
Abstract
Myelin damage and abnormal remyelination processes lead to central nervous system dysfunction. Glial activation-induced microenvironment changes are characteristic features of the diseases with myelin abnormalities. We previously showed that ginsenoside Rg1, a main component of ginseng, ameliorated MPTP-mediated myelin damage in mice, but the underlying mechanisms are unclear. In this study we investigated the effects of Rg1 and mechanisms in cuprizone (CPZ)-induced demyelination mouse model. Mice were treated with CPZ solution (300 mg· kg−1· d−1, ig) for 5 weeks; from week 2, the mice received Rg1 (5, 10, and 20 mg· kg−1· d−1, ig) for 4 weeks. We showed that Rg1 administration dose-dependently alleviated bradykinesia and improved CPZ-disrupted motor coordination ability in CPZ-treated mice. Furthermore, Rg1 administration significantly decreased demyelination and axonal injury in pathological assays. We further revealed that the neuroprotective effects of Rg1 were associated with inhibiting CXCL10-mediated modulation of glial response, which was mediated by NF-κB nuclear translocation and CXCL10 promoter activation. In microglial cell line BV-2, we demonstrated that the effects of Rg1 on pro-inflammatory and migratory phenotypes of microglia were related to CXCL10, while Rg1-induced phagocytosis of microglia was not directly related to CXCL10. In CPZ-induced demyelination mouse model, injection of AAV-CXCL10 shRNA into mouse lateral ventricles 3 weeks prior CPZ treatment occluded the beneficial effects of Rg1 administration in behavioral and pathological assays. In conclusion, CXCL10 mediates the protective role of Rg1 in CPZ-induced demyelination mouse model. This study provides new insight into potential disease-modifying therapies for myelin abnormalities.
Similar content being viewed by others


Introduction
Demyelination causes serious damage to the central nervous system (CNS), leading to a broad spectrum of diseases, such as multiple sclerosis (MS) [1] and acute disseminated encephalomyelitis (ADEM) [2]. Decreased oligodendrocytes (OLs) and myelin loss have also been found in the pathogenesis and progression of neurodegenerative diseases, such as Alzheimer’s disease (AD) [3] and Parkinson’s disease (PD) [4], which aggravate the degeneration and impede the recovery of damaged nerves [5]. Therefore, finding strategies that can effectively reduce myelin loss and promote myelin regeneration is important for treatment [6].
Activation and proliferation of glial cells are often accompanied by myelin loss [7]. The proliferation of astrocytes and microglia, as well as the release of pro-inflammatory cytokines and chemokines, mediate OLs apoptosis and myelin vacuolization and further cause damage to the nervous system [8]. However, the interaction between glial cells also participates in the process of regeneration [9]. Both astrocytes and microglia can secrete IL-10, BDNF and other regulatory factors to promote the repair of the myelin sheath [10, 11]. Microglia have a phagocytic effect on myelin sheath fragments in the demyelinated region, which also provides a favorable environment for remyelination [12]. Moreover, astrocytes recruit microglia and oligodendrocyte precursor cells (OPCs) for myelin fragmentation phagocytosis and promote the maturation of new OLs after injury. The interaction between these glial cells constitutes a microenvironment for myelin repair [13]. However, in many diseases, myelin regeneration cannot proceed successfully due to an inappropriate glial microenvironment. Therefore, studying the regulation of glial cells in the CNS is one of the important prerequisites to reduce the loss of the myelin sheath and promote myelin regeneration [13]. Clarner et al. found that oligodendrocyte apoptosis paralleled by early microglial activation is mediated in part by the chemokine CXCL10 [14]. In preclinical studies, CXCL10 was reported to be involved in MS pathology. CXCL10 and its receptor CXCR3 increase in the cerebrospinal fluid (CSF) during acute relapse in MS patients [15,16,17]. Histopathological research suggests that CXCL10 is mainly associated with astrocytes around inflammatory lesions. During the early oligodendrocyte injury period, CXCL10 displays increased expression after 2 days of cuprizone treatment, and induces a detrimental phenotype in microglia, which mediates the complex intercellular communication of astrocytes, microglia, and potentially OLs [14].
Ginsenoside Rg1, one of the most active components of ginseng, has been reported to be protective against many CNS neurodegenerative diseases. In our previous studies, we found that Rg1 could ameliorate MPTP-mediated myelin damage [4]. However, the mechanism by which Rg1 attenuates demyelination is not fully understood. In addition, Rg1 can also attenuate concanavalin A-induced hepatitis in mice by inhibiting CXCL10, thereby reducing the inflammatory response in the peripheral system [18]. Thus, we suspect that CXCL10 is associated with the role of Rg1 in demyelination regulation in the CNS.
The toxic demyelination model induced by cuprizone (CPZ) is commonly used to study the process of myelin loss and regeneration [19]. Since this model does not destroy the blood–brain barrier, peripheral immune cells are not involved in the process of myelin demyelination [20]. Therefore, the CPZ model is an appropriate method to study the effects of Rg1 regulation on CNS glial cell interactions and the improvement of myelin loss. In this study, we found that Rg1 treatment significantly alleviated the behavior and pathological damage caused by CPZ, which was associated with the regulation of CXCL10, and thus the glia response and the associated microenvironment.
Materials and methods
Animals and reagents
Male C57BL/6 N mice (7 weeks old, 21.5 ± 1.5 g) were purchased from SPF Biotechnology Co., Ltd (Beijing, China). All mice were raised in an environment with a 12:12 h light/dark cycle under constant temperature and humidity. All animal care and experimental procedures were complied with the principles outlined in the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Peking Union Medical College and Chinese Academy of Medical Sciences. Animal studies reported are in compliance with the 3Rs concept.
Ginsenoside Rg1 (Fig. 1a) was purchased from Yuanye Corporation (Shanghai, China). CPZ and lipopolysaccharide (LPS) were purchased from Sigma Corporation (Sigma-Aldrich, St Louis, MO, USA). CXCL10 recombinant protein was purchased from PeproTech Corporation (USA). CPZ was administered by intragastric gavage (300 mg/kg, dissolved in 0.5% CMC-Na at a concentration of 30 mg/mL) [21,22,23,24]. Ginsenoside Rg1 was diluted to concentration of 0.5 mg/mL (5 mg/kg), 1 mg/mL (10 mg/kg), and 2 mg/mL (20 mg/kg) by the cuprizone solution, which was administered by intragastric gavage at the same time as CPZ.

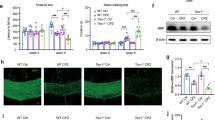
a The chemical structure of ginsenoside Rg1. b Animal experimental group and procedures. c Weight changes in the experiment. d Pole test in week 5. The time taken for each group to climb down the pole was tested. e Rotarod test. The latency time of each group to stay on the rotarod was recorded. Data were presented as mean ± SD, n = 7. #P < 0.05; ##P < 0.01; ###P < 0.001 vs. the control group; *P < 0.05; **P < 0.01; ***P < 0.001 vs. the model group.
