images of myocardial tissues stained by TTC in different groups. The infarct area was stained pale white and non-infarct area was stained brick red. f Quantification of infarct size by TTC staining in different groups, lncRNA-6395-knockout decreased the infarct size after MI. n = 8, **P < 0.01 vs WT, ##P < 0.01 vs I/R. g Expression of Bcl-2 by Western blot analysis in different groups. β-Actin served as a loading control. n = 5, **P < 0.01 vs WT, ##P < 0.01 vs I/R. h Activities of LDH in different groups of mice. n = 5, **P < 0.01 vs WT, ##P < 0.01 vs I/R. i, j Expression of Bax and p53 by Western blot analysis in different groups. β-Actin served as a loading control. n = 5, **P < 0.01 vs WT, ##P < 0.01 vs I/R. k P53 expression at mRNA level in lncRNA-6395+/− mice subjected to I/R injury by qRT-PCR. n = 4, *P < 0.05 vs WT. Data are expressed by mean ± SEM.
Discussion
In the present study, we functionally characterized that lncRNA-6395 promoted cardiomyocytes apoptosis and deteriorated I/R injury through interacting with p53. LncRNA-6395 was increased in I/R injured hearts and H2O2-treated NMVCs. Overexpression of lncRNA-6395 had deleterious effects in NMVCs, while knockdown of lncRNA-6395 did the opposite. LncRNA-6395 promoted apoptosis and deteriorated I/R injury through enhancing p53 nuclear transportation by preventing ubiquitination-mediated p53 degradation. Knockdown of lncRNA-6395 decreased cardiac infarct size and improved heart function in mice with I/R injury.
Cardiomyocyte loss induced by I/R injury is the major challenge in the treatment of MI, and inhibition of cardiomyocyte apoptosis is considered to be a critical therapeutic strategy [16]. LncRNAs have been proved to possess critical functions in regulating biological and pathological processes. Recently, several lncRNAs have been reported to be involved in I/R injury. Liu et al. found that lncRNA CAIF alleviates I/R injury by inhibiting cardiac autophagy [17]. Knockdown of lncRNA NRF attenuates I/R injury by targeting miR-873 [18]. LncRNA Neat1 aggravates myocardial I/R injury via promoting apoptosis and autophagy in diabetic rats [19]. Our previous study demonstrated that LncRNA-ACART alleviated cardiomyocyte apoptosis via the PPARγ/Bcl-2 pathway [20]. In this study, we found that lncRNA-6395 was notably upregulated in I/R injured hearts and H2O2-treated NMVCs. Forced expression of lncRNA-6395 promoted NMVC apoptosis and reduced cell viability; knockdown of lncRNA-6395 attenuated the detrimental effects induced by H2O2, alleviated myocardial I/R injury and improved heart function in mice.
LncRNAs regulate gene expression at the epigenetic, transcriptional and posttranscriptional levels by binding or interacting with DNAs, RNAs or proteins. LncRNA CCRR improves cardiac conduction by directly binding with Cx43-interacting protein CIP85 and preventing CIP85-mediated Cx43 endocytic trafficking and degradation [4]. LncRNA ZFAS1 directly binds to a critical Ca2+ handling protein SERCA2a and represses its expression, and therefore impairs cardiac contractile function during MI [9]. In this study, we found that lncRNA-6395 exerts its effects by directly binding to p53 and therefore preventing its ubiquitination-mediated degradation and promoting its nuclear translocation. Song et al. found that p53 expression is upregulated in the myocardium of patients with congestive heart failure, and overexpression of p53 promotes cardiomyocyte apoptosis [21]. It has been reported that lysine methyltransferase Smyd2 can methylate p53, which leads to p53-mediated cardiomyocytes apoptosis decrease [22]. Accumulation of p53 leads to myocardial apoptosis, promoting p53 degradation can protect the heart from ischemia injury [23]. Our experimental results showed that overexpression of lncRNA-6395 increased the protein level of p53, while knockdown of lncRNA-6395 resulted in the downregulation of p53 and protected the hearts against the I/R injury.
Ubiquitin-proteasome degradation pathway is the main metabolic pathway of endogenous protein. Several lines of evidence indicated that lncRNAs are involved in this process. LncRNA TROJAN directly binds to ZMYND8 and promotes ZMYND8 ubiquitin-proteasome degradation in triple-negative breast cancer [24]. UPAT binds to the ubiquitination site of protein uhrf1, and thus protects uhrf1 from being degraded by ubiquitination [25]. Our data showed that the ubiquitin-proteasome inhibitor MG132 counteracted the downregulation of p53 by lncRNA-6395 knockdown, indicating that regulation of lncRNA-6395 on p53 expression involves ubiquitin-proteasome system and lncRNA-6395 might serve as a protein stabilizer for p53.
