
Abstract
Vascular calcification (VC) is characterized by pathological depositions of calcium and phosphate in the arteries and veins via an active cell-regulated process, in which vascular smooth muscle cells (VSMCs) transform into osteoblast/chondrocyte-like cells as in bone formation. VC is associated with significant morbidity and mortality in chronic kidney disease (CKD) and cardiovascular disease, but the underlying mechanisms remain unclear. In this study we investigated the role of large-conductance calcium-activated potassium (BK) channels in 3 experimental VC models. VC was induced in vascular smooth muscle cells (VSMCs) by β-glycerophosphate (β-GP), or in rats by subtotal nephrectomy, or in mice by high-dosage vitamin D3. We showed that the expression of BK channels in the artery of CKD rats with VC and in β-GP-treated VSMCs was significantly decreased, which was functionally confirmed by patch-clamp recording. In β-GP-treated VSMCs, BK channel opener NS1619 (20 μM) significantly alleviated VC by decreasing calcium content and alkaline phosphatase activity. Furthermore, NS1619 decreased mRNA expression of ostoegenic genes OCN and OPN, as well as Runx2 (a key transcription factor involved in preosteoblast to osteoblast differentiation), and increased the expression of α-SMA protein, whereas BK channel inhibitor paxilline (10 μM) caused the opposite effects. In primary cultured VSMCs from BK−/− mice, BK deficiency aggravated calcification as did BK channel inhibitor in normal VSMCs. Moreover, calcification was more severe in thoracic aorta rings of BK−/− mice than in those of wild-type littermates. Administration of BK channel activator BMS191011 (10 mg· kg−1 ·d−1) in high-dosage vitamin D3-treated mice significantly ameliorated calcification. Finally, co-treatment with Akt inhibitor MK2206 (1 μM) or FoxO1 inhibitor AS1842856 (3 μM) in calcified VSMCs abrogated the effects of BK channel opener NS1619. Taken together, activation of BK channels ameliorates VC via Akt/FoxO1 signaling pathways. Strategies to activate BK channels and/or enhance BK channel expression may offer therapeutic avenues to control VC.
Similar content being viewed by others


Introduction
Vascular calcification (VC) is a serious kidney complication that affects 47%–92% of patients with chronic kidney disease (CKD) [1,2,3]. VC is associated with significant morbidity and mortality in these patients, predominantly due to cardiovascular events [4, 5]. VC is characterized by pathological calcium and phosphate depositions in the arteries and veins [6] via an active cell-regulated process, during which vascular smooth muscle cells (VSMCs) transform into osteoblast/chondrocyte-like cells similar to those observed in skeletal bone formation [2, 7]. Although the VC phenotype in CKD is well described, the mechanism of VSMC transdifferentiation is not fully understood.
The large-conductance calcium-activated potassium channels (also known as BK, BKCa, KCa1.1, or MaxiK) are distributed in various tissues and cells and are especially highly expressed in VSMCs [8]. They play a pivotal role in regulating important physiological functions related to vascular tone and myocardial perfusion by integrating changes in intracellular Ca2+ and membrane potential [9, 10]. Emerging evidence indicates that BK channels may be involved in the pathogenesis of cardiovascular diseases [11, 12]. Diabetes mellitus-induced vascular dysfunction is associated with abnormalities in BK channel function and expression [13], and BK channel function is markedly depressed by metabolic syndrome in coronary smooth muscles [14]. Additionally, the inhibition of the intermediate-conductance channels (IKCa, a Ca2+-activated K+ channel, similar to BK channels) prevents the phenotypic transition and VC of VSMCs [15]. In line with these observations, our previous studies showed that the BK channels were involved in the proliferation and mineralization of osteoblasts, which resemble the VC process [16, 17]. The role of the BK channels in CKD-associated VC is still unclear; thus, we designed a study to test whether BK channels are involved in the calcification and osteogenic differentiation of VSMCs in the context of CKD.
Materials and methods
Reagents
β-Glycerophosphate (#50020), BMS191011 (#SML0866), NS1619 (#N170), and alizarin red S (#A5533) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Paxilline was purchased from Tocris Bioscience (#2006, Bristol, UK). MK2206 was from Selleck Chemicals (#S1078, Houston, Texas, USA). Calcitriol was from MedChemExpress (#HY-10002, Monmouth Junction, NJ, USA). Primary antibodies for Runx2(#sc-101145) and GAPDH (#sc-32233) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); p-AKT (#4691), AKT (#9271), FoxO1 (#2880) and P-FoxO1 (#9464) were from Cell Signaling Technology (Denvers, MA, USA). The antibodies also include anti-BK (#APC-107, Alomone Labs, Israel), and α-SMA (#ab32575, abcam, USA). The anti-mouse and anti-rabbit IgG secondary antibodies were from Yeasen (#33101ES60, #33201ES60, Shanghai, China). Other chemicals and reagents used were of analytical grade.
Animals
All animals were supplied by the Animal Center at the School of Pharmacy, Fudan University (Shanghai, China). All animal experiments were approved by the Animal Experimentation Ethics Committee of Fudan University, and performed in compliance with the Animal Management Rule of the Ministry of Health, China (documentation No. 55, 2001) as well as the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All animals were housed in individual cages at 25 °C and 25% humidity on a 12/12-h light/dark cycle and fed a standard powder diet. The BK channel knockout mice were generated in our lab using a previously established protocol [17].