The primary antibodies used in this research were as follows, anti-APP (AB32136, Abcam, 1:500), anti-Olig-2 (AB9610, Millipore,1:200), anti-GFAP (Z0334, Dako, 1:250), anti-Iba-1 (019-19741, Wako,1:500), anti-CXCL10 (sc-374092, Santa,1:100), anti-TNF-α (ab199013, Abcam,1:500 Cambridge, UK), anti-IL-1β (PA5-46956, Thermo Invitrogen, 1:500), anti-iNOS (D6B6S, CST, 1:500), anti-BDNF (sc-546, Santa Cruz, 1:200), anti-IL-10 (sc-365858, Santa Cruz, 1:200). The horseradish peroxidase (HRP)-conjugated secondary antibodies (KPL,1:200) were used in Western blot. Alexa FluorTM 488-conjugated donkey anti-rabbit IgG (A21206, Invitrogen, 1:500), Alexa FluorTM 546-conjugated donkey anti-rabbit IgG (A10036, Invitrogen, 1:500) were used along with Hoechst 33342 solution (H342, Dojindo,1:1000) in the fluorescent staining.
Animal experiments
The first set of animal experiments
The experiment was performed according to the procedures shown in Fig. 1b. The mice were randomly divided into five groups 7 days after acclimatization (n = 7/group). The mice in the control group were treated with 0.5% CMC-Na by intragastric gavage for 5 weeks. The mice in the CPZ group were administered with CPZ solution (300 mg/kg) by gavage administration for 5 weeks to make demyelination model. The mice of CPZ + Rg1 groups were pretreated with CPZ solution for 1 week, then treated with Rg1 by gavage for 4 weeks (5, 10, and 20 mg/kg). Intragastric administration was performed once daily for all groups of mice. Body weight was measured every 3 days. Behavioral tests were performed during the week of administration, including pole test and rotarod test. All mice were sacrificed at the end of 5 weeks of CPZ administration.
The second set of animal experiments
The experiment was performed according to the procedures. The mice were randomly divided into the following four groups: AAV-NC shRNA + vehicle, AAV-NC shRNA + CPZ, AAV-CXCL10 shRNA + CPZ, and AAV-CXCL10 shRNA + CPZ + Rg1 (n = 6/group). Three weeks after the administration of AAV in the lateral ventricles, CPZ was given to the associated groups, while the control group was treated with the vehicle. Rg1 was treated 1 week after the initiation of CPZ. Pole test and rotarod test were performed in week 5. All mice were sacrificed at the end of 5 weeks of CPZ administration.
Neurological behavioral tests
Pole test
The pole test was used to detect the dyskinesia of animals in the CPZ model group as described in our previous work [25]. Briefly, a vertical wooden stick with a rough surface of 50 cm in length and 1 cm in diameter was prepared, and a wooden ball with a diameter of 2.5 cm was installed with the same rough surface at the top of the stick to assist positioning the animals on the top. The wooden stick was attached to a home cage and the bottom of the cage was covered with bedding. The time taken for the animal to climb down the pole was measured. Each animal was tested three times at 30 min intervals. The mean value of three trials obtained from each animal was used for statistical analysis.
Rotarod test
The rotarod test was performed to assess motor coordination and balance as previously described [26]. The detection was addressed by using a rotarod equipment (IITC Life Science, CA, USA). The animals were first trained on the rolling rotarod with a constant speed of 5 rpm for 300 s, and then tested for their latency time on the rolling under a linear accelerating speed from 5 to 30 rpm in 300 s. Each animal was tested three times with an interval time of at least 30 min. The average of three trials was calculated and used for statistical analysis.
Tissue preparation
All animals were anesthetized by intraperitoneal injection of 1% sodium pentobarbital at a dose of 50 mg/kg bw. Brains were separated, and the corpus callosum (CC) was obtained, quickly frozen in liquid nitrogen and stored at −80 °C for biochemical analysis. For histological analysis, mice were anesthetized and perfused with phosphate-buffered saline (PBS), and then perfused with paraformaldehyde. Brains were dissected, postfixated overnight, and then embedded in paraffin. Coronal sections of 5 μm were cut for pathological testing of Luxol fast blue (LFB) staining or immunohistochemistry (IHC).
Luxol fast blue (LFB) staining
Myelin structures in the tissue sections were stained with LFB (Servicebio, China). Paraffin sections were dewaxed, washed with water and treated with LFB solution at 60 °C for 4 h. The slices were then cooled for 15 min, washed with water, soaked in 70% ethanol for 1 min, and washed with running water for 1 min. The sections were immersed in lithium carbonate solution for 15 s for color development and then stopped by running water for 1 min. This distinguishable process was repeated four times until the blue color faded in the unmyelinated area. Finally, the sections were dehydrated and sealed with neutral balsam. Images were taken with a light microscope (Olympus, Japan)
Immunohistochemistry (IHC) and immunofluorescence (IF)
Paraffin sections from the coronal of the CC were selected, and then dewaxed and cleared in dimethylbenzene and different concentrations of ethanol for rehydration before washing with water. Afterwards, sections were boiled in citrate acid buffer solution for antigen retrieval. After washing with PBS, the paraffin sections were treated with Triton X-100, and then blocked with 5% BSA for 40 min at room temperature. The sections were incubated with the primary antibodies overnight at 4 °C. For IHC, the paraffin sections were soaked with PBST three times, and incubated for 2 h with HRP-conjugated secondary antibodies. The sections were washed three times with PBST, covered with 3, 3′-diamino-benzidine (DAB) solution, and stopped when a color change was observed. Finally, the sections were dehydrated and sealed with neutral balsam. Images were taken with a light microscope (Olympus, Japan). For IF, the paraffin sections were soaked three times with PBST and incubated for 2 h with Alexa Fluor conjugated secondary antibodies. The sections were washed three times with PBST, sealed with glycerol, and observed by a laser scanning confocal microscope (TCSSP2, Leica).
Western blot
Western blot analysis was performed as described [27]. Briefly, tissue (0.1 mL lysis buffer/10 mg protein) or cells (0.1 mL lysis buffer/106 cells) were lysed in cold RIPA lysates (C1053, Applygen) with protease inhibitors. The concentrations of extracted protein were quantified with a BCA kit (P1511, Applygen). The protein samples (30 μg protein/sample) were electrophoretically separated on SDS-PAGE gels at 10% or 12% concentration and transferred onto PVDF membranes by wet electroblotting. The membrane was blocked with 5% BSA solution for 2 h, and then incubated overnight at 4 °C with the primary antibody. After washing with TBST three times (5 min per wash), the membrane was incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature, washed and detected with Super Enhanced Chemiluminescence Detection Reagent (P1050, Applygen). The image was processed with an Enhanced Chemiluminescence Plus detection system (Image Quant Las4000, GE Healthcare).
