
Abstract
Heart aging is characterized by structural and diastolic dysfunction of the heart. However, there is still no effective drug to prevent and treat the abnormal changes in cardiac function caused by aging. Here, we present the preventive effects of emodin and its derivative Kanglexin (KLX) against heart aging. We found that the diastolic dysfunction and cardiac remodeling in mice with D-galactose (D-gal)-induced aging were markedly mitigated by KLX and emodin. In addition, the senescence of neonatal mouse cardiomyocytes induced by D-gal was also reversed by KLX and emodin treatment. However, KLX exhibited better anti-heart aging effects than emodin at the same dose. Dysregulated mitophagy was observed in aging hearts and in senescent neonatal mouse cardiomyocytes, and KLX produced a greater increase in mitophagy than emodin. The mitophagy-promoting effects of KLX and emodin were ascribed to their abilities to enhance the protein stability of Parkin, a key modulator in mitophagy, with different potencies. Molecular docking and SPR analysis demonstrated that KLX has a higher affinity for the ubiquitin-like (UBL) domain of Parkin than emodin. The UBL domain might contribute to the stabilizing effects of KLX on Parkin. In conclusion, this study identifies KLX and emodin as effective anti-heart aging drugs that activate Parkin-mediated mitophagy and outlines their putative therapeutic importance.
Similar content being viewed by others


Introduction
Cardiovascular disease (CVD) is the leading cause of mortality in elderly individuals [1]. With increasing age, cardiac senescence results in structural and functional changes in the heart that are commonly associated with an increased risk of CVD and impaired functional capacity in elderly individuals. Accordingly, anti-heart aging therapies would be effective for preventing health deterioration during aging. Despite recent progress in the treatment of cardiovascular diseases, research on delaying heart aging to inhibit further CVD progression is still urgently needed [2].
Mitochondria are not only the primary sources of energy in the heart but also key regulators of cardiomyocyte survival [3]. Accumulation of damaged mitochondria is the core feature of cardiac aging, which results in excessive generation of reactive oxygen species (ROS), leading to oxidative damage and further cardiac dysfunction [4, 5]. However, damaged mitochondria are normally eliminated through mitophagy, a type of selective autophagy of mitochondria, to maintain cellular energetic and metabolic homeostasis [6]. Notably, defective mitophagy is commonly observed in senescent cardiomyocytes, and amelioration of the decreased mitophagy is effective in preventing heart aging and cardiac dysfunction in aging animals [7,8,9]. To date, several mitophagy pathways have been discovered, among which the PINK1/Parkin-mediated ubiquitin-dependent pathway is the predominant pathway [10,11,12]. Growing evidence has indicated that targeting the PINK1/Parkin pathway is beneficial for treatment of diverse cardiac pathological conditions, such as cardiac hypertrophy, heart failure, and myocardial infarction [13, 14]. This prompted us to propose that the PINK1/Parkin pathway might be a pharmacological target for some therapeutic agents.
Emodin (1,3,8-trihydroxy-6-methylanthraquinone) is a natural anthraquinone compound extracted from Rheum palmatum and Polygonum cuspidatum. It is known to exert a variety of pharmacological effects, such as anti-inflammatory, antitumor, antiviral, antibacterial, antiallergic, antidiabetic, and hepatoprotective effects [15,16,17,18]. Our previous studies have demonstrated that emodin can alleviate cardiac fibrosis by upregulating MTA3 and ameliorate hyperglycemia and dyslipidemia in rats with type II diabetes by targeting microRNA-20b [19, 20]. In addition, our group synthesized a novel anthraquinone compound, 4,5-dihydroxy-7-methyl-9,10-anthraquinone-2-ethyl succinate, via chemical modification of emodin on the hydroxyl group of the anthracene ring that exhibited better solubility and bioactivity and lower toxicity than emodin and named it Kanglexin (KLX). Our previous studies have shown that KLX has protective effects on the vascular endothelium and myocardium. It can reduce blood pressure [21], attenuate lipid accumulation [22], accelerate diabetic wound healing [23], and prevent MI-induced cardiac dysfunction [24]. Therefore, more rigorous investigations of its potential use for alleviating heart aging are warranted.
The present study aimed to compare the effects of KLX and emodin on cardiac function in aging mice and elucidate the underlying molecular mechanisms in modulating cardiac senescence. Specifically, pharmacological modulation of PINK1/Parkin-mediated mitophagy by KLX and emodin was investigated in depth as a potential strategy for heart aging-related degenerative processes.
Materials and methods
Materials and animals
Kanglexin was obtained from the Department of Pharmaceutical Chemistry, Harbin Medical University. Emodin and rapamycin were purchased from Solarbio Co. (Beijing, China). Three-month-old female C57BL/6 mice were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd. and housed under standard animal room conditions (temperature 22 ± 1 °C and humidity 55% ± 5%). The use of animals was approved by the Ethics Committees of Harbin Medical University, and the protocols conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The mice were randomly divided into six groups: a control group, a D-gal-induced aging group, D-gal + KLX groups (low- and high-dose groups that received 10 and 20 mg · kg−1 · d−1 KLX, respectively), a D-gal + emodin (20 mg · kg−1) group, and a D-gal + rapamycin (3 mg · kg−1) group. The mouse aging model was established by subcutaneously injecting mice with 120 mg · kg−1 D-gal in the back for 8 weeks. Simultaneously, Kanglexin, emodin and rapamycin (dissolved in distilled water with 0.5% sodium carboxymethylcellulose) were administered via gastric gavage for 8 weeks.
Echocardiographic assessment of cardiac function
After anesthetization with 0.2 g · kg−1 avertin (Sigma, St. Louis, MO, USA), the mice were subjected to echocardiography measurement using a Vevo® 2100 High-Resolution Imaging System (VisualSonics, Toronto, Canada) ultrasound machine equipped with a 10.0-MHz phase-array transducer. Cardiac diastolic function was evaluated by the ratio of the E peak (left ventricular (LV) diastolic maximum blood flow) to the A peak (atrial systolic maximum blood flow through the mitral valve). The LV ventricular posterior wall diastole (LVPWD) was measured. The LV mass, LV ejection fraction (EF), and fractional shortening (FS) were calculated from M-mode recordings. Images of the heart used for quantification were obtained in both parasternal long-axis and short-axis views.