Animal model of vascular calcification
Twenty male Sprague-Dawley (SD) rats (weighing 200 ± 20 g) were randomly divided into two groups. In the CKD calcification group, rats were subjected to subtotal nephrectomy, i.e. the removal of the right kidney and infarction of approximately 2nd/3rd of the left kidney as described by Alesutan et al. [18], which was performed under isoflurane anesthesia (1.5%–2%). To improve the calcification-molding rate in CKD, the rats were fed a diet supplemented with 1.8% phosphorous and 4% calcium, and calcitriol (1 μg/kg, every 2 days) was intraperitoneally administered. The control group rats were maintained on a standard diet. After 12 weeks, the animals were sacrificed, and the thoracic aortas, abdominal aortas, and blood, were harvested for further analysis.
To induce VC in mice, ten 6-week-old C57BL/6 male mice were subcutaneously injected with cholecalciferol (Vitamin D3, #C9756, Sigma-Aldrich) at a dose of 500,000 IU/kg body weight, and another five mice were administered physiological saline for the first 3 days as described by Kwon et al. [19]. Five of ten mice were intraperitoneally injected with 10 mg/kg BMS191011 for the first 5 days. On day 9, the mice were euthanized under isoflurane anesthesia, and tissues were collected in liquid nitrogen for further study.
VSMC culture and treatment
VSMCs were isolated from rat or murine thoracic aorta by rinsing off blood from the aorta with PBS, cutting the aorta into small pieces (after removal of endothelium and adventitia), and plating it in a cell culture flask with Dulbecco’s modified Eagle’s medium (DMEM, #11995115, Gibco, USA) containing 20% fetal bovine serum (FBS, #10091148, Gibco) at 37 °C in an incubator containing 95% air and 5% CO2 for 7–14 days. VSMCs migrating from explants were collected and maintained in a growth medium (DMEM containing 10% FBS). The VSMCs obtained between passages 4 and 8 were used in the experiments. For calcification, confluent VSMCs were treated with calcifying media (2.5 mM CaCl2 and 10 mM β-glycerophosphate) for 6 days. The murine VSMC cell line MOVAS (ATCC® CRL-2797TM) was purchased from Shanghai Sixin Biotechnology Co., Ltd. (Shanghai, China). To explore the effect of BK channels on VSMC calcification, the calcifying medium was supplemented with the BK channel inhibitor paxilline (10 μM) or the agonist NS1619 (20 μM). After 6 days of treatment, cells were collected for further study. To explore the signaling pathway of BK channel-mediated VC, the Akt inhibitor MK2206 (1 μM) and forkhead box O1 (FoxO1) inhibitor AS1842856 (3 μM) were administered with or without NS1619 in the calcifying medium. Each experiment was performed at least thrice, unless stated otherwise.
Aortic ring calcification
The thoracic aortas were extracted from rats or mice following removal of the endothelium, and the adventitia was cut into 2–3 mm sections. The vascular rings were cultured in DMEM with 10% FBS in an incubator with 5% CO2 at 37 °C. Similar to the VSMCs, vascular rings were incubated in DMEM or calcifying media, which were changed every 2 days. After 6 days, the vascular rings were fixed in formalin for further analysis.
Quantification of calcium content
Cells or tissues were rinsed thrice with ice-cold PBS and decalcified with 0.6 M HCl for 24 h at 4 °C. Calcium content in the supernatant was examined using the Calcium Assay Kit (#C004-2-1, Jiancheng Bioengineering Institute, Nantong, China). Values were normalized to the levels of total protein or tissue weight.
Alkaline phosphatase (ALP) activity assay
ALP activity was measured using an ALP Colorimetric assay kit according to the manufacturer’s instructions (#P0321S, Beyotime Biotechnology, Shanghai, China). Briefly, the cultured VSMCs were trypsinized, washed thrice with ice-cold PBS, and lysed with RIPA Lysis Buffer (#P0013C, Beyotime Biotechnology). After centrifugation at 8000 × g for 10 min at 4 °C, the cell supernatants were collected for detecting ALP activity and total protein content by the bicinchoninic acid (BCA) assay. Values were normalized to the total protein levels.
Alizarin red S staining
Calcification was assessed by alizarin red S staining, as previously described [20]. Briefly, VSMCs grown in six-well plates were fixed with 4% formaldehyde for 45 min at 4 °C after which they were washed with distilled water, exposed to alizarin red S (2% aqueous) for 5 min, and washed again with distilled water. Stained cells were first observed under a light microscope (Olympus BX 50, Olympus Optical Tokyo, Japan) for positive red/purple staining. For quantification of the Alizarin Red S staining, cells were incubated with 10% HDPD for 15 min and the positive density was measured at 540 nm using a TECAN infinite 200 PRO spectrophotometer (Tecan Systems Inc., Austria). Values were normalized to the total protein concentration measured using the BCA assay.
Von Kossa staining
Calcification of aortas was analyzed by von Kossa staining [21]. To obtain paraffinized sections of aortic tissue, a 1-cm segment of the aorta was fixed with 10% formalin (pH 7.4, 0.1 M) for 24 h at room temperature (RT). After dehydration in an ethanol gradient, the aorta tissue was embedded in paraffin. Some aortic tissue sections (6-μm thick) were stained with hematoxylin-eosin (H&E), while others were deparaffinized, dehydrated, and then treated with 5% AgNO3 for 30 min at RT, followed by exposure to ultraviolet light for 1.5–2 h, and then washed thrice with deionized water. Specimens were counterstained with eosin and examined under a light microscope (Olympus BX 50).
Western blotting
Immunoblotting was performed to assess the protein content. Briefly, the proteins were separated by electrophoresis on 10% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (# IPV00010, Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk (prepared in 1×Tris-buffered saline Tween [TBST]) for 1 h at RT, and then incubated with primary antibodies against BK channels (1:1000), Runx2 (1:1000), alpha-smooth muscle actin (α-SMA; 1:1000), Akt (1:1000), p-Akt (1:1000), FoxO1 (1:1000), p-FoxO1(1:1000), and GAPDH (1:10,000) overnight at 4 °C. The membranes were then washed thrice with 1× TBST, and incubated with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit IgG, 1:10,000) for over 1.5 h at RT. After three washes with 1× TBST, the protein content was analyzed by Image Lab and normalized to GAPDH levels. All experiments were repeated at least thrice.