Quantitative PCR analysis
Total RNA was isolated (0.1 mL TRIzol/10 mg tissue) using TRIzol reagent (15596018, Invitrogen, USA). The cDNA was reversed using a One-Step gDNA Removal and cDNA Synthesis SuperMix Kit from 1 μg total RNA (AT311-03, TransGen, China) according to the manufacturer’s instructions. Primers used for qPCR are as follows. A TransStart Tip Green qPCR Supermix Kit (AQ601-02, TransGen, China) was used to amplify and check with a LineGene 9600 Quantitative PCR System (FQD-96A, Bioer, China). The mRNA levels were analyzed and normalized with comparative CT (2−△△CT) methods. The primers were: CXCL10-Forward: 5′-CTGAGTCCTCGCTCAAGTGG-3′, CXCL10-Reverse: 5′-GTCGCACCTCCACATAGCTT-3′; GAPDH-Forward: 5′-GGAGAGTGTTTCCTCGTCCC-3′, GAPDH-Reverse: 5′-ATCCGTTCACACCGACCTTC-3′.
Cell culture and treatment
The CTX-TNA2 murine astrocyte cell line was obtained from ATCC. Cells were cultured with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). In the experiment, CTX cells were plated in 6-well plates (2 × 105 cells/well). The culture medium was changed to DMEM without FBS for 8 h, and then divided into the following three groups: the control, LPS and LPS + Rg1 groups. The LPS group was treated with DMEM containing 1 μg/mL LPS for 24 h. The Rg1 group was treated with DMEM containing LPS plus 40 μM Rg1 for 24 h, and the culture medium of all groups was replaced with DMEM after 6 h. Then, the supernatant and protein of each well were extracted for use.
The BV-2 murine microglial cell line was purchased from the Cell Resource Center of Peking Union Medical College, and cultured in DMEM/F12 plus 10% FBS. For the CXCL10 treatment experiment, BV-2 cells were plated into 6-well plates (1 × 106 cells/well), and then divided into three groups. The control group was treated with 1 mL DMEM/F12 without 10% FBS for 24 h, and the CXCL10 group was treated with DMEM containing CXCL10 recombinant protein (125 ng/mL) for 24 h, and the Rg1 + CXCL10 group was treated with DMEM containing CXCL10 recombinant protein plus 40 μM Rg1 for 24 h. For the CTX cell culture supernatant experiment, BV-2 cells were divided into three groups. The untreated CTX cell culture supernatant was added to the control CTX supernatant group. The LPS-treated CTX cells supernatant was added to the LPS-treated CTX supernatant BV-2 group, and the supernatant of Rg1 + LPS-treated CTX cells was added to the Rg1 + LPS CTX supernatant BV-2 group. Twenty-four hours later, the protein of each well was extracted to check its expression.
Primary microglial culture was performed as previously described. Brain tissue (excluding the cerebellum and meninges) of C57BL/6 mouse pups (P0–P2) was mashed, strained, and centrifuged (300 × g, 10 min) in cold MEM. The pellet was resuspended in 20 mL MEM supplemented with 10% FBS and 1% penicillin–streptomycin, and then seeded in 75 cm2 flasks precoated with poly-D-lysine (20 mg/mL). Cells were incubated at 37 °C with 5% CO2, and after 48 h, the medium was changed to remove cellular debris and nonadherent cells. After 10–14 days, microglia were harvested by shaking the flasks (5 h, 65 rpm) on an orbital shaker in an incubator (37 °C, 5% CO2). The supernatant containing microglia was collected, centrifuged, resuspended in fresh MEM, and then seeded in 6-well plates containing BV-2 cells for a CXCL10-treated experiment.
Cell migration
The migration ability of BV-2 cells was detected with a 6.5 mm Transwell chamber with an 8.0 μm crater as described previously [28]. Then, 600 μL CXCL10 recombinant protein solution (50 ng/mL) was added to the lower chambers. BV-2 cells were transferred into the top chamber (4 × 103 cells/chamber) and allowed to migrate at 37 °C in a 5% CO2 atmosphere. To evaluate the intervention effect of Rg1, 40 μM Rg1 was added to the CXCL10 protein in the lower chambers. After 48 h, the upper chamber was immersed in methanol for 30 min to immobilize the cells. The fixed cells in the upper chamber were stained with crystal violet. Cells on the upper surface were removed with a cotton swab. The upper chamber was immersed in PBS, and the number of cells that migrated to the lower surface was counted under a light microscope (Eclipse Ti-U, Nikon).
Phagocytosis assay with fluorescence beads
Green fluorescence Zymosan A BioParticles (#Z-23373; Thermo Fisher Scientific) were used to assay the phagocytosis of BV-2 cells. At the end of the administration, BV-2 cells were plated in a 96-well plate (1 × 104 cells/well), and exposed to fluorescence microsphere solution (104 particles/mL) for 30 min. After removing the bead solution and washing with PBS for three times, 100 μL DMEM/F12 medium was added to each well. Live cell imaging was carried out using a fluorescence microscope, and images were obtained every 5 min using a 488 nm laser.
Image analysis
The integrated optical density of LFB staining and the number of Olig-2 cells, cells migrating to the lower layer in Transwell assay and cells engulfing fluorescent bioparticles were evaluated in full microscopic fields of CC by ImagePro Plus (Media Cybernetics, USA). The cell fluorescence of GFAP or Iba-1 positive signals were measured by Image J by calculating the corrected total cell fluorescence (CTCF). CTCF = integrated density − (area of selected cell × mean fluorescence of background readings). The particle mean is the average CTCF value of the control group. The value of the other group was normalized to the average value of the control group. The observed-field was chosen randomly.
Oligonucleotide synthesis and vector construction
The CXCL10 promoter (−2000 to +100) and CXCL10 promoter mutant were synthesized and cloned into the pGL3-basic dual-luciferase vector. The above vectors were transfected with Lipofectamine 2000 according to the manufacturer’s instructions.
The CXCL10 shRNA AAV vector was constructed and packaged by Obio Technology Corp., Ltd. (Shanghai, China). The RNA interference sequence for mouse CXCL10 (5′-TAGATTCCGGATTCAGACATC-3′) [29] and the negative control (5′-TTCTCCGAACGTGTCAGACGT-3′) were constructed into the AAV vector and packaged as pAKD-CMV-bGlobin-mCherry-H1-shRNA (CXCL10) (AAV-CXCL10 shRNA, AAV2/9, titer 4.5 × 1012), and pAKD-CMV-bGlobin-mCherry-H1-shRNA (AAV-NC shRNA, AAV2/9, titer 4.5 × 1012).
Dual-luciferase reporter assay
Cells were seeded in 96-well plates and incubated as described. Twenty-four hours later, cells were transfected with constructs as indicated. Six hours after transfection, the treatment was performed as described. Twenty-four hours after transfection, cells were lysed with passive lysis buffer for 20 min. Firefly and Renilla luciferase activities were determined in lysates using a Dual-Luciferase Assay System (Promega, USA) in GloMax®-96 GloMax luminometer (Promega, USA).