Histopathological and morphometric analysis
Histopathological changes and collagen accumulation were evaluated by hematoxylin and eosin (HE) and Masson’s trichrome staining according to the procedures described in our previous study [25]. Heart tissue was fixed in 4% paraformaldehyde, embedded in paraffin and cut cross-sectionally into 5 μm thick sections. The tissue sections were deparaffinized and stained with HE or Masson’s trichrome reagent. Images of the heart were captured with a light microscope (Carl Zeiss Microscopy, Jena, Germany).
β-galactosidase staining (SA-β-Gal)
A Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, Boston, USA) was used to evaluate senescence according to the manufacturer’s instructions. Briefly, for cell staining, the cells were removed from the growth medium and plated into a 35-mm well plate containing 1 mL of 1× Fixative Solution. The cells were fixed at room temperature for 10–15 min. The plate was rinsed twice with phosphate-buffered saline (PBS), after which 1 mL of the β-Galactosidase Staining Solution was added to each 35-mm well. The plate was incubated in a dry incubator (no CO2) at 37 °C overnight. The cells were examined under a light microscope (Carl Zeiss Microscopy, Jena, Germany).
Primary culture of neonatal mouse cardiomyocytes
Cardiomyocytes were isolated from 1- to 3-day-old neonatal Kunming mice according to the procedures described in our previous study [26]. In brief, after dissection and washes, the hearts were finely minced, and the pieces were placed in 0.25% trypsin. The pooled cell suspension was centrifuged and resuspended in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U · mL−1 penicillin and 100 μg · mL−1 streptomycin. The resuspended solution was plated onto a culture flask at 37 °C for 90 min to enable preferential attachment of fibroblasts to the bottom. The nonadherent and weakly attached cells, mainly cardiomyocytes, were removed and seeded onto plates. The cells were incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. After 48 h, cardiomyocytes that adhered to the culture dish were used for subsequent experimental procedures.
The cardiomyocyte aging model was established via D-gal (40 μM) treatment for 24 h. KLX-L (10 μM), KLX-H (20 μM), and emodin (20 μM) were dissolved in DMSO and added to the medium together with D-gal.
Culturing of an adult human ventricular cardiomyocyte cell line (AC16)
Human AC16 cells, which possess many of the biochemical and morphological properties of cardiomyocytes, were cultured as previously described [27]. Briefly, nondifferentiated AC16 cells were maintained in DMEM: F12 medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin and grown at 37 °C in a humidified atmosphere of 5% CO2 + 95% air.
Transmission electron microscopy examinations
Selected samples were fixed in 2.5% glutaraldehyde in 0.1 mol · L−1 PBS (pH 7.4). The specimens were then rinsed and postfixed in PBS-buffered 1% OsO4 for 1–2 h, stained en bloc in uranyl acetate, dehydrated in ethanol, and embedded in epoxy resin via standard procedures. The ultrathin sections were electron-stained and examined under an electron microscope (JEOL Ltd., Tokyo, Japan).
Cell cycle progression assay
Cell cycle distribution was determined by flow cytometric analysis. AC16 cells were harvested by trypsinization, washed three times with PBS, centrifuged at 1000×g for 5 min and fixed gently with 70% cold alcohol at 4 °C overnight. Before detection, the cells were washed twice with ice-cold PBS and incubated with RNase and propidium iodide (PI) (Invitrogen, Carlsbad, CA, USA) at 4 °C in the dark for 30 min. Finally, cell cycle phase analysis was performed using a flow cytometer (BD Bioscience, Franklin Lakes, USA).
mRFP-GFP-LC3 staining
An mRFP-GFP-LC3 adenovirus construct was obtained from Hanbio (autophagy-adv-GFP-RFP-LC3B-1000, Hanbio, Shanghai, China). The fluorescence of this construct depends on the difference in pH between acidic autolysosomes and neutral autophagosomes. The red/green (yellow) or red fluorescence enables researchers to monitor the progression of autophagic flux. The cells infected with the adenovirus were imaged using confocal fluorescence microscopy (Carl Zeiss Microscopy, Jena, Germany).
Western blotting
Protein samples were loaded into a 12% sodium dodecyl sulfate polyacrylamide gel (80 μg · well−1). After electrophoresis, the proteins were transferred onto a nitrocellulose filter membrane, which was subsequently blocked with 5% nonfat milk dissolved in PBS for 2 h. The membrane was probed with primary antibodies against Parkin (Invitrogen, Carlsbad, CA, USA), LC3 (CST, Boston, USA), p21 (Abcam, Cambridge, UK), p53 (Abcam, Cambridge, UK), and p62 (CST, Boston, USA) diluted at 1:1000 in PBS buffer at 4 °C overnight. The membrane was then washed in PBS with 0.05% Tween 20 three times and incubated with a fluorescence-conjugated anti-rabbit or anti-mouse IgG secondary antibody at room temperature for 1 h (1:10,000; LI-COR, Lincoln, NE, USA). GAPDH (Zhongshanjinqiao, Beijing, China) was used as an internal control. The Western blot bands were quantified using an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE, USA) by measuring the band intensity (area × OD).
RNA isolation and quantitation
Total RNA was extracted from cardiomyocytes and myocardial tissues by using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The quality of the RNA samples was measured with a NanoDrop ND-8000 (Thermo Fisher Scientific, Waltham, MA, USA) to ensure an A260/A280 ratio of 1.8–2.0. The RNA was reverse-transcribed into cDNA using a Reverse Transcription Kit (Takara, Dalian, China). Real-time PCR was then performed with SYBR Green (04913914001; Roche, Basel, Switzerland). GAPDH was used as an internal control for Parkin in myocardial tissue and cardiomyocytes. The sequences of the primer pairs are as follows:
Parkin: forward, 5′-GGGGCTGGCGGTCATT-3′, and reverse, 5′-GGTGAGGGTTGCTTGTTTGC-3′;
GAPDH: forward, 5′-AGTTCAACGGCACAGTCAAG-3′, and reverse, 5′-TACTCAGCACCAGCATCACC-3′.