Quantitative PCR
Gene expression was assessed by qPCR as described by Wang et al. [22]. Briefly, total RNA was extracted from the cells or aortas using RNAiso Plus reagent (# 9109, TaKaRa, Japan) and reverse transcribed into cDNA using PrimeScript RT Master Mix (#RR036A, TaKaRa). qPCR was performed using 2 μL of the cDNA mixed with SYBR® Premix Ex Taq™ solution (#RR0420A, TaKaRa). Each reaction was performed in triplicate and analyzed on a BIO-RAD CFX Connect Real-time PCR system and GAPDH was used for normalization. The specific primers are as follows: OCN, 5′-ATGTCCAAGCAGGAGGGCAGTA-3′ (forward) and 5′-CCAAGTCCATTGTTGAGGTAGCG-3′ (reverse); OPN, 5′-ATCTCACCATTCGGATGAGTCT-3′ (forward) and 5′-TGTAGGGACGATTGGAGTGAAA-3′ (reverse); Runx2, 5′-CAAGCACAAGTGATTGGCCGAACT-3′ (forward) and 5′-CTCAACCACGAAGCCTGCAATTT-3′ (reverse); BK, 5′-AGGAATGCATCTTGGCGTCACTC-3′ (forward) and 5′-CCTCGAAGTGCATTCTCCTCAGC-3′ (reverse); α-SMA, 5′-GTCCCAGACATCAGGGAGTAA-3′ (forward) and 5′-TCGGATACTTCAGCGTCAGGA -3′ (reverse); GAPDH, 5′-GGCACAGTCAAGGCTGAGAATG-3′ (forward) and 5′-ATGGTGGTGAAGACGCCAGTA-3′ (reverse).
Immunohistochemistry
Freshly harvested aortas were embedded in paraffin as described by Zebger-Gong et al. [23]. Subsequently, the paraffin sections were first deparaffinized and rehydrated at 60 °C for 2 h, incubated in ethanol with gradient concentrations of 100%, 90%, 80%, 70%, and 50% for 5 min each, and washed in PBS for 5 min. To block endogenous catalase activity, the sections were incubated with 0.5% hydrogen peroxide at RT for 10 min. The sections were then incubated with 5% normal non-immune serum for 10 min at RT, followed by incubation with primary antibodies against Runx2 (1:200), α-SMA (1:200), and BK channels (1:200) or PBS at 4 °C overnight to prevent non-specific binding of immunoglobulin. After washing with PBS thrice (5 min each time), all sections were incubated with the respective secondary antibodies for 1 h at RT, followed by incubation with diaminobenzidine and counterstaining with H&E. Finally, the sections were analyzed by a light microscope (Olympus BX 50).
Whole-cell patch clamping
Whole-cell patch clamping was performed as described in detail [22, 24] using a Multiclamp 700B amplifier (Molecular Devices, USA) at RT. The patch pipettes were fabricated from glass capillary tubes using a PC-10 Puller (Narishige, Japan) with a resistance of 4–5 MΩ. Data acquisition and stimulation protocols were controlled by pCLAMP 10.5 (Molecular Devices, CA, USA). The capacitance transients were canceled in the patch-clamp recordings. Cells with a seal resistance (Rs) of less than 1 GΩ were omitted. To minimize voltage errors, Rs was compensated to 80%–85%, and cells with uncompensated Rs higher than 10 MΩ were discarded; all patch-clamping experiments met this criterion with a holding potential of −80 mV. The outward currents of murine smooth muscle cells were elicited by step pulses ranging from −50 to +80 mV for 200 ms with increments of 10 mV. The paxilline-sensitive currents evoked using these protocols were entirely attributable to the BK channels.
The standard external solution for BK channels consisted of 150 mM NaCl, 0.8 mM MgCl2, 5.4 mM KCl, 5.4 mM CaCl2, and 10 mM HEPES. The pH of the solution was adjusted to 7.4 by adding NaOH. The internal solution consisted of 10 mM NaCl, 125 mM KCl, 10 mM HEPES, 6.2 mM MgCl2, and 5 mM free Ca2+, and the pH of the solution was adjusted to 7.2 by adding KOH. The total Ca2+ was increased to yield the desired free concentration, which was calculated using the program Maxchelator [25].
Statistical analysis
All experimental results are presented as the mean ± standard error of the mean (SEM). Statistical significance was assessed by two-tailed unpaired Student’s t-test for comparison between two groups and one-way ANOVA for multiple comparisons. Values with P < 0.05 were considered statistically significant. All data analyses were performed using GraphPad PRISM 7.0 (GraphPad Software, Inc., USA).
Results
BK channels are downregulated under calcification in vitro and in vivo
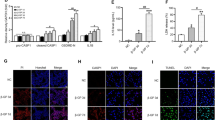
We first tested whether calcification affects the expression of BK channels by exposing cultured primary rat VSMCs to a calcified medium (10 mM β-glycerophosphate, β-GP, 2.5 mM Ca2+, and 2% FBS) for 7 days, which is a commonly used in vitro model to induce calcification. Subsequent experiments showed that β-GP-induced calcification of rat VSMCs decreased the expression (assessed by immunoblotting) of the BK channels and α-SMA but increased that of Runx2 (Fig. 1a), a key transcription factor involved in preosteoblast to osteoblast differentiation [26]. In the same cells, the mRNA levels of Runx2, osteocalcin (OCN), and osteopontin (OPN), which are involved in osteogenesis, were also increased in the presence of β-GP (Fig. 1b).