Stereotaxic injection of AAV
Animals were anaesthetized with 1.5% isoflurane mixed with medical air (20% oxygen, 80% nitrogen) at a flow rate of 0.8 L/min and placed in a stereotaxic apparatus fixed with a mouse adaptor. AAV-CXCL10 shRNA or AAV-NC shRNA was injected into the lateral ventricles at a flow rate of 0.1 μL/min with a final volume of 0.5 μL. The needle was left in place for 5 min after the injection. The stereotaxic coordinates for the lateral ventricle were as follows: 0.14 mm from bregma, 0.75 or −0.75 mm to the midline and 2.25 mm below the subdural matter. The animals were allowed to recover for 3 weeks after injection, before treatment with CPZ or vehicle.
Electron microscopy (EM)
EM was performed as previously described [30]. Briefly, anesthetized animals were perfused. Brains were trimmed and postfixed with 2.5% glutaraldehyde for 2 h, washed with 0.1 M PBS, and then exposed to 1% osmium tetraoxide for 2 h. After several subsequent washes with water, the tissues were dehydrated with gradient alcohol and embedded in Epon resin. Randomly selected ultrathin sections were stained with uranyl acetate and lead citrate and examined using a transmission electron microscope (H-7650, HITACHI, Tokyo, Japan). The G-ratio of the neuronal myelinated fibers in the CC was quantified using Image J software implemented with a plug-in (http://gratio.efil.de/) to allow the semiautomated analysis of randomly selected sets of axons [31, 32].
Statistical analysis
The researchers assessing the assays were blinded to the respective treatment groups. All data are presented as the mean ± SD. The data were statistically analyzed using one-way ANOVA followed by Dunnett’s test or Newman-Keuls test for multiple comparisons in different groups or Student’s t test for comparisons of two groups with GraphPad Prism 7.0 software (GraphPad, USA). P < 0.05 was regarded as statistically significant.
Results
Rg1 ameliorates impaired behavior caused by CPZ
CPZ treatment for 5 weeks caused demyelination. To determine the role of Rg1 in this model, Rg1 intervention started at the second week (Fig. 1b). CPZ treatment caused a significant decrease in body weight in the first 13 days followed by an increase. Compared with the model group, Rg1 treatment alleviated body weight loss and increased the rate of weight gain, especially in the 20 mg/kg group, which was significant on day 22 (Fig. 1c). The pole test and the rotarod test were employed to check the improvement of behavior disorders with Rg1. In the pole test, the CPZ-treated mice took more time to climb down the pole than the control mice. Bradykinesia was markedly ameliorated in the 20 mg/kg Rg1 group (Fig. 1d). Motor coordination ability is shown in Fig. 1e. With Rg1 treatment, the latency time to fall off the rotarod was increased. The results above indicated that Rg1 could significantly reduce the motor and coordination impairment caused by CPZ administration.
Rg1 reduces demyelination and axonal injury caused by CPZ
Demyelination was evaluated using LFB staining, which revealed a large area of myelin loss in the CC after CPZ administration, while Rg1 intervention reduced the degree of demyelination (Fig. 2a, b). The myelin sheath is formed by OLs. Demyelination causes the loss of OLs. With immunostaining for Olig-2, a marker of OLs, we found that Rg1 significantly alleviated the CPZ-induced loss of Olig-2+ cells (Fig. 2c, d). The structural and functional integrities of the myelin sheath are essential for maintaining the normal transmission of nerve signals. Demyelination can lead to abnormal axonal function, and eventually damage nerve fiber structures. We tested the IHC staining of amyloid precursor protein (APP), and the APP+ bulbs (arrows) represent axonal damage induced by cuprizone. We found that axonal damage was evident in the CC of the model group, and was almost absent in the Rg1-treated group, especially in the 10 and 20 mg/kg treated groups (Fig. 2e).

a Representative images of LFB staining of each group in corpus callosum. Upper scale bar = 500 μm, lower scale bar = 50 μm. b A histogram of quantitative data of LFB positive area in CC. c Representative IHC images of Olig-2+ cells of each group. d Histogram of quantitative data of the number of Olig-2+ cells. Upper scale bar = 200 μm, lower scale bar = 50 μm. e Representative IHC images of APP+ nerve fibers of each group in the corpus callosum. Scale bar = 50 μm. Data are presented as mean ± SD, n = 3. #P < 0.05; ##P < 0.01 vs. the control group; *P < 0.05; **P < 0.01 vs. the model group.
Rg1 modulates the glial response induced in the CPZ model
OL damage and myelin loss are accompanied by activation of microglia and astrocytes days after CPZ treatment [33]. To investigate the effect of Rg1 on glial cell activation induced by CPZ, we used IF to label astrocytes and microglia in CC with the marker proteins GFAP and Iba-1, respectively. Compared with the control group, the CPZ group had significantly increased GFAP-positive astrocytes in the CC of mice, and the cell body was enlarged. The number of astrocytes decreased significantly with Rg1 intervention (Fig. 3a, b). Similarly, Rg1 intervention significantly reversed the increase in the number of Iba-1-positive microglia induced by CPZ administration (Fig. 3c, d). We also tested other pro-inflammatory factors and anti-inflammatory cytokines. The expression of the pro-inflammatory factors iNOS, TNF-α, and IL-1β was compared. We found that Rg1 had no obvious effect on iNOS (Fig. 3e, f), while the inhibitory effect on TNF-α and IL-1β was significant (Fig. 3g, h). BDNF was also investigated in our experiment. There was no apparent effect with the CPZ treatment. Rg1 had a partial tendency to enhance BDNF, although there was no significant statistic difference (Fig. 3i). IL-10, a marker of M2 microglial cells, was downregulated in the CPZ model, which was reversed by Rg1 treatment (Fig. 3j). These results suggest that Rg1 intervention modulated the levels of inflammatory factors and the glial microenvironment in the CPZ model.

a Representative immunofluorescent images of GFAP+ cells in the CC of the CPZ model. Scale bar = 20 μm. b Histogram of quantitative data of GFAP+ particles. c Representative immunofluorescent images of Iba-1+ cells of each group in the CC of the CPZ model. Scale bar = 20 μm. d Histogram of quantitative data of Iba-1+ particles. e Representative protein bands of inflammatory and regulatory cytokines in CC. f–h Histogram of quantitative data of the protein expression levels of the inflammatory factors, iNOS, TNF-α, and IL-1β. i, j Histogram of quantitative data of BDNF and IL-10. Data are presented as mean ± SD, n = 3. #P < 0.05; ##P < 0.01; ###P < 0.001 vs. the control group; *P < 0.05; **P < 0.01; ***P < 0.001 vs. the model group.