Molecular docking
Molecular docking was performed as described in our previous study [28]. The crystal structure of Parkin (PDB: 1ify) was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/), and the structure of Parkin was prepared with the biopolymer tool SYBYL-X 2.0 (Tripos, USA). Hydrogen atoms were added, and the protein was subjected to the AMBER FF99 force field. The water in the protein was then removed. A 1000-iteration minimization of the hydrogen atoms was followed by a 100 ps molecular dynamics simulation to refine the positions of the targets. The input ligand (KLX) file format was mol2 for all docking programs used. An SFXC file was built using the PDB-prepared protein structure. The protocol was generated using the ligand with a threshold of 0.50 and the bloat set to 0 (default settings). CScore calculations were enabled on the Surflex docking runs. The number of additional starting conformations per molecule was set to 10, and the number of angstroms used to expand the search grid was set to 6. The maximum number of conformations per fragment was 30, the maximum number of rotatable bonds per molecule was 150, and the maximum number of poses per ligand was 50. The minimum RMSD between final poses was set to 0.05.
Surface plasmon resonance
Ligand/protein binding analysis was performed using an OpenSPR localized SPR biosensor (Nicoya Life Science Inc., Kitchener, Canada).
Parkin served as a receptor and was immobilized to gold nanoparticles on a COOH sensor chip using standard EDC/NHS (1:1) solution. Then, 200 μL of Parkin ligand (R&D Systems, USA) in activation buffer was introduced into the sensor chip at a flow rate of 20 µL · min−1 with 20 mM Tris-HCl (pH 7.4) as the running buffer for 4 min. After the ligand-chip interaction was complete, the upper sample inlet was rinsed with buffer and air-dried. The baseline was observed for 5 min to ensure stability. Finally, the analyte (100 μL, KLX 0-50 μM) was introduced into the receptor-bound sensor chip at a flow rate of 20 µL · min−1. The binding time for binding of the analyte to the ligand was 240 s, and the natural dissociation time was 480 s. The kinetic parameters of the binding between KLX/emodin and Parkin were calculated and analyzed with Trace Drawer software (Ridgeview Instruments AB, Sweden).
Statistical analysis
The data in this study are shown as the mean ± SEM. Differences among groups were analyzed using one-way ANOVA for multiple comparisons. Two-tailed Student’s t test was used for comparisons between two groups (GraphPad Prism version 7.0). P < 0.05 was considered to indicate statistical significance.
Results
KLX improves diastolic dysfunction in aging mice
To explore whether KLX (Fig. 1a) and emodin (Fig. 1b) have preventive effects on cardiac dysfunction in aging mice, we established a subacute aging mouse model via subcutaneous injection of D-gal for 8 weeks, which mice exhibited natural aging phenotypes [29]. Simultaneously, KLX and emodin were administered to different groups via gastric gavage for 8 weeks. Rapamycin has been suggested to have an anti-aging effect; therefore, we used it as a positive control. Echocardiographic measurements showed that compared with 3-month-old young mice, mice with D-gal-induced aging exhibited markedly weakened cardiac diastolic function, as measured by the ratio of the E peak to the A peak (the E/A ratio), and markedly increased LV mass; these changes were effectively corrected by emodin and KLX administration (Fig. 1c–f). There was no statistically significant difference between KLX (20 mg · kg−1) and rapamycin (3 mg · kg−1), but the effects of KLX (20 mg · kg−1) were greater than those of emodin at the same dose. The EF% (Fig. 1g), FS% (Fig. 1h), LV end diastolic dimension (LVEDD), LV end systolic dimension (LVESD) and LVPWD value (Supplementary Table. 1) had no significant differences among groups. These results indicated that diastolic function was deteriorated and that systolic function was preserved in mice with D-gal-induced aging, consistent with the changes observed in naturally aging mice, more importantly, KLX and emodin significantly improved diastolic dysfunction in aging mice [30, 31].

D-gal was used to establish a mouse model of cardiac aging. a Chemical structure of KLX (C21H18O8). b Chemical structure of emodin (C15H10O5). c Representative echocardiographic images showing the effects of KLX and emodin on the ratio of the E peak to the A peak (E/A ratio) for evaluation of cardiac diastolic function in mice with D-gal-induced aging. d Statistical results for the E/A ratio. n = 7. e Echocardiographic images showing no significant changes in cardiac systolic function in mice with D-gal-induced aging. f Statistical results for the LV mass. g Statistical results for the left ventricular ejection fraction (EF%). h Statistical results for the left ventricular fractional shortening (FS%). ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. D-gal; &P <0.05, &&P < 0.01 between the groups at both ends of the line. The data are expressed as the mean ± SEM.
KLX delays cardiac senescence and improves cardiac remodeling in aging mice
We then tested the influences of KLX and emodin on cardiac senescence and cardiac remodeling. p53 and p21 are commonly used indicators of aging [32]. Our results showed that D-gal significantly increased the protein levels of p53 and p21 in the heart tissues of aging mice relative to those of young control mice, while emodin or KLX administration reversed these effects. Moreover, KLX elicited stronger downregulatory effects than emodin on p53 and p21 (Fig. 2a, b). A marked increase in positive staining for senescence-associated β-galactosidase (SA-β-Gal; a marker of aging) was consistently observed in aging mouse hearts but was suppressed by emodin and KLX treatment (Fig. 2c). To further explore the effects of KLX and emodin on cardiac remodeling, we performed HE staining of heart cross-sections. The results demonstrated that the heart tissue was severely damaged in the D-gal treatment group, but the detrimental changes were reversed by KLX and emodin, as indicated by improved myocardial organization and reduced myocardial fiber breakage, decreased nuclear size irregularity, reduced nuclear fragmentation, and reduced cardiomyocyte shrinkage and dissolution (Fig. 2d). In addition, Masson staining showed that emodin and KLX reduced collagen deposition and fibrosis in aging hearts (Fig. 2e, f). These results suggested that emodin and KLX could alleviate cardiac senescence and improved cardiac remodeling in aging mice.