VC was induced either by β-GP in rat VSMCs (a, b, n = 3) and murine VSMCs (MOVAS, g–i, n = 6) or by subtotal nephrectomy in rats (c–f, n = 3). a Representative original Western blots and semi-quantitative analysis of α-SMA, Runx2, and BK proteins. b Transcript-level expression of osteogenic genes determined by quantitative real-time PCR (qPCR) followed by normalization. c Von Kossa staining of calcium nodules and quantitative analysis of calcium content in the thoracic aortas from vehicle and calcification groups. d Expression and semi-quantitative analysis of Runx2, α-SMA, and BK channel proteins during calcification further verified by Western blotting. e Immunohistochemistry for α-SMA, Runx2, and BK channels. f Relative amounts of Runx2, OCN, α-SMA, and BK mRNA were determined by qPCR following normalization. g Representative traces of whole-cell outward currents underlying β-GP (−) or β-GP (+) MOVAS before and after treatment with 10 μM paxilline (PAX). The holding potential was −80 mV, and the currents were evoked using step pulses ranging from −50 to +80 mV for 200 ms in increments of 10 mV. h Quantification of mean current densities of endogenous BK channels in β-GP (−) and β-GP (+) MOVAS cells. i Quantification of the mean proportion of PAX-sensitive currents to the total outward currents of the β-GP (−) and β-GP (+) MOVAS. Data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 indicate significant differences between treatment and control-treated groups (β-GP (−) or vehicle).
To confirm our in vitro observations, we tested whether the CKD-related VC in rats affected the abundance of BK channels. CKD-related VC was induced in rats by subtotal nephrectomy [27] via intraperitoneal injection of calcitriol combined with a high phosphorus diet. To prove that calcification animal model in CKD rats works, the serum levels of biochemical parameters like blood urea nitrogen (BUN), creatinine (Cr), calcium (Ca) and phosphate (Pi), and ALP activity and calcium content in CKD rat aorta were observed (Supplementary Fig. S1). The calcium content in the thoracic aortas was measured using von Kossa staining (Fig. 1c). Similar to the results obtained in the in vitro experiments, immunoblotting, immunohistochemistry, and qPCR revealed that CKD-VC was associated with a significant reduction in the expression of α-SMA and BK channels at the transcript and protein levels in the rat aorta (Fig. 1d–f), while mRNA levels of Runx2 and OCN were increased in the presence of CKD-VC (Fig. 1f).
BK channel function is impaired under VC in vitro
To further explore the effect of calcification on the function of BK channels in vitro, we tested the currents of BK channels in β-GP (−) or β-GP (+)-treated MOVAS using whole-cell patch clamping. The currents of the MOVAS BK channels could be evoked by depolarizing the voltage steps from a holding potential of −80 mV. The outward currents of the β-GP (−) MOVAS were markedly blocked by the high-affinity inhibitor paxilline [28, 29] at all membrane potentials (Fig. 1g), which implies that the blocked outward currents of MOVAS cells are endogenous BK channel currents. However, compared with those of the β-GP (−) MOVAS, the outward currents of the β-GP (+) MOVAS were more insensitive to PAX (Fig. 1g). Therefore, the current density of endogenous BK channels (PAX-sensitive channels) in the β-GP (−) group was higher than that in the β-GP (+) group (Fig. 1h and Supplementary Table S1). The proportion of currents in the PAX-sensitive BK channels to total outward currents underlying the β-GP (−) VSMCs was ~70% under each voltage stimulation, while the proportion of the β-GP (+) group was ~40% (Fig. 1i and Supplementary Table S2).
These in vitro observations indicate that the expression and function of BK channels are negatively regulated by β-GP-induced calcification, and changes in BK channels are accompanied by altered expression of the selected osteogenic transcription factors.
BK channels control calcification in vitro
To examine the effect of BK channels on vascular osteo/chondrogenesis and calcification in vitro, we pre-incubated rat VSMCs exposed to β-GP with NS1619 (20 μM, a specific BK channel opener) or the inhibitor paxilline (10 μM). Activation of the BK channels abolished the β-GP-induced calcification in rat VSMCs, as NS1619-treated cells showed a marked reduction in Ca2+-enriched deposition (Fig. 2a), calcium content, ALP activity (Fig. 2b), gene expression of osteogenic markers (OCN and OPN) (Fig. 2c) but showed increased α-SMA protein levels (Fig. 2d, e). In contrast, inhibition of the BK channels with paxilline exacerbated the osteo-/chondrogenic differentiation and mineralization in rat VSMCs (Fig. 2a). Furthermore, cells treated with paxilline exhibited increased calcium content, ALP activity (Fig. 2b), and OCN and OPN gene expression (Fig. 2c) and reduced α-SMA protein content (Fig. 2d, e).

a Representative original images of alizarin red S staining and quantification analysis in VSMCs. Calcified areas are shown in red. NS1619 (BK channel opener, 20 μM), paxilline (BK channel inhibitor, 10 μM). b Normalized calcium content and ALP activity in rat VSMCs. c Relative mRNA levels of the osteogenic genes OCN and OPN were determined by quantitative real-time PCR (qPCR) following normalization. d, e Representative Western blots and semi-quantitative analysis of Runx2, α-SMA, and BK channel proteins. f Representative traces of outward currents of β-GP (+)-treated MOVAS before and after administration of 20 μM NS1619. g Quantification of mean current densities of outward currents underlying β-GP (+)-treated MOVAS before and after administration of 20 μM NS1619. h Quantification of the NS1619-induced increase in the rate of outward currents of β-GP (+)-treated MOVAS. Data are expressed as the mean ± SEM. a–d, n = 3; e–h, n = 6, *P < 0.05, **P < 0.01, ***P < 0.001.