Rg1 inhibits the CXCL10 level in the CPZ model
Since Rg1 has a positive effect on the glial environment, we tested its role in CXCL10, an effective mediator of OLs, microglia and the glial environment. We found that the mRNA and protein levels of CXCL10 were both upregulated in the CPZ group, and downregulated upon Rg1 treatment (Fig. 4a–c). Further, we verified this result with IF staining of CXCL10 and Iba-1 to determine the interaction between CXCL10 and microglia. As shown in Fig. 4d, in the model group and the Rg1 low dose group, more CXCL10+ particles were colocalized with microglia. CXCL10 was obviously lower in the 10 and 20 mg/kg Rg1-treated groups, and thus, there was an interaction with microglia.

a The transcriptional level of CXCL10 was tested using qPCR in CC. b CXCL10 protein level was tested using Western blot. c Quantitative data of the Western blot of CXCL10. d Immunofluorescence images of the CXCL10+ particles of each group in CC. e Quantitative data of CXCL10+ particles. f Quantitative data of the number of CXCL10+ and Iba-1+ cells in CC. Data are presented as mean ± SD, n = 3. #P < 0.05; ##P < 0.01; ###P < 0.001 vs. the control group; *P < 0.05; **P < 0.01; ***P < 0.001 vs. the model group. Scale bar = 100 μm.
Rg1 modulates NF-κB activation and the transcription of CXCL10
As Rg1 inhibited the upregulation of CXCL10 in the CPZ model, we aimed to explore the molecular mechanism by which Rg1 regulates CXCL10, and the promoter region of CXCL10 was analyzed with JASPAR. NF-κB binding sites were found in the promoter region. Whether Rg1 could regulate the nuclear translocation of NF-κB in the CPZ model is unknown. As shown in Fig. 5a, with Rg1 treatment, nuclear NF-κB was obviously downregulated compared with that in the CPZ group (Fig. 5a, b). To further elucidate the regulation of the CXCL10 promoter, we constructed luciferase vectors containing the CXCL10 promoter (Luc-CXCL10 Pro) or the NF-κB binding site mutant CXCL10 promoter (Luc-CXCL10 Pro-Mut) (Fig. 5c). LPS treatment significantly enhanced the luciferase activity, which was reduced with Rg1 treatment (Fig. 5d). In the Luc-CXCL10 Pro-Mut group, Rg1 treatment and LPS treatment were not obviously different (Fig. 5e), confirming that Rg1 modulated CXCL10 transcription by regulating NF-κB.

a, b Representative blots and analysis of the nuclear/cytoplasm NF-κB ratio in CC. c Sequence logo of NF-κB binding sites from JASPAR. Schematic model of NF-κB binding sites in the CXCL10 promoter region and schematic of Luc-CXCL10 Pro and Luc-CXCL10 Pro-Mut vectors. d, e Luciferase activity in CTX cells treated with LPS or LPS + Rg1. Data are presented as mean ± SD, n = 3. ###P < 0.001 vs. the control group; *P < 0.05; ***P < 0.001 vs. the model group (CPZ group in (b), LPS group in (d, e).
Rg1 reverses the changes in microglia induced by CXCL10
The expression level of CXCL10 was upregulated as described above. We also wanted to check the pure role of Rg1 in the effect of CXCL10. Thus, we used recombinant CXCL10 protein (125 ng/mL) to treat primary microglial cells and the microglial cell line BV-2, and detected changes in cytokines. Interestingly, we found the addition of CXCL10 induced the expression of the pro-inflammatory cytokines TNF-α and IL-1β in microglial cells, which were also downregulated by Rg1 (40 μM) (Fig. 6a–f). This result suggests that in addition to the transcriptional regulation of CXCL10, Rg1 can also affect the proinflammation effect purely caused by CXCL10. Since the results showed the same tendency in primary cells (Fig. 6a–c) and BV-2 cells (Fig. 6d–f), we employed BV-2 cells to perform the later in vitro experiments. Through Transwell experiment, we found that CXCL10 in the lower chamber significantly promoted the downward migration of BV-2 cells from the upper chamber. However, when Rg1 was added to the lower chamber with CXCL10, the migration of BV-2 cells to the lower chamber was significantly reduced. Thus, Rg1 also inhibited the CXCL10 induced migration of BV-2 cells (Fig. 6g, h). Next, we assessed the effect of CXCL10 administration and Rg1 intervention on the phagocytic function of BV-2 cells by measuring the number of fluorescent bioparticles per unit time taken by BV-2 cells. The experimental results showed that the phagocytosis of fluorescence microparticles by microglia was not significantly different among the three groups, which indicates that CXCL10 has no effect on phagocytosis (Fig. 6i, j).

a–c Representative protein bands and quantitative analysis of the pro-inflammatory factors TNF-α and IL-1β in primary microglial cells after the stimulation with CXCL10 (125 ng/mL) with or without Rg1 (40 μM). d–f Representative protein bands and quantitative analysis of the pro-inflammatory factors TNF-α and IL-1β in BV-2 cells after the stimulation with CXCL10 (125 ng/mL) with or without Rg1 (40 μM). g, h Transwell assay of BV-2 cells treated with CXCL10 with or without Rg1. Scale bar = 50 μm. Quantitative analysis of the number of BV-2 cells migrating to the lower layer. i, j Phagocytic function test and quantitative analysis of each group. Scale bar = 20 μm. Data are presented as mean ± SD, n = 3. #P < 0.05; ##P < 0.01; ###P < 0.001 vs. the control group; *P < 0.05; ***P < 0.001 vs. the CXCL10 group.
Rg1 improves the phagocytosis of microglia without the role of CXCL10
To determine the effect of Rg1 on glial microenvironment, we used LPS to stimulate astroglia cell line CTX cells with or without Rg1 treatment. The supernatant was collected to treat BV-2 cells. CXCL10 was upregulated in LPS-treated CTX cells (Fig. 7a, b). Treatment with the CTX supernatant upregulated the pro-inflammatory cytokines TNF-α and IL-1β in BV-2 cells. Rg1 inhibited the pro-inflammatory phenotype of BV-2 cells (Fig. 7c–e). However, the phagocytosis phenotypes were not the same as those of CXCL10-treated cells, as shown in Fig. 6i. Compared with the untreated CTX supernatant group, LPS-treated CTX supernatant interfered with the phagocytic function. The presence of phagocytic BV-2 cells was observed with the LPS-treated CTX supernatant, which could phagocytize more fluorescence microspheres and faster than the control group. Rg1 further improved the phagocytic ability of BV-2 cells (Fig. 7f), which suggests that the phagocytic effect of Rg1 on microglia was not related to CXCL10.