a Western blot analysis of the protein level of the senescence marker p53. n = 5. b Western blot analysis of the protein level of the senescence marker p21. n = 5. c β-Galactosidase staining (×200) images showing the inhibitory effects of KLX and emodin on heart aging, as indicated by decreased positive staining. n = 5. Scale bar: 200 μm. d HE staining (×400) showing the inhibitory effects of KLX and emodin on the pathological and morphological changes of aging hearts. The white dashed box indicates the myocardial fiber arrangement, and the yellow arrow indicates myocardial fiber breakage. n = 5. Scale bar: 100 μm. e Masson trichrome staining (×400) showing the effects of KLX and emodin on reducing collagen deposition and fibrosis in heart tissue. The black rectangular box indicates the myocardial fiber arrangement, and the yellow asterisk indicates collagen fiber deposition. n = 5. f Statistical results for fibrosis as revealed by Masson staining. Scale bar: 100 μm. ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. D-gal; &P < 0.05, &&P < 0.01, &&&P < 0.001 between the groups at both ends of the line. The data are expressed as the mean ± SEM.
KLX inhibits cardiomyocyte senescence
To further validate the suppressive effects of KLX on cardiac senescence, we cultured primary cardiomyocytes from mice and induced cell senescence with D-gal (40 μM). We found that KLX and emodin treatment markedly inhibited the upregulation of p21 and p53 protein levels induced by D-gal (Fig. 3a, b). Consistently, SA-β-gal staining showed that KLX and emodin reversed D-gal-induced senescence of primary cardiomyocytes (Fig. 3c, d). More importantly, KLX exerted a better anti-senescent effect than emodin at the same concentration (Fig. 3a–d). The accumulation of reactive oxygen species (ROS) in senescent cells is a well-recognized indicator and inducer of aging [33]. We also found obvious accumulation of ROS in senescent primary cardiomyocytes that was significantly reduced by emodin and KLX treatment (Fig. 3e, f). Flow cytometry results showed that most AC16 cells treated with D-gal were arrested in the G0/G1 phase and that the arrest was accompanied by a decrease in the proportion of cells in the G2/M phase, but these changes were effectively abrogated by emodin and KLX (Fig. 3g, h).

D-gal (40 μM) was used to establish a senescence model with cardiomyocytes. a Representative Western blot and quantitation results for the levels of the senescence marker p53 in primary neonatal mouse cardiomyocyte lysates. n = 5. b Representative Western blot and quantitation results for the levels of the senescence marker p21 in primary neonatal mouse cardiomyocyte lysates. n = 5. c SA-β-galactosidase staining images showing the inhibitory effects of KLX and emodin on primary neonatal mouse cardiomyocyte senescence, as indicated by decreased positive staining. n = 5. Scale bar: 50 μm. d Statistical results for SA-β-galactosidase staining. e ROS production in primary neonatal mouse cardiomyocytes was evaluated by using H2DCFDA probe staining. n = 5. Scale bar: 20 μm. f Statistical results for ROS production. g The cell cycle distribution of AC16 cells was measured by flow cytometry analysis following D-gal treatment and cotreatment with KLX, emodin and rapamycin. n = 5. h Statistical results for flow cytometry. ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. D-gal; &P < 0.05, &&P < 0.01, &&&P < 0.001 between the groups at both ends of the line. The data are expressed as the mean ± SEM.
KLX promotes Parkin-mediated mitophagy in aging hearts
To further elucidate the subcellular mechanisms underlying the anti-aging effects of KLX and emodin, we performed transmission electronic microscopy (TEM) to observe the ultrastructural changes in cardiac tissue. The images revealed that D-gal treatment resulted in mitochondrial swelling, myofilament rupture, nuclear constriction, and mitophagy inhibition. However, emodin and KLX treatment markedly ameliorated the mitochondrial structure damage and increased mitophagy (Fig. 4a). In the process of mitophagy, microtubule-associated protein 1 light chain 3-I (LC3-I) is conjugated to phosphatidylethanolamine to form an LC3-phosphatidylethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes. The protein sequestosome 1 (SQSTM1/p62) can bind ubiquitin and LC3, thereby targeting the autophagosome and facilitating clearance of ubiquitinated proteins [34]. The Western blot results showed decreased LC3 II/I protein levels and elevated p62 protein levels in the hearts of D-gal-treated mice, which suggested decreased mitophagy. Nevertheless, emodin and KLX mitigated the defects in mitophagy induced by D-gal (Fig. 4b–d). Cargo ubiquitination by the E3 ubiquitin ligase Parkin is important for triggering of selective mitophagy [35]. We, therefore, tested whether Parkin was involved in the mitophagy-promoting effects of emodin and KLX. As illustrated in Fig. 4b, e, the expression of Parkin was significantly decreased in the hearts of aging mice, but emodin and KLX administration abolished this effect. Immunofluorescence staining also demonstrated the enhancing effects of emodin and KLX on Parkin expression in aging hearts (Fig. 4f).

a Transmission electron microscopy (TEM) images showing the changes in mitochondria and autophagy in cardiac tissue. The red asterisks indicate autophagosomes, and the red arrow indicates pyknosis. Scale bars: 2 and 0.4 μm. b–e Western blot analysis of the autophagy marker proteins LC3, p62, and Parkin. n = 5. f Immunofluorescence staining of frozen heart sections showing the enhancing effects of KLX and emodin on Parkin (green) expression in aging hearts. α-actin staining was used to label myocardial tissue (red), and DAPI staining was used to label cell nuclei (blue). Scale bar: 20 μm. ***P < 0.001 vs. control; ##P < 0.01, ###P < 0.001 vs. D-gal; &&P < 0.01, &&&P < 0.001 between the groups at both ends of the line. The data are expressed as the mean ± SEM.