We also tested whether NS1619 could reverse the decrease in BK channel currents in the β-GP (+) MOVAS. In the patch-clamp recording, the outward currents of the β-GP (+) MOVAS could be potently enhanced by the administration of NS1619 (Fig. 2f). The current density also increased at membrane potentials ranging from +10 to +80 mV (Fig. 2g and Supplementary Table S3). The rate of NS1619-induced increase in the outward currents of β-GP (+) MOVAS was ~200% under voltage stimulations ranging from 0 to +80 mV (Fig. 2h and Supplementary Table S4). These results suggest that BK channels are potent negative regulators of VC in vitro.
Calcification is aggravated in primary cultured BK−/− VSMCs
To further explore the role of BK channels in calcification and osteo-/chondrogenic transdifferentiation of VSMCs exposed to β-GP, primary cultured VSMCs were isolated from the global BK knockout (BK−/−) male mice or their wild-type littermates (BK+/+) [17]. As shown in Fig. 3a–c, calcification and Runx2 protein abundance were increased in BK−/− VSMCs compared with those in BK+/+ VSMCs; however, the expression of α-SMA was decreased (Fig. 3c, d). Genetic deletion of the BK channels had no effect on OPN and OCN mRNA levels under non-β-GP condition (Fig. 3d), while OPN, OCN, and Runx2 mRNA expression (Fig. 3d) and ALP activity (Fig. 3e) were higher in the BK−/− VSMCs with β-GP. Similarly, increased Ca2+ deposition in the thoracic aortas of BK−/− mice was observed post von Kossa staining (Fig. 3f). These results demonstrate that BK channel deficiency is sufficient to promote osteogenic-like differentiation of VSMCs in vitro.

a, b Representative images showing alizarin red S staining and quantification analysis in primary cultured VSMCs derived from the BK knockout mice (BK−/−) or their wild-type littermates (BK+/+) with or without induced calcification. Calcified areas are shown in red. c Representative original Western blots and semi-quantitative analysis of Runx2 and α-SMA proteins. d The relative mRNA levels of α-SMA, OPN, OCN, and Runx2 in BK+/+ or BK−/− VSMCs. e Bar chart showing the quantification of ALP activity after normalization. f Hematoxylin and eosin (H&E) and von Kossa staining of calcium nodules in thoracic aorta rings from BK+/+ or BK−/− mice. Data are expressed as the mean ± SEM. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001.
BK channel activation ameliorates vitamin D3-induced calcification in mice
Previous studies have shown that vitamin D3 induces calcification and mineralization in vivo [19, 30, 31]. Thus, we tested whether the activation of BK channels would be effective in preventing vitamin D3-induced VC in mice in vivo. The activation of the BK channels with BMS191011 (10 mg/kg), a selective activator of BK channels [32], obviously reduced calcium deposition and content in the arteries of the calcified mice induced by vitamin D3, as observed post von Kossa staining (Fig. 4a, b). Additionally, the beneficial effects of BK channel activation were paralleled by the decrease in the expression of Runx2 at mRNA and protein levels (Fig. 4c, e), ALP activity in the arteries (Fig. 4d), and the increase in α-SMA mRNA and protein levels (Fig. 4c, e). Taken together, these observations indicate that the activation of the BK channels with BMS191011 reduces VC and osteogenic differentiation in vitamin D3-treated mice.

a Calcification in the thoracic and abdominal aortas was analyzed by von Kossa staining. BMS191011 (BMS, BK channel opener, 10 mg/kg). b Quantification of the calcium content in the aortas normalized by tissue weight. c The mRNA levels of Runx2 and α-SMA were analyzed by quantitative real-time PCR (qPCR). d Bar chart showing the quantification results of ALP activity after normalization in mice aorta. e Representative original Western blots and semi-quantitative analysis of Runx2 and α-SMA. Data are expressed as the mean ± SEM. n = 5, *P < 0.05, **P < 0.01, ***P < 0.001.
BK channels regulate calcification via the Akt/FoxO1 signaling pathway
Next, we investigated the mechanisms underlying the beneficial effects of BK channels on rat VSMC calcification. By screening for several pathways involved in calcification with Western blotting (data not shown), we found that the expression of Akt was decreased in the calcified thoracic aorta of CKD-related VC rats (Supplementary Fig. S2a) and calcified VSMCs (Supplementary Fig. S2b). Moreover, paxilline treatment markedly reduced the expression of p-Akt in VSMCs with or without induction by β-GP (Supplementary Fig. S2b). Additionally, decreased p-Akt protein and Akt mRNA levels were observed in the VSMCs of BK−/− mice compared with those in the VSMCs of BK+/+ mice (Supplementary Fig. S2c and S2d). Thus, we tested whether the Akt pathway is involved in the progression of osteoblast-like differentiation of rat VSMCs following BK channel activation. We observed that the disruption of the Akt signaling pathway with a potent allosteric Akt inhibitor MK2206 (1 μM) [33] abolished the effect of NS1619 on calcified VSMCs induced by β-GP (Fig. 5). Compared with the effect of NS1619 addition, Akt inhibition led to increased calcification (assessed by Alizarin Red S staining, Fig. 5a, b), ALP activity (Fig. 5c) and Runx2, OCN, and OPN mRNA expression (Fig. 5d). However, Akt inhibition led to reduced expression of α-SMA and BK channel and phosphorylation of Akt and FoxO1 (Fig. 5e).