a, b The expression of CXCL10 was tested in LPS (1 μg/mL) treated CTX cells with or without Rg1 (40 μM). c–e The expression levels of pro-inflammatory cytokines TNF-α and IL-1β in CTX supernatant treated BV-2 cells. Data are presented as mean ± SD, n = 3. #P < 0.05; ##P < 0.01 vs. the control group; *P < 0.05; **P < 0.01 vs. the model group. f Time-lapse results of fluorescence microsphere phagocytosis of BV-2 cells with CTX supernatant treatment. Phagocytosis of BV-2 cells in the control CTX supernatant group, in the LPS treatment CTX supernatant group and in the LPS and Rg1-treated CTX supernatant. Scale bar = 20 μm.
CXCL10 inhibition abolishes the protective role of Rg1 on demyelization
To further check the relationship between Rg1 and CXCL10, we first constructed and packaged the AAV-CXCL10 shRNA and the NC shRNA AAV virus, which was injected into the lateral ventricles for 3 weeks to check the inhibitory effect of CXCL10. As shown in Fig. 8a, AAV-CXCL10 shRNA abolished almost 80% of CXCL10 expression. Further, we injected AAV before CPZ administration. One week after the initiation of CPZ administration, 20 mg/kg Rg1 was administered once a day for 4 weeks. The pole test and rotarod test suggested that the motor coordination ability of the CXCL10 inhibition group (AAV-CXCL10 shRNA + CPZ) was improved compared with that of the AAV-NC shRNA + CPZ group (***P < 0.001 in Fig. 8b, c, **P < 0.01 in Fig. 8d). However, treatment with or without Rg1 was not significantly different than the AAV-CXCL10 shRNA + CPZ group, which indicates that the protective role of Rg1 mostly relies on the downregulation of CXCL10. We further employed EM to verify the ultrastructure of myelin in each group. The structures of myelin in the control group were complete, clear and well organized. With CPZ treatment, these structures were destroyed, where the myelin sheath did not tightly enwrap axons but disorganized, vacuolated and contained many electron dense deposits (Fig. 8f). The myelin status was enhanced in the CXCL10 inhibition groups regardless of Rg1 administration. The microstructure associated with g-ratio analysis and LFB staining results confirm that CXCL10 inhibition abolished the protective role of Rg1 in demyelization (Fig. 8f–i).

a The inhibitory effect of CXCL10 with AAV-CXCL10 shRNA in vivo (Student’s t test, n = 3). b Experimental procedures. c Pole test in the fifth week of CPZ treatment. The time taken for each group to climb down the pole was tested. d The rotarod test. The latency time of each group to stay on the rotarod was recorded. Data are presented as mean ± SD, n = 6. e qPCR was used to test the expression of CXCL10 in CC (n = 3). f EM test of the ultrastructure of myelin in each group (scale bar = 2 μm). The details circled by red dotted lines were amplified as follows (scale bar = 500 nm). g G-ratio quantification in CC. h, i LFB staining of CC. ##P < 0.01; ###P < 0.001 vs. AAV-NC shRNA+vehicle group; **P < 0.01; ***P < 0.001 vs. AAV-NC shRNA + CPZ group.
Discussion
Demyelination and the accompanying microenvironmental changes aggravate degeneration and impede the recovery of damaged nerves. CPZ administration is recognized as a suitable model to study demyelination, remyelination and glial microenvironmental changes in demyelination diseases without disrupting the blood–brain barrier or interfering with T-cell infiltration [34, 35]. Studies have shown that OLs loss reaches its peak at the third week of CPZ administration. OLs apoptosis, myelin sheath injury and activation of microglial cells can also induce astrocyte activation and proliferation [36], and promote both microglial migration to the injured site and phagocytosis by secreting inflammatory factors or chemokines, which aggravate demyelination and nerve fiber injury [37]. Sustained OLs damage and myelin loss in the CPZ model are further mediated by glial cells [14]. Thus, glial environmental modification is necessary during demyelination damage.
As reported, during demyelination, myelin fragments activate microglia and produce chemokines. The upregulated expression of chemokines has been reported in many cases of demyelination related disease. Studies have found that the specific chemokine CXCL10 in CSF is upregulated in myelin demyelination lesions in MS patients [38]. CXCL10 is an important chemokine that is significantly upregulated in the CPZ model [39]. Studies have demonstrated that under homeostatic conditions, the expression of the CXCL10 gene is low, but significantly increased under inflammatory conditions. In the peripheral system, CXCL10 is able to chemotaxis Th1 lymphocytes to migrate to the site of inflammation, resulting in an immune response. In addition, a significant increase in the CXCL10 level was also observed in EAE model [40]. Furthermore, inflammatory factors such as IL-1β are involved in regulating the expression of CXCL10 and other chemokines in the peripheral system [41]. In our research, we found that CXCL10 could activate microglial cells to produce IL-1β (Fig. 7), which suggests that there might be crosstalk between CXCL10 and IL-1β to aggravate the microenvironment after demyelination.
Ginsenoside Rg1, a main active ingredient of ginseng, has a variety of central nervous protective effects. Rg1 intervention can effectively improve glial hyperplasia, reduce the level of inflammation, increase the number of mature OLs at the damaged site, and ameliorate myelin loss [42]. In recent years, the inhibitory effect of Rg1 on chemokine function has also been discovered in peripheral inflammation [43]. It has been found that Rg1 can reduce the expression of CXCL10 and alleviate the liver injury induced by calcanin A [18]. However, the mechanism by which Rg1 regulates CXCL10 is still unclear. In addition, it is unclear whether Rg1 affects CXCL10 in the CNS. In our previous study, we reported that Rg1 could regulate the NF-κB pathway caused by oxidative stress in the CNS [44]. NF-κB plays a vital role in the process of TNF-α mediated synthesis and secretion of CXCL10 [45]. In this study, we found that Rg1 alleviates behavior and pathological myelin damage in a CPZ-induced model. Rg1 reduces the level of CXCL10 by regulating the nuclear translocation of NF-κB (Fig. 5), thus modulating the consequential changes of microglial cells. Interestingly, Jin et al. found that after CXCL10 treatment of 4T1 cells, the expression level of p65 was significantly increased, while the protein synthesis of IκB was inhibited [46]. Taken together, CXCL10 functions as a mediator between the damage cycles, revealing new insight into the treatment of diseases associated with myelin loss. In addition to transcriptional regulation, in this study, we found that Rg1 also had an effect on the pure treatment of CXCL10, seen as reversing the inflammatory phenotype of microglial cells and weakening the migration of microglial cells to the demyelinating lesion site (Fig. 6).