KLX promotes Parkin-mediated mitophagy in senescent cardiomyocytes
To verify the promotive effects of emodin and KLX on Parkin-mediated mitophagy in senescent cardiomyocytes, we used D-gal (40 μM) to induce cell senescence in primary cardiomyocytes with or without emodin or KLX treatment. LC3 II/I and Parkin protein levels were downregulated in response to D-gal treatment. An increase in the p62 protein level also suggested that D-gal treatment suppressed mitophagy (Fig. 5a–d). Emodin and KLX significantly reversed these alterations. Moreover, TEM provided additional evidence that emodin and KLX positively regulated mitophagy, as indicated by the improved mitochondrial structure and increased autophagosomes in primary cardiomyocytes (Fig. 5e). In addition, we infected cardiomyocytes with an adenovirus expressing a tandem GFP-mRFP-LC3 fluorescent protein to assess the influences of emodin and KLX on autophagic flux. The results demonstrated that D-gal diminished the red fluorescent protein (RFP) signal, indicating that it suppressed autolysosome formation. Conversely, emodin and KLX treatment promoted the formation of autolysosomes (Fig. 5f, g).

a–d Representative Western blot and quantitation results for p62, LC3 and Parkin in primary neonatal mouse cardiomyocyte lysates showing the reversal of D-gal-induced downregulation of Parkin-mediated mitophagy by KLX and emodin. n = 5. e Representative transmission electron microscopy (TEM) images showing the changes in mitochondria and decreases in mitophagy in cardiomyocytes with D-gal-induced senescence. Emodin and KLX treatment markedly ameliorated mitochondrial structural changes and increased mitophagy. Scale bars: 20 and 0.4 μm. The red asterisks indicate autophagosomes, and the red arrows indicate swollen mitochondria. f, g Representative images showing LC3 staining in different groups infected with the GFP-RFP-LC3 adenovirus for 24 h. Autophagosomes are indicated by yellow puncta in the merged images, and autolysosomes are indicated by red puncta in the merged images. The mean numbers per cell were calculated. Scale bars: 50 and 16.7 μm. n = 5. ***P < 0.001 vs. control; #P < 0.05, ###P < 0.001 vs. D-gal; &&P < 0.01, &&&P < 0.001 between the groups at both ends of the line. The data are expressed as the mean ± SEM.
KLX promotes Parkin protein stability
Next, we wanted to elucidate how KLX regulates Parkin expression and Parkin-mediated mitophagy. As shown in Fig. 6a, D-gal treatment reduced mitochondrial translocation of Parkin protein, as indicated by decreased colocalization of Parkin protein and Mito Tracker, whereas emodin and KLX treatment increased the protein levels and mitochondrial translocation of Parkin. To investigate whether emodin and KLX acted by stabilizing Parkin mRNA, we used 10 μg · mL−1 actinomycin D (Act D) to block overall transcriptional activity and then quantified Parkin mRNA levels at varying time points ranging from 0 to 24 h. The results showed no significant differences in the half-life of Parkin mRNA among the control, KLX and emodin groups (Fig. 6b). Then, we tested the influences of emodin and KLX on the stability of the Parkin protein. Cycloheximide (CHX; 25 μg · mL−1) was utilized to block Parkin protein synthesis, and Parkin protein levels were detected at different time points. We found that the degradation rate of Parkin protein was delayed in the KLX and emodin treatment groups compared to the control group. Moreover, KLX treatment had a greater maintaining effect on Parkin protein stability than emodin at 12 and 24 h (Fig. 6c, d).

a Representative immunofluorescence staining showing the reduced colocalization (yellow arrows) of Parkin and mitochondria (Mito Tracker signal) in cardiomyocytes with D-gal-induced aging and the increases in Parkin mitochondrial translocation induced by emodin and KLX administration. Scale bars: 50 and 12.5 μm. b KLX and emodin did not significantly influence Parkin mRNA degradation induced by the RNA synthesis inhibitor Act D (10 μg · mL−1), as measured by qPCR. n = 3. c, d Western blot analysis of the effects of KLX and emodin on Parkin protein degradation induced by the protein synthesis inhibitor CHX (25 μg · mL−1). The relative intensity of the Parkin band was normalized to the intensity of the GAPDH band, n = 5. ***P < 0.001 vs. control. The data are expressed as the mean ± SEM.
To further confirm whether the anti-heart aging role of KLX relies on Parkin, we transfected Parkin siRNA (siParkin) into primary cardiomyocytes. Six hours later, D-gal was used to induce senescence. The results demonstrated that KLX treatment reversed D-gal-induced cardiomyocyte senescence, as indicated by decreased expression of p21 and p53. However, knockdown of Parkin abolished the anti-senescence effects of KLX (Fig. S1a, b). Moreover, siParkin reduced the promotive effects of KLX on LC3 II/I and Parkin expression but elevated p62 levels (Fig. S1c–e), suggesting that knockdown of Parkin diminished the mitophagy-promoting effect of KLX on senescent cardiomyocytes. The above results further confirm that the anti-aging effect of KLX is mediated by Parkin-induced mitophagy.
KLX directly interacts with Parkin protein
To further investigate the possible mechanism by which KLX and emodin regulate Parkin protein stability, molecular docking simulation was performed with KLX, emodin and Parkin using SYBYL 2.0 software. The docking results indicated that KLX tightly fitted to the UBL domain of Parkin (PDB: 1ify) and could form H-bonds with residues Arg33 and Gln34 (Fig. S2a). The calculated CScore of 6.75 and polar value of 2.33 indicated that optimal polar interaction occurred between KLX and the UBL domain of Parkin. The crash value of −1.77 reflected the degree of intermolecular collision. In contrast, the CScore, polar value and crash value in the emodin-Parkin docking program were 4.64, 2.09, and −0.78, indicating a lower binding affinity between emodin and Parkin than between KLX and Parkin (Fig. S2c). Previous studies have supported the idea that the UBL domain of Parkin functions to inhibit its intrinsic autoubiquitination activity. Pathogenic mutations in the UBL domain result in degradation of the Parkin protein through autoubiquitination, leading to proteasomal degradation and inactivation of Parkin [36]. We tested this hypothesis using surface plasmon resonance (SPR) analysis to determine the binding affinity between KLX/emodin and Parkin. The SPR data clearly showed that KLX interacted with Parkin to form a stable complex with an affinity constant (KD) of 1.52 × 10−5 M. The association rate constant (Ka) was 1.08 × 103 M−1 · s−1, and the dissociation rate constant (Kd) was 1.65 × 10−2 s−1 (Fig. S2b). However, the binding affinity between emodin and Parkin was markedly lower than that of the KLX-Parkin complex, with a KD value of 7.22 × 10−5 M, a Ka value of 1.62 × 102 M−1 · s−1 and a Kd value of 1.17 × 10−2 s−1 (Fig. S2d), which might contribute in part to its weaker influence on parkin-mediated mitophagy than KLX. Therefore, the above results revealed that direct interaction between KLX and Parkin might prevent the ubiquitinated proteins from recognizing and binding with Parkin and resulted in the delay of Parkin protein degradation.