a, b Representative images showing alizarin red S staining and quantification analysis of VC. NS1619 (BK channel opener, 20 μM), MK2206 (Akt inhibitor, 1 μM). c Bar chart showing the quantification of ALP activity. d Bar chart showing the quantification the relative mRNA expression of α-SMA, Runx2, OCN, and OPN in rat VSMCs. e Representative original Western blots and semi-quantitative analysis of α-SMA, BK, p-Akt (ser473), Akt, p-FoxO1 (Thr24), and FoxO1 proteins. Data are expressed as the mean ± SEM. n = 3, *P < 0.05, **P < 0.01, ***P < 0.001.
FoxO1 is a downstream target regulated by Akt and may be involved in calcification [34]. To test this hypothesis, VSMCs exposed to β-GP/NS1619 were pre-incubated with AS1842856 (3 µM), a selective inhibitor of FoxO1 [35]. Alizarin red S staining showed that FoxO1 inhibition counteracted the β-GP/NS1619-induced calcification, which was confirmed by increased Ca2+ deposition (Fig. 6a, b), calcium content (Fig. 6c), ALP activity (Fig. 6d), and OCN and OPN mRNA levels (Fig. 6g) in VSMCs. These changes were accompanied by a decrease in the levels of α-SMA, BK channel, and FoxO1 proteins as well as a reduction in FoxO1 phosphorylation (Fig. 6e, f). Moreover, to confirm the exact role of FoxO1 in this process, the localization of FoxO1 in cytoplasm and nucleus in rat VSMCs after NS1619 treatment was observed. We found that NS1619 increased the distribution of FoxO1 in the cytoplasm compared to that in the VC group (Supplementary Fig. S3), which was consistent with increased p-FoxO1 levels (Fig. 5e) in the NS1619-treated group in presence of β-GP compared to that in the VC group. These results indicate that the observed BK channel-dependent effects on cell transformation and osteoblast-like differentiation in rat VSMCs in vitro may be mediated, at least in part, by the Akt-FoxO1 axis.

a, b Representative images showing alizarin red S staining and quantification analysis of VC. NS1619 (BK channel opener, 20 μM), AS1842856 (AS, FoxO1 inhibitor, 3 μM). c, d Bar chart and quantification of calcium content and ALP activity. e, f Representative Western blots and semi-quantitative analysis of α-SMA, BK channel, p-FoxO1, and FoxO1 proteins. g The relative mRNA expression of α-SMA, OCN, OPN, and Runx2 in rat VSMCs. Data are expressed as the mean ± SEM. n = 3, **P < 0.01, ***P < 0.001.
Discussion
The present study shows that BK channels play an important role in the regulation of VC in CKD. We observed that the activation of the BK channels inhibits the calcification of VSMCs in vivo and in vitro, while BK inhibition or genetic deletion exacerbates VC. Mechanistic studies suggest that the beneficial effects of BK channels on VC are mediated, at least in part, via the Akt/FoxO1 pathway.
BK channels are crucial in VSMCs, especially as they maintain the arterial tone in smooth muscle. Moreover, several studies have shown that BK channel dysfunction is involved in various cardiovascular pathologies, such as subarachnoid hemorrhage [36], hypertension [37], and left ventricular hypertrophy [38]. However, the possible involvement of BK channels in VC has not yet been explored. Here, we found that the expression of BK channels is decreased under calcifying conditions in VSMCs and mice with CKD-associated VC. The reduction in BK channels is accompanied by a decrease in the levels of α-SMA and an increase in the expression of the markers of osteo-/chondrogenic transformation and calcification (Runx2, OCN, and OPN). Thus, we hypothesized that BK channels are involved in the progression of calcification. Previous loss-of-function studies performed in vitro have also demonstrated the beneficial effect of BK channels on VC. Pharmacological inhibition or genetic silencing of the BK channels under conditions of calcification exacerbated the calcification process and increased the expression of Runx2, a transcription factor, and OCN, which regulates mineralization by inhibiting the formation of apatite crystals and promoting osteoclast function [39]. Notably, BK channel inhibition enhances OPN mRNA expression simultaneously, indicating that BK channels may be involved in the compensatory state of VSMCs induced by β-GP, where OPN may be upregulated to counteract the progression of VC [40]. This hypothesis was confirmed by the observed increase in ALP activity in paxilline-treated cells. ALP inhibits hydroxyapatite formation and deposition in the vascular wall through pyrophosphate hydrolysis [27].
In contrast, activation of the BK channels with two specific openers, NS1619 and BMS191011, alleviated VC. These findings indicate that BK channel activation may represent a potential therapeutic strategy in the clinical management of VC.
Notably, paxilline suppressed the expression of the BK channels; however, previous reports have shown that paxilline reduces BK channel activity but not its expression [28]. This phenomenon may be attributed to the β-GP-induced VC, rather than being secondary to the effect of paxilline. Therefore, paxilline inhibits the activity of BK channels, a phenomenon that in turn aggravates the β-GP-induced VC and causes a reduction in the expression of BK channels. However, the reason underlying reduced BK expression in presence of calcification warrants further research.
It had been previously demonstrated that Akt phosphorylation was reduced significantly in insulin-induced BK knockout adipocytes or in paxilline-treated WT adipocytes [41]; a similar phenomenon was observed in our study. Paxilline treatment or BK channel deficiency in VSMCs reduced the expression of phosphorylated Akt. These results suggest a correlation between BK channels and Akt, which is a downstream of calcification signaling. Moreover, we found that BK channel opener NS1619 induced the phosphorylation of Akt in the presence of β-GP. As a BK channel agonist, NS1619 has been reported to activate BK channels by increasing sarcoplasmic reticulum calcium release [42]. And calcium could affect the phosphorylation of Akt [43], a phenomenon that may be associated with the dysregulated calcium levels during calcification. Additionally, the inhibition of Akt counteracted the effects of the BK channel activator, thereby promoting osteo-/chondrogenic transdifferentiation in calcification. Thus, we hypothesize that the beneficial effect of BK channels on VC is mediated partly by Akt. Surprisingly, the inhibition of Akt was also accompanied by a decrease in the expression of the BK channels, which is consistent with a previous report showing that suppression of Akt signaling facilitates BK degradation in oxidative-stress induced diabetic vessels [44]. We speculated that the inhibition of Akt exacerbated calcification, which in turn resulted in decreased BK channel expression. However, the mechanisms by which BK channels affect Akt expression or Akt inhibition results in downregulated expression of BK channels are still unclear; further research is needed to determine the exact relationships.