When myelin loss occurs, a large amount of myelin fragments was produced, which impairs myelin regeneration. Phagocytosis of the myelin fragments damaged by microglia is an indispensable process of myelin regeneration. In this experiment, the phagocytic activity of microglia was not significantly influenced by CXCL10 (Fig. 6i), which was consistent with the report of Clarner et al. [14]. Since our previous research found that Rg1 could modulate the M1/M2 polarization of microglial cells [47], we also checked the role of Rg1 in LPS-treated supernatant model. With the stimulation from the treated supernatant, the phagocytosis of microglia was upregulated, especially upon Rg1 treatment (Fig. 7f). Although the expression of CXCL10 was also stimulated (Fig. 7a), we suspect that cytokines other than CXCL10 influence the phagocytic activity of microglia. Although the phagocytosis of microglia in the LPS-treated supernatant was obviously more active than that in the control group, in the Rg1 group, the phagocytic speed was faster (Fig. 7f). Rg1 can trigger the upregulation of IL-10, an M2 marker of microglia, which might contribute to its phagocytic activity (Fig. 3e, j). Furthermore, with the inhibition of CXCL10, the protective role of Rg1 was no better than that of CXCL10 treatment (Fig. 8). We speculate that the inhibition rate conceals the role of Rg1 in phagocytosis, which also indicates that CXCL10 targeted anti-inflammation is the main protective factor.
In addition, we also checked the number of Olig-2 and APC-positive OLs. The total number of OLs decreased after CPZ modeling. With the administration of Rg1, the number of olig-2+/APC+ OLs in the CC significantly increased (Fig. 2c and Supplementary Fig. S3), which suggests that Rg1 might increase the number of newly mature OLs, and further proves that Rg1 contributes to myelin sheath regeneration.
To characterize the source of CXCL10 during initial microglial activation, Clarner et al. performed double IHC staining of GFAP and CXCL10 in animals fed CPZ for 1 week. We also performed an additional experiment of animals to label CXCL10 with GFAP and Iba-1 in CC after 1 week of CPZ treatment according to Clarner et al. From the result of Supplementary Fig. S1, we could see the colocalization of CXCL10 with GFAP, but not Iba-I at the initial treatment of CPZ. The above result suggests our result is consistent with that of the former researcher. Fig. 4d shows the results from animals fed CPZ for 5 weeks (Fig. 1b, experimental diagram). Crosstalk between astrocytes and microglia would be helpful to elucidate the role of glial cells in pathological conditions. There have been no reports of CXCL10 and microglia in the CPZ model; thus, we investigated this possibility with IF staining of CXCL10 and Iba-1 to determine the interaction between CXCL10 and microglia during this period. CXCR3, which is expressed specifically on the surface of microglia [13, 48], is the receptor of CXCL10. The colocalization might provide an indication of the interaction between CXCL10 and CXCR3 on microglial cells. Thus, our result does not conflict with the former report, since the test time of Fig. 4d is different. Moreover, we confirmed previous research that CXCL10 release from astrocytes is an early response, that triggers the subsequent glial response.
In conclusion, our study demonstrates that Rg1 could significantly improve the behavioral abnormalities, myelin loss, axonal injury, and oligodendrocyte loss caused by CPZ administration. Rg1 relieves myelin injury and the pathological microenvironment through the NF-κB translocational regulation of CXCL10, a chemokine synthesized by astroglia cells. Our research suggests that the regulation of Rg1 on proinflammation and migration of microglial cells in the CPZ model is associated with CXCL10, and this inhibition abolishes the protective role of Rg1 in the CPZ model (Fig. 9).

Rg1 relieves myelin injury and the pathological microenvironment by inhibiting the NF-κB transcriptional activation of CXCL10, a chemokine synthesized by astroglial cells. The regulation by Rg1 on proinflammation and migration of microglia cells in CPZ model was associated with CXCL10, and this inhibition abolished the protective effect of Rg1 in the CPZ model.
References
Scolding NJ, Pasquini M, Reingold SC, Cohen JA, International Conference on Cell-Based Therapies for Multiple Sclerosis, International Conference on Cell-Based Therapies for Multiple Sclerosis, et al. Cell-based therapeutic strategies for multiple sclerosis. Brain. 2017;140:2776–96.
Samra K, Boon IS, Packer G, Jacob S. Lethal high: acute disseminated encephalomyelitis (ADEM) triggered by toxic effect of synthetic cannabinoid black mamba. BMJ Case Rep. 2017;2017:bcr2016218431.
Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, Anthony J, et al. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–90.
Heng Y, Zhang QS, Mu Z, Hu JF, Yuan YH, Chen NH. Ginsenoside Rg1 attenuates motor impairment and neuroinflammation in the MPTP-probenecid-induced parkinsonism mouse model by targeting alpha-synuclein abnormalities in the substantia nigra. Toxicol Lett. 2016;243:7–21.
Gao D, Tang T, Zhu J, Tang Y, Sun H, Li S. CXCL12 has therapeutic value in facial nerve injury and promotes Schwann cells autophagy and migration via PI3K-AKT-mTOR signal pathway. Int J Biol Macromol. 2019;124:460–8.
Tahmasebi F, Pasbakhsh P, Mortezaee K, Madadi S, Barati S, Kashani IR. Effect of the CSF1R inhibitor PLX3397 on remyelination of corpus callosum in a cuprizone-induced demyelination mouse model. J Cell Biochem. 2019;120:10576–86.
Gudi V, Gingele S, Skripuletz T, Stangel M. Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front Cell Neurosci. 2014;8:73.
Nicaise C, Marneffe C, Bouchat J, Gilloteaux J. Osmotic demyelination: from an oligodendrocyte to an astrocyte perspective. Int J Mol Sci. 2019;20:1124.
Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci. 2008;65:2702–20.
Yang J, Jiang Z, Fitzgerald DC, Ma C, Yu S, Li H, et al. Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. J Clin Invest. 2009;119:3678–91.
Su WF, Wu F, Jin ZH, Gu Y, Chen YT, Fei Y, et al. Overexpression of P2X4 receptor in Schwann cells promotes motor and sensory functional recovery and remyelination via BDNF secretion after nerve injury. Glia. 2019;67:78–90.
Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–95.
Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, et al. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain. 2013;136:147–67.
Clarner T, Janssen K, Nellessen L, Stangel M, Skripuletz T, Krauspe B, et al. CXCL10 triggers early microglial activation in the cuprizone model. J Immunol. 2015;194:3400–13.
Krauthausen M, Saxe S, Zimmermann J, Emrich M, Heneka MT, Muller M. CXCR3 modulates glial accumulation and activation in cuprizone-induced demyelination of the central nervous system. J Neuroinflammation. 2014;11:109.
Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103:807–15.
Sorensen TL, Trebst C, Kivisakk P, Klaege KL, Majmudar A, Ravid R, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol. 2002;127:59–68.
Cao L, Zou Y, Zhu J, Fan X, Li J. Ginsenoside Rg1 attenuates concanavalin A-induced hepatitis in mice through inhibition of cytokine secretion and lymphocyte infiltration. Mol Cell Biochem. 2013;380:203–10.
Zhang Y, Zhang H, Wang L, Jiang W, Xu H, Xiao L, et al. Quetiapine enhances oligodendrocyte regeneration and myelin repair after cuprizone-induced demyelination. Schizophr Res. 2012;138:8–17.
Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. 2015;129:429–47.
Abo Taleb HA, Alghamdi BS. Neuroprotective effects of melatonin during demyelination and remyelination stages in a mouse model of multiple sclerosis. J Mol Neurosci. 2020;70:386–402.