Discussion
In the present study, we investigated the roles of the Parkin-mitophagy axis in the anti-heart aging effects of KLX and emodin in a mouse model of D-gal-induced aging and in senescent cardiomyocytes. First, KLX and emodin improved diastolic dysfunction and cardiac remodeling in aging mice and attenuated cardiomyocyte senescence, similar to rapamycin. Second, KLX and emodin significantly reversed the abnormal downregulation of Parkin-mediated mitophagy caused by heart aging and cardiomyocyte senescence. Third, we experimentally identified Parkin as a direct target of KLX and emodin and further revealed the promotive effects of KLX and emodin on Parkin protein stabilization. More importantly, we confirmed that KLX is more effective than emodin. This difference might be partially due to the different binding affinities of KLX and emodin for the Parkin protein and result in distinct regulation of protein stability.
Heart aging primarily manifests as cardiac remodeling and LV diastolic dysfunction, which lay the pathological foundation for further deterioration of cardiac function [37, 38]. The Framingham Heart Study and the Baltimore Longitudinal Study on Aging have demonstrated that aging results in a decline in diastolic function accompanied by slight LV hypertrophy and relatively preserved systolic function at rest, even in healthy individuals without concomitant cardiovascular diseases [39]. These features are also observed in aging mice. Liang et al. evaluated cardiac structure and function in 4- and 24-month-old male mice and found significantly decreased E/A ratios in aged hearts that were not accompanied by significant differences in EF% or FS% [14]. In our study, we found that the E/A ratio was decreased by 59% in D-gal-administered mice but that high-dose KLX administration increased the index to nearly normal levels. Although emodin and a low dose of KLX markedly improved the E/A ratios of aging mice, the effects were less pronounced than those of high-dose KLX. In addition, our results suggested that EF% and FS% were slightly decreased in the D-gal group but were not significantly different from those in the control group. LVESD, LVEDD, and LVPWD were also preserved. These effects further suggested that emodin and KLX moderately ameliorated heart aging. Regarding the pathology of aging hearts, cardiomyocyte hypertrophy, myocardial fiber breakage [40], fibroblast proliferation and increased collagen deposition [41] have also been identified. All these changes may form the structural basis for the reduced compensatory capacity of aging hearts. Moderate cardiac hypertrophy appears to be an adaption to maintain normal cardiac volume and pump function, but the progression of hypertrophy causes heart failure. We also found that KLX and emodin significantly improved cardiac damage and cardiac fibrosis in aging hearts. The decreased LV mass in the KLX and emodin groups further confirmed the anti-hypertrophic effects of these compounds on aging hearts. These findings are consistent with the effects observed in an MI mouse model in our previously published study [27].
Some pharmacological agents that exert anti-oxidant and anti-inflammatory effects have been explored for their usefulness in anti-heart aging. Metformin increases AMPK activity, and statins reduce the levels of ROS [42]. Moreover, ACE inhibitors and angiotensin receptor blockers have been found to extend lifespan and attenuate age-associated CVD in murine models [43]. Rapamycin, an inhibitor of the mTOR pathway, can extend the lifespans of both male and female mice and inhibit cell senescence [44, 45]. Although there have been multiple preclinical studies on anti-heart aging drugs, clinical investigations in human subjects are still lacking. Therefore, identification of more candidates for heart aging treatment is urgently needed. Emodin is a naturally occurring anthraquinone from the traditional Chinese medicine rhubarb with a variety of biological activities and has been used primarily as an anti-inflammatory, antibacterial, anticancer, diuretic, and cardiovascular protection agent [17, 46,47,48]. One study has shown that emodin extends the lifespan of Caenorhabditis elegans through the insulin/IGF-1 signaling pathway [49]. However, the role of emodin in regulating heart aging has not been previously reported; the present study is thus the first to unravel the anti-heart aging properties of emodin and KLX in animal and cellular models. In our previous study, we found that KLX prevented MI-induced cardiac contractile dysfunction, as indicated by the reversal of decreases in EF% and FS% [24]; in this study, we found that KLX had better ameliorative effects on deteriorating heart function in aging mice than emodin, as evidenced by the results of both in vivo and in vitro experiments. The doses and concentrations of KLX and emodin used for the in vivo and in vitro experiments were selected based on our published pharmacological studies and were within the safe range (Fig. S3) [19, 20, 24].