Furthermore, the expression of FoxO1 was altered upon Akt activation, indicating that the Akt/FoxO1 signaling cascade may mediate the effects of BK channels on VC. Some studies have shown that Akt activation may inhibit FoxO1/3 expression, resulting in increased Runx2 expression in H2O2- or glucose-oxidase-exposed calcified VSMCs [26, 45]. However, this effect may be attributed to the different models used to study VC, suggesting that Akt signaling may be regulated in a context-specific and model-specific manner. Nonetheless, our report suggests a possible mechanism underlying the role of BK channels in calcification.
In summary, our study demonstrates a novel role of BK channels in VC. BK channel activation inhibits VC, at least in part, via the Akt/FoxO1 pathway. Strategies to activate BK channel signaling may open a new therapeutic avenue for VC.
References
Peeters MJ, van den Brand JA, van Zuilen AD, Koster Y, Bots ML, Vervloet MG, et al. Abdominal aortic calcification in patients with CKD. J Nephrol. 2017;30:109–18.
Byon CH, Chen Y. Molecular mechanisms of vascular calcification in chronic kidney disease: the link between bone and the vasculature. Curr Osteoporos Rep. 2015;13:206–15.
Disthabanchong S. Vascular calcification in chronic kidney disease: pathogenesis and clinical implication. World J Nephrol. 2012;1:43–53.
Mizobuchi M, Towler D, Slatopolsky E. Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol. 2009;20:1453–64.
Vervloet M, Cozzolino M. Vascular calcification in chronic kidney disease: different bricks in the wall? Kidney Int. 2017;91:808–17.
Voelkl J, Cejka D, Alesutan I. An overview of the mechanisms in vascular calcification during chronic kidney disease. Curr Opin Nephrol Hypertens. 2019;28:289–96.
Shroff RC, Shanahan CM. The vascular biology of calcification. Semin Dial. 2007;20:103–9.
Latorre R, Castillo K, Carrasquel-Ursulaez W, Sepulveda RV, Gonzalez-Nilo F, Gonzalez C, et al. Molecular determinants of BK channel functional diversity and functioning. Physiol Rev. 2017;97:39–87.
Dopico AM, Bukiya AN, Jaggar JH. Calcium- and voltage-gated BK channels in vascular smooth muscle. Pflug Arch. 2018;470:1271–89.
Jia X, Yang J, Song W, Li P, Wang X, Guan C, et al. Involvement of large conductance Ca2+-activated K+ channel in laminar shear stress-induced inhibition of vascular smooth muscle cell proliferation. Pflug Arch. 2013;465:221–32.
Rusch NJ. BK channels in cardiovascular disease: a complex story of channel dysregulation. Am J Physiol Heart Circ Physiol. 2009;297:H1580–2.
Zhu Y, Ye P, Chen SL, Zhang DM. Functional regulation of large conductance Ca2+-activated K+ channels in vascular diseases. Metabolism. 2018;83:75–80.
Lu T, Chai Q, Jiao G, Wang XL, Sun X, Furuseth JD, et al. Downregulation of BK channel function and protein expression in coronary arteriolar smooth muscle cells of type 2 diabetic patients. Cardiovasc Res. 2019;115:145–53.
Borbouse L, Dick GM, Asano S, Bender SB, Dincer UD, Payne GA, et al. Impaired function of coronary BK(Ca) channels in metabolic syndrome. Am J Physiol Heart Circ Physiol. 2009;297:H1629–37.
Freise C, Querfeld U. Inhibition of vascular calcification by block of intermediate conductance calcium-activated potassium channels with TRAM-34. Pharmacol Res. 2014;85:6–14.
Hei H, Gao J, Dong J, Tao J, Tian L, Pan W, et al. BK knockout by TALEN-mediated gene targeting in osteoblasts: KCNMA1 determines the proliferation and differentiation of osteoblasts. Mol Cells. 2016;39:530–5.
Wang Y, Guo Q, Hei H, Tao J, Zhou Y, Dong J, et al. BK ablation attenuates osteoblast bone formation via integrin pathway. Cell Death Dis. 2019;10:738.
Alesutan I, Feger M, Tuffaha R, Castor T, Musculus K, Buehling SS, et al. Augmentation of phosphate-induced osteo-/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc Res. 2016;110:408–18.
Kwon DH, Eom GH, Ko JH, Shin S, Joung H, Choe N, et al. MDM2 E3 ligase-mediated ubiquitination and degradation of HDAC1 in vascular calcification. Nat Commun. 2016;7:10492.
Song Y, Hou M, Li Z, Luo C, Ou JS, Yu H, et al. TLR4/NF-κB/Ceramide signaling contributes to Ox-LDL-induced calcification of human vascular smooth muscle cells. Eur J Pharmacol. 2017;794:45–51.
Lee K, Kim H, Park HS, Kim KJ, Song H, Shin HI, et al. Targeting of the osteoclastogenic RANKL-RANK axis prevents osteoporotic bone loss and soft tissue calcification in coxsackievirus B3-infected mice. J Immunol. 2013;190:1623–30.
Wang Y, Tao J, Wang M, Yang L, Ning F, Xin H, et al. Mechanism of regulation of big-conductance Ca2+-activated K+ channels by mTOR complex 2 in podocytes. Front Physiol. 2019;10:167.