Generally, protein exerts its function in certain sub-cellular compartments of cells. P53 is normally held in the cytoplasm, under stress condition p53 will be transported into the nucleus by a microtubule-dependent dynein [26]. Studies demonstrated that lncRNAs could regulate the sub-cellular localization of proteins. Imamura et al. demonstrated that lncRNA NEAT1 binds to protein SFPQ which leads SFPQ to separate from IL-8 promoter, and relocate to paraspeckles [27]. Cytosolic lncRNA P53RRA interacts with G3BP1 and causes p53 to separate from a G3BP1 complex, resulting in p53 retention in the nucleus and further leading to cell cycle arrest, apoptosis, and ferroptosis [28]. Our results showed that enforced expression of lncRNA-6395 led to the upregulation and translocation of p53 into the nucleus, while knockdown of lncRNA-6395 attenuated p53 level and nuclear translocation induced by H2O2.
In normal circumstances, p53 is maintained at low levels by its negative regulator murine double minute-2 (MDM2) a ubiquitin ligase. MDM2 and p53 form a negative-feedback loop, in which p53 induces the expression of MDM2, which in turn promotes the ubiquitination-mediated degradation of p53 and decreases cellular p53 level [29]. In response to cellular stresses including DNA damage, hypoxia, oncogene activation, etc., the protein stability and abundance of p53 are increased and the p53 transcriptional response is augmented [10]. Wang et al. reported that lncRNA-UCA1 knockdown inhibited MDM2 level by interacting with its direct target miR-143 thereby inducing the activation of p53 in H/R-induced cardiomyocyte apoptosis [30]. Our results showed that lncRNA-6395 did not yield significant effects on MDM2 expression (Supplementary Fig. S5), indicating that MDM2 pathway may not be involved in p53 regulation by lncRNA-6395.
In summary, we demonstrated that lncRNA-6395 promotes apoptosis and deteriorates I/R injury through preventing ubiquitination-mediated p53 degradation and enhancing p53 nuclear transportation; knockdown of lncRNA-6395 decreased cardiac infarct size and improved heart function in mice with I/R injury. Our study provides a novel insight into the understanding of the role of lncRNA in I/R injury.
References
Li J, Xuan Z, Liu C. Long non-coding RNAs and complex human diseases. Int J Mol Sci. 2013;14:18790–808.
Huang H, Bu YZ, Zhang XY, Liu J, Zhu LY, Fang Y. LINC01433 promotes hepatocellular carcinoma progression via modulating the miR-1301/STAT3 axis. J Cell Physiol. 2019;234:6116–24.
Pan B, Zhao M, Xu L. Long noncoding RNA gastric cancer-associated transcript 3 plays oncogenic roles in glioma through sponging miR-3127-5p. J Cell Physiol. 2019;234:8825–33.
Zhang Y, Sun L, Xuan L, Pan Z, Hu X, Liu H, et al. Long non-coding RNA CCRR controls cardiac conduction via regulating intercellular coupling. Nat Commun. 2018;9:4176.
Wang YZ, Zhu DY, Xie XM, Ding M, Wang YL, Sun LL, et al. EA15, MIR22, LINC00472 as diagnostic markers for diabetic kidney disease. J Cell Physiol. 2019;234:8797–803.
Bian EB, Xiong ZG, Li J. New advances of lncRNAs in liver fibrosis, with specific focus on lncRNA-miRNA interactions. J Cell Physiol. 2019;234:2194–203.
Lan Y, Xiao XW, He ZC, Luo Y, Wu CF, Li L, et al. Long noncoding RNA OCC-1 suppresses cell growth through destabilizing HuR protein in colorectal cancer. Nucleic Acids Res. 2018;46:5809–21.
Li ZW, Hou PF, Fan DM, Dong MC, Ma MS, Li HY, et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017;24:59–71.
Zhang Y, Jiao L, Sun LH, Li YR, Gao YQ, Xu CQ, et al. LncRNA ZFAS1 as a SERCA2a inhibitor to cause intracellular Ca2+ overload and contractile dysfunction in a mouse model of myocardial infarction. Circ Res. 2018;122:1354–68.
Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20:199–210.
Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9:e1003368.
Mahmoudi S, Henriksson S, Corcoran M, Mendez-Vidal C, Wiman KG, Farnebo M. Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol Cell. 2009;33:462–71.
Sun F, Zhuang Y, Zhu H, Wu H, Li D, Zhan L, et al. LncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. Mol Cell Cardiol. 2019;133:188–98.