Mitochondrial dysfunction is a central contributor to aging and aging-related disorders, such as Alzheimer’s disease [50], Parkinson’s disease [51], and heart failure [52]. Efficient removal of dysfunctional mitochondria through mitophagy is crucial for mitochondrial maintenance and cell survival. Disruption of mitophagy has been implicated in aging [8]. Consistently, our study also supports the role of mitophagy in regulating cardiac senescence and indicates that KLX and emodin are positive regulators of mitophagy. The PINK1/Parkin pathway has been demonstrated to be critical for mitophagy and thereby the maintenance of mitochondrial integrity and function. Elimination of damaged mitochondria is predominantly achieved via Parkin-mediated mitophagy. Previously published studies have demonstrated that Parkin levels are downregulated in cardiac disorders such as ischemia/reperfusion injury, hypoxic injury, diabetic cardiomyopathy, cardiac hypertrophy, and heart failure [53]. More importantly, Parkin has been found to be downregulated in the hearts of aged (24-month-old) mice [54]. In another study, the hearts of 15-month-old Parkin−/− mice showed declines in the cardiac functional reserve, as assessed by the responses of maximal and minimal dp/dt to dobutamine infusion, and exhibited enhanced SA-β-gal activity. Moreover, overexpression of Parkin ameliorated the functional declines in aged hearts [7]. Ren et al. [55] reported that the expression of Parkin and Pink1 was suppressed in a senescence-like cardiomyocyte model induced by D-gal. Moreover, restoration of mitophagy via overexpression of Parkin prevents mitochondrial dysfunction and induction of senescence [56]. Moreover, a study has demonstrated that 12-month-old Parkin-deficient POLG mice (premature aging model mice) also have normal cardiac function under baseline conditions [57]. These different findings are likely due to alterations in Parkin expression with increasing age and to the different genetic backgrounds of the mice. Our data provide additional evidence that Parkin is downregulated in hearts with D-gal-induced aging and in senescent cardiomyocytes and that this abnormal downregulation is reversed by KLX and emodin. As a consequence, Parkin translocation from the cytoplasm to mitochondria is increased, which further leads to restoration of mitophagy. In addition, knocking down Parkin markedly reduced the positive effects of KLX on Parkin and mitophagy, which further confirms Parkin as a valid drug target for anti-heart aging.
Parkin is an E3 ubiquitin-protein ligase consisting of an N-terminal ubiquitin-like domain (UBL) followed by RING0, RING1, In Between Ring (IBR) and RING2 domains [58]. The UBL domain is critical for substrate recognition, proteasome association and regulation of cellular Parkin protein levels, thereby influencing Parkin protein activity and function [58,59,60]. In the present study, the results of molecular docking simulations with SYBYL software showed that both KLX and emodin could insert themselves into the UBL domain of Parkin by forming hydrogen bonds with different amino acid residues. Some studies have revealed that binding to the UBL domain of the Parkin protein increases protein stability by preventing the recognition of parkin by ubiquitinated proteins and subsequently preventing proteasomal degradation. For example, Jacob et al. found that destabilization of the UBL domain resulted from rearrangements to hydrophobic core packing, resulting in conformational changes and impaired autoinhibition of Parkin phosphorylation [59]. Both UIMs of EPS15 have been shown to be required for interaction with the UBL domain, and partner binding can relieve the inhibition and activate Parkin [36, 61]. Thus, the UBL domain could be a viable therapeutic target, and as a rescue therapy to increase the stability of Parkin. Notably, in our study, we found that the binding residues of Parkin were different between KLX and emodin. The SPR results also revealed a stronger binding affinity between KLX and Parkin than between emodin and Parkin, which further resulted in different promotive effects on Parkin protein stability. As a consequence, KLX exhibited better anti-heart aging effects than emodin.
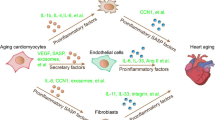
In summary, our study found that KLX and emodin improved cardiac remodeling and deterioration of heart function caused by aging by increasing Parkin-mediated mitophagy. More importantly, KLX displays stronger efficacy than emodin because of its more potent binding affinity for Parkin. Our findings indicate that KLX and emodin should be considered as potential candidates for the prevention and treatment of heart aging (Fig. 7).

Underlying mechanisms of the protective effect of KLX against cardiac aging (created with BioRender.com).
References
Sattar N, Gill JMR, Alazawi W. Improving prevention strategies for cardiometabolic disease. Nat Med. 2020;26:320–5.
Dhingra R, Vasan RS. Age as a risk factor. Med Clin North Am. 2012;96:87–91.
Srinivasan S, Guha M, Kashina A, Avadhani NG. Mitochondrial dysfunction and mitochondrial dynamics—the cancer connection. Biochim Biophys Acta Bioenerg. 2017;1858:602–14.
Song J, Huang Y, Zheng W, Yan J, Cheng M, Zhao R, et al. Resveratrol reduces intracellular reactive oxygen species levels by inducing autophagy through the AMPK-mTOR pathway. Front Med. 2018;12:697–706.
Zhang Y, Du W, Yang B. Long non-coding RNAs as new regulators of cardiac electrophysiology and arrhythmias: molecular mechanisms, therapeutic implications and challenges. Pharmacol Ther. 2019;203:107389.
Strack R. A clearer view of mitophagy. Nat Methods. 2020;17:656.
Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308.
Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–6.
Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85:257–73.
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298.
Shao D, Kolwicz SC, Wang P, Roe ND, Villet O, Nishi K, et al. Increasing fatty acid oxidation prevents high fat diet induced cardiomyopathy through regulating parkin mediated mitophagy. Circulation. 2020;142:983–97.
Zhang ZL, Wang NN, Ma QL, Chen Y, Yao L, Zhang L, et al. Somatic and germline mutations in the tumor suppressor gene PARK2 impair PINK1/Parkin-mediated mitophagy in lung cancer cells. Acta Pharmacol Sin. 2020;41:93–100.
Yang M, Linn BS, Zhang Y, Ren J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2293–302.
Liang W, Moyzis AG, Lampert MA, Diao RY, Najor RH, Gustafsson AB. Aging is associated with a decline in Atg9b-mediated autophagosome formation and appearance of enlarged mitochondria in the heart. Aging Cell. 2020;19:e13187.
Wang Z, Chen H, Chen J, Hong Z, Liao Y, Zhang Q, et al. Emodin sensitizes human pancreatic cancer cells to EGFR inhibitor through suppressing Stat3 signaling pathway. Cancer Manag Res. 2019;11:8463–73.
Dong X, Fu J, Yin X, Cao S, Li X, Lin L, et al. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother Res. 2016;30:1207–18.
Du C, Shi L, Wang M, Mao P, Wang J, Wei Y, et al. Emodin attenuates Alzheimer’s disease by activating the protein kinase C signaling pathway. Cell Mol Biol. 2019;65:32–7.
Cui Y, Chen LJ, Huang T, Ying JQ, Li J. The pharmacology, toxicology and therapeutic potential of anthraquinone derivative emodin. Chin J Nat Med. 2020;18:425–35.