Zebger-Gong H, Müller D, Diercke M, Haffner D, Hocher B, Verberckmoes S, et al. 1,25-Dihydroxyvitamin D3-induced aortic calcifications in experimental uremia: up-regulation of osteoblast markers, calcium-transporting proteins and osterix. J Hypertens. 2011;29:339–48.
Zhang S, Zhang Z, Shen Y, Zhu Y, Du K, Guo J, et al. SCN9A epileptic encephalopathy mutations display a gain-of-function phenotype and distinct sensitivity to oxcarbazepine. Neurosci Bull. 2020;36:11–24.
Chen Q, Tao J, Hei H, Li F, Wang Y, Peng W, et al. Up-regulatory effects of curcumin on large conductance Ca2+-activated K+ channels. PLoS One. 2015;10:e0144800.
Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–27.
Voelkl J, Luong TT, Tuffaha R, Musculus K, Auer T, Lian X, et al. SGK1 induces vascular smooth muscle cell calcification through NF-κB signaling. J Clin Invest. 2018;128:3024–40.
Knaus HG, McManus OB, Lee SH, Schmalhofer WA, Garcia-Calvo M, Helms LM, et al. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry. 1994;33:5819–28.
Choi TY, Lee SH, Kim SJ, Jo Y, Park CS, Choi SY. BK channel blocker paxilline attenuates thalidomide-caused synaptic and cognitive dysfunctions in mice. Sci Rep. 2018;8:17653.
Ha CM, Park S, Choi YK, Jeong JY, Oh CJ, Bae KH, et al. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vascul Pharmacol. 2014;63:29–36.
Price PA, Faus SA, Williamson MK. Warfarin-induced artery calcification is accelerated by growth and vitamin D. Arterioscler Thromb Vasc Biol. 2000;20:317–27.
Romine JL, Martin SW, Meanwell NA, Gribkoff VK, Boissard CG, Dworetzky SI, et al. 3-[(5-Chloro-2-hydroxyphenyl)methyl]-5-[4-(trifluoromethyl)phenyl]-1,3,4-oxadiazol-2(3H)-one, BMS-191011: opener of large-conductance Ca2+-activated potassium (maxi-K) channels, identification, solubility, and SAR. J Med Chem. 2007;50:528–42.
Xiang RF, Wang Y, Zhang N, Xu WB, Cao Y, Tong J, et al. MK2206 enhances the cytocidal effects of bufalin in multiple myeloma by inhibiting the AKT/mTOR pathway. Cell Death Dis. 2017;8:e2776.
Biggs WH 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–6.
Yu F, Wei R, Yang J, Liu J, Yang K, Wang H, et al. FoxO1 inhibition promotes differentiation of human embryonic stem cells into insulin producing cells. Exp Cell Res. 2018;362:227–34.
Aihara Y, Jahromi BS, Yassari R, Nikitina E, Agbaje-Williams M, Macdonald RL. Molecular profile of vascular ion channels after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:75–83.
Pabbidi MR, Roman RJ. Elevated K+ channel activity opposes vasoconstrictor response to serotonin in cerebral arteries of the Fawn Hooded Hypertensive rat. Physiol Genomics. 2017;49:27–36.
Kim N, Chung J, Kim E, Han J. Changes in the Ca2+-activated K+ channels of the coronary artery during left ventricular hypertrophy. Circ Res. 2003;93:541–7.
Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am J Physiol Ren Physiol. 2014;307:F891–900.
Kaartinen MT, Murshed M, Karsenty G, McKee MD. Osteopontin upregulation and polymerization by transglutaminase 2 in calcified arteries of Matrix Gla protein-deficient mice. J Histochem Cytochem. 2007;55:375–86.
Ren J, Cheng Y, Wen X, Liu P, Zhao F, Xin F, et al. BK(Ca) channel participates in insulin-induced lipid deposition in adipocytes by increasing intracellular calcium. J Cell Physiol. 2021;236:5818–31.
Bentzen BH, Osadchii O, Jespersen T, Hansen RS, Olesen SP, Grunnet M. Activation of big conductance Ca2+-activated K+ channels (BK) protects the heart against ischemia-reperfusion injury. Pflug Arch. 2009;457:979–88.
Wang Y, Ali Y, Lim CY, Hong W, Pang ZP, Han W. Insulin-stimulated leptin secretion requires calcium and PI3K/Akt activation. Biochem J. 2014;458:491–8.
Lu T, Chai Q, Yu L, d’Uscio LV, Katusic ZS, He T, et al. Reactive oxygen species signaling facilitates FOXO-3a/FBXO-dependent vascular BK channel β1 subunit degradation in diabetic mice. Diabetes. 2012;61:1860–8.
Deng L, Huang L, Sun Y, Heath JM, Wu H, Chen Y. Inhibition of FOXO1/3 promotes vascular calcification. Arterioscler Thromb Vasc Biol. 2015;35:175–83.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81773801 and No. 81973385 to XMZ; No. 82074162 to JT); Shanghai Science and Technology Innovation (No. 20410713300 to XMZ); and the Experimental Animal Project of Shanghai Science and Technology Commission (No. 201409004700).
Author information
Authors and Affiliations
Contributions
XMZ and HX designed the study; FLN, JT, DDL, LLT, and MLW performed the experiments; FLN, JT, and LLT performed data analysis; FLN and JT wrote the manuscript. SR, CL, and HC revised the manuscript. All authors read and approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Ning, Fl., Tao, J., Li, Dd. et al. Activating BK channels ameliorates vascular smooth muscle calcification through Akt signaling. Acta Pharmacol Sin 43, 624–633 (2022). https://doi.org/10.1038/s41401-021-00704-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00704-6