Li X, Luo S, Zhang J, Yuan Y, Jiang W, Zhu H, et al. lncRNA H19 alleviated myocardial I/RI via suppressing miR-877-3p/Bcl-2-mediated mitochondrial apoptosis. Mol Ther Nucleic Acids. 2019;17:297–309.
Wang ZH, Liu JL, Wu L, Yu Z, Yang HT. Concentration-dependent wrestling between detrimental and protective effects of H2O2 during myocardial ischemia/reperfusion. Cell Death Dis. 2014;5:e1297.
Li M, Ding W, Tariq MA, Chang W, Zhang X, Xu W, et al. A circular transcript of ncx1 gene mediates ischemic myocardial injury by targeting miR-133a-3p. Theranostics. 2018;8:5855–69.
Liu CY, Zhang YH, Li RB, Zhou LY, An T, Zhang RC, et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun. 2018;9:29.
Wang K, Liu F, Liu CY, An T, Zhang J, Zhou LY, et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. 2016;23:1394–405.
Ma M, Hui J, Zhang QY, Zhu Y, He Y, Liu XJ. Long non-coding RNA nuclear-enriched abundant transcript 1 inhibition blunts myocardial ischemia reperfusion injury via autophagic flux arrest and apoptosis in streptozotocin-induced diabetic rats. Atherosclerosis. 2018;277:113–22.
Wu H, Zhu H, Zhuang Y, Zhang J, Ding X, Zhan L, et al. LncRNA ACART protects cardiomyocytes from apoptosis by activating PPAR-gamma/Bcl-2 pathway. J Cell Mol Med. 2020;24:737–46.
Song H, Conte JV Jr., Foster AH, McLaughlin JS, Wei C. Increased p53 protein expression in human failing myocardium. J Heart Lung Transplant. 1999;18:744–9.
Sajjad A, Novoyatleva T, Vergarajauregui S, Troidl C, Schermuly RT, Tucker HO, et al. Lysine methyltransferase Smyd2 suppresses p53-dependent cardiomyocyte apoptosis. Biochim Biophys Acta. 2014;1843:2556–62.
Naito AT, Okada S, Minamino T, Iwanaga K, Liu ML, Sumida T, et al. Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury. Circ Res. 2010;106:1692–702.
Jin X, Xu XE, Jiang YZ, Liu YR, Sun W, Guo YJ, et al. The endogenous retrovirus-derived long noncoding RNA TROJAN promotes triple-negative breast cancer progression via ZMYND8 degradation. Sci Adv. 2019;5:eaat9820.
Taniue K, Kurimoto A, Sugimasa H, Nasu E, Takeda Y, Iwasaki K, et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc Natl Acad Sci USA. 2016;113:1273–8.
Vousden KH, Vande Woude GF. The ins and outs of p53. Nat Cell Biol. 2000;2:E178–180.
Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, et al. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL-8 expression upon immune stimuli. Mol Cell. 2014;53:393–406.
Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78:3484–96.
Nag S, Qin J, Srivenugopal KS, Wang M, Zhang R. The MDM2-p53 pathway revisited. J Biomed Res. 2013;27:254–71.
Wang QS, Zhou J, Li X. LncRNA UCA1 protects cardiomyocytes against hypoxia/reoxygenation induced apoptosis through inhibiting miR-143/MDM2/p53 axis. Genomics. 2020;112:574–80.
Acknowledgements
The work was supported partly by the National Natural Science Foundation of China (No. 81872871, 82073844, 82070283), Postdoctoral Science Foundation of China (2021T140172) and National Science and Technology Major Project (2018ZX10101003-003-003).
Author information
Authors and Affiliations
Contributions
MYZ, YJL, ZWP, and CQX designed and supervised this study; LFZ, QZ, LZ, XD, and XWZ performed the cell experiments; XYP, LLP, BM, and WDS performed the animal experiments; LFZ, Q,Z and LZ performed the statistical analysis; YJL, MYZ, LFZ and QZ wrote the manuscript; All authors contributed to manuscript revision, and read and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhan, Lf., Zhang, Q., Zhao, L. et al. LncRNA-6395 promotes myocardial ischemia-reperfusion injury in mice through increasing p53 pathway. Acta Pharmacol Sin 43, 1383–1394 (2022). https://doi.org/10.1038/s41401-021-00767-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00767-5
Keywords
This article is cited by
-
The Role of P53 in Myocardial Ischemia-Reperfusion Injury
Cardiovascular Drugs and Therapy (2023)
-
Long Noncoding RNAs Involved in Cardiomyocyte Apoptosis Triggered by Different Stressors
Journal of Cardiovascular Translational Research (2022)