Reiszadeh-Jahromi S, Sepand MR, Ramezani-Sefidar S, Shahlaei M, Moradi S, Yazdankhah M, et al. Sepantronium bromide (YM155), a small molecule survivin inhibitor, promotes apoptosis by induction of oxidative stress, worsens the behavioral deficits and develops an early model of toxic demyelination: in vivo and in-silico study. Neurochem Res. 2019;44:2482–98.
Wang SS, Bi HZ, Chu SF, Dong YX, He WB, Tian YJ, et al. CZ-7, a new derivative of Claulansine F, promotes remyelination induced by cuprizone by enhancing myelin debris clearance. Brain Res Bull. 2020;159:67–78.
Zhen W, Liu A, Lu J, Zhang W, Tattersall D, Wang J. An alternative cuprizone-induced demyelination and remyelination mouse model. ASN Neuro. 2017;9:1759091417725174.
Zhang Z, Chu SF, Wang SS, Jiang YN, Gao Y, Yang PF, et al. RTP801 is a critical factor in the neurodegeneration process of A53T alpha-synuclein in a mouse model of Parkinson’s disease under chronic restraint stress. Br J Pharmacol. 2018;175:590–605.
Luchtman DW, Shao D, Song C. Behavior, neurotransmitters and inflammation in three regimens of the MPTP mouse model of Parkinson’s disease. Physiol Behav. 2009;98:130–8.
Zhang Z, Chu SF, Mou Z, Gao Y, Wang ZZ, Wei GN, et al. Ganglioside GQ1b induces dopamine release through the activation of Pyk2. Mol Cell Neurosci. 2016;71:102–13.
Omar Zaki SS, Kanesan L, Leong MYD, Vidyadaran S. The influence of serum-supplemented culture media in a transwell migration assay. Cell Biol Int. 2019;43:1201–4.
Wightman SC, Uppal A, Pitroda SP, Ganai S, Burnette B, Stack M, et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br J Cancer. 2015;113:327–35.
Sun JD, Liu Y, Yuan YH, Li J, Chen NH. Gap junction dysfunction in the prefrontal cortex induces depressive-like behaviors in rats. Neuropsychopharmacology. 2012;37:1305–20.
Cruz-Martinez P, Gonzalez-Granero S, Molina-Navarro MM, Pacheco-Torres J, Garcia-Verdugo JM, Geijo-Barrientos E, et al. Intraventricular injections of mesenchymal stem cells activate endogenous functional remyelination in a chronic demyelinating murine model. Cell Death Dis. 2016;7:e2223.
Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci. 2010;30:8953–64.
Buschmann JP, Berger K, Awad H, Clarner T, Beyer C, Kipp M. Inflammatory response and chemokine expression in the white matter corpus callosum and gray matter cortex region during cuprizone-induced demyelination. J Mol Neurosci. 2012;48:66–76.
Kramann N, Menken L, Hayardeny L, Hanisch UK, Bruck W. Laquinimod prevents cuprizone-induced demyelination independent of Toll-like receptor signaling. Neurol Neuroimmunol Neuroinflamm. 2016;3:e233.
Vega-Riquer JM, Mendez-Victoriano G, Morales-Luckie RA, Gonzalez-Perez O. Five decades of cuprizone, an updated model to replicate demyelinating diseases. Curr Neuropharmacol. 2019;17:129–41.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–7.
Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW 2nd, et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. 2019;22:1635–48.
Cui LY, Chu SF, Chen NH. The role of chemokines and chemokine receptors in multiple sclerosis. Int Immunopharmacol. 2020;83:106314.
Shen Q, Zhang R, Bhat NR. MAP kinase regulation of IP10/CXCL10 chemokine gene expression in microglial cells. Brain Res. 2006;1086:9–16.
Klein RS, Izikson L, Means T, Gibson HD, Lin E, Sobel RA, et al. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol. 2004;172:550–9.
Kaffe E, Fiorotto R, Pellegrino F, Mariotti V, Amenduni M, Cadamuro M, et al. beta-Catenin and interleukin-1beta-dependent chemokine (C-X-C motif) ligand 10 production drives progression of disease in a mouse model of congenital hepatic fibrosis. Hepatology. 2018;67:1903–19.
Zong Y, Ai QL, Zhong LM, Dai JN, Yang P, He Y, et al. Ginsenoside Rg1 attenuates lipopolysaccharide-induced inflammatory responses via the phospholipase C-gamma1 signaling pathway in murine BV-2 microglial cells. Curr Med Chem. 2012;19:770–9.
Shim JY, Kim MH, Kim HD, Ahn JY, Yun YS, Song JY. Protective action of the immunomodulator ginsan against carbon tetrachloride-induced liver injury via control of oxidative stress and the inflammatory response. Toxicol Appl Pharmacol. 2010;242:318–25.
Liu Q, Kou JP, Yu BY. Ginsenoside Rg1 protects against hydrogen peroxide-induced cell death in PC12 cells via inhibiting NF-kappaB activation. Neurochem Int. 2011;58:119–25.
Harris DP, Bandyopadhyay S, Maxwell TJ, Willard B, DiCorleto PE. Tumor necrosis factor (TNF)-alpha induction of CXCL10 in endothelial cells requires protein arginine methyltransferase 5 (PRMT5)-mediated nuclear factor (NF)-kappaB p65 methylation. J Biol Chem. 2014;289:15328–39.
Jin WJ, Kim B, Kim D, Park Choo HY, Kim HH, Ha H, et al. NF-kappaB signaling regulates cell-autonomous regulation of CXCL10 in breast cancer 4T1 cells. Exp Mol Med. 2017;49:e295.
Liu JQ, Zhao M, Zhang Z, Cui LY, Zhou X, Zhang W, et al. Rg1 improves LPS-induced Parkinsonian symptoms in mice via inhibition of NF-kappaB signaling and modulation of M1/M2 polarization. Acta Pharmacol Sin. 2020;41:523–34.
Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95:10896–901.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81973499, 81873026, 81730096, 82074044, 81773924, and 81960729), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2M-1-004), the Opening Program of the Shanxi Key Laboratory of Chinese Medicine Encephalopathy (CME-OP-2017001), and the High-End Foreign Experts Introduction Program (G20200001485).
Author information
Authors and Affiliations
Contributions
NHC and ZZ designed the study. YXD, ZZ, SFC, SSW, YJT, YSD, and XY performed the experiments and drafted the paper; SFC and NHC participated in data analysis; WBH and ZZW was involved in discussion of the experiments. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary Information
Rights and permissions
About this article

Cite this article
Dong, Yx., Chu, Sf., Wang, Ss. et al. Rg1 exerts protective effect in CPZ-induced demyelination mouse model via inhibiting CXCL10-mediated glial response. Acta Pharmacol Sin 43, 563–576 (2022). https://doi.org/10.1038/s41401-021-00696-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00696-3