Xiao D, Hu Y, Fu Y, Wang R, Zhang H, Li M, et al. Emodin improves glucose metabolism by targeting microRNA-20b in insulin-resistant skeletal muscle. Phytomedicine. 2019;59:152758.
Xiao D, Zhang Y, Wang R, Fu Y, Zhou T, Diao H, et al. Emodin alleviates cardiac fibrosis by suppressing activation of cardiac fibroblasts via upregulating metastasis associated protein 3. Acta Pharm Sin B 2019;9:724–33.
Zhao Y, Zhu J, Liang H, Yang S, Zhang Y, Han W, et al. Kang Le Xin reduces blood pressure through inducing endothelial-dependent vasodilation by activating the AMPK-eNOS pathway. Front Pharmacol. 2019;10:1548.
Li X, Hu X, Pan T, Dong L, Ding L, Wang Z, et al. Kanglexin, a new anthraquinone compound, attenuates lipid accumulation by activating the AMPK/SREBP-2/PCSK9/LDLR signalling pathway. Biomed Pharmacother. 2020;133:110802.
Zhao Y, Wang X, Yang S, Song X, Sun N, Chen C, et al. Kanglexin accelerates diabetic wound healing by promoting angiogenesis via FGFR1/ERK signaling. Biomed Pharmacother. 2020;132:110933.
Bian Y, Li X, Pang P, Hu XL, Yu ST, Liu YN, et al. Kanglexin, a novel anthraquinone compound, protects against myocardial ischemic injury in mice by suppressing NLRP3 and pyroptosis. Acta Pharmacol Sin. 2020;41:319–26.
Zhang Y, Liu X, Bai X, Lin Y, Li Z, Fu J, et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J Pineal Res. 2018;64:0-e12449.
Zhang Y, Sun L, Xuan L, Pan Z, Hu X, Liu H, et al. Long non-coding RNA CCRR controls cardiac conduction via regulating intercellular coupling. Nat Commun. 2018;9:4176.
Zhang Y, Jiao L, Sun L, Li Y, Gao Y, Xu C, et al. LncRNA ZFAS1 as a SERCA2a inhibitor to cause intracellular Ca2+ overload and contractile dysfunction in a mouse model of myocardial infarction. Circ Res. 2018;122:1354–68.
Liu X, Wei Y, Bai X, Li M, Li H, Wang L, et al. Berberine prevents primary peritoneal adhesion and adhesion reformation by directly inhibiting TIMP-1. Acta Pharm Sin B 2020;10:812–24.
Guo B, Guo Q, Wang Z, Shao JB, Liu K, Du ZD, et al. D-Galactose-induced oxidative stress and mitochondrial dysfunction in the cochlear basilar membrane: an in vitro aging model. Biogerontology 2020;21:311–23.
Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428–38.
Wierich MC, Schipke J, Brandenberger C, Abdellatif M, Eisenberg T, Madeo F, et al. Cardioprotection by spermidine does not depend on structural characteristics of the myocardial microcirculation in aged mice. Exp Gerontol. 2019;119:82–8.
Kim YY, Jee HJ, Um JH, Kim YM, Bae SS, Yun J. Cooperation between p21 and Akt is required for p53-dependent cellular senescence. Aging Cell 2017;16:1094–103.
Labuschagne CF, Brenkman AB. Current methods in quantifying ROS and oxidative damage in Caenorhabditis elegans and other model organism of aging. Ageing Res Rev. 2013;12:918–30.
Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17:262–75.
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–14.
Safadi SS, Shaw GS. Differential interaction of the E3 ligase parkin with the proteasomal subunit S5a and the endocytic protein Eps15. J Biol Chem. 2010;285:1424–34.
Kim HL, Lim WH, Seo JB, Chung WY, Kim SH, Kim MA, et al. Association between arterial stiffness and left ventricular diastolic function in relation to gender and age. Medicines. 2017;96:e5783.
Shah AM, Claggett B, Kitzman D, Biering-Sorensen T, Jensen JS, Cheng S, et al. Contemporary assessment of left ventricular diastolic function in older adults: the Atherosclerosis Risk in Communities Study. Circulation. 2017;135:426–39.
Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–54.
Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560–8.
Chiao YA, Kolwicz SC, Basisty N, Gagnidze A, Zhang J, Gu H, et al. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging. 2016;8:314–27.
Alfaras I, Di Germanio C, Bernier M, Csiszar A, Ungvari Z, Lakatta EG, et al. Pharmacological strategies to retard cardiovascular aging. Circ Res. 2016;118:1626–42.
Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA, Inserra F. Protective effect of long-term angiotensin II inhibition. Am J Physiol Heart Circ Physiol. 2007;293:H1351–8.
Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46.
Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S, et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017;16:564–74.
Monisha BA, Kumar N, Tiku AB. Emodin and its role in chronic diseases. Adv Exp Med Biol. 2016;928:47–73.
Ho TY, Wu SL, Chen JC, Li CC, Hsiang CY. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir Res. 2007;74:92–101.
Li Z, Lin Y, Zhang S, Zhou L, Yan G, Wang Y, et al. Emodin regulates neutrophil phenotypes to prevent hypercoagulation and lung carcinogenesis. J Transl Med. 2019;17:90.
Zhao X, Lu L, Qi Y, Li M, Zhou L. Emodin extends lifespan of Caenorhabditis elegans through insulin/IGF-1 signaling pathway depending on DAF-16 and SIR-2.1. Biosci Biotechnol Biochem. 2017;81:1908–16.
Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, et al. The mitochondrial deubiquitinase USP30 opposes Parkin-mediated mitophagy. Nature. 2014;510:370–5.
Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem. 2016;139:216–31.
Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, et al. AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res. 2018;122:712–29.
Mukherjee UA, Ong SB, Ong SG, Hausenloy DJ. Parkinson’s disease proteins: novel mitochondrial targets for cardioprotection. Pharmacol Ther. 2015;156:34–43.
Wang S, Ge W, Harns C, Meng X, Zhang Y, Ren J. Ablation of toll-like receptor 4 attenuates aging-induced myocardial remodeling and contractile dysfunction through NCoRI-HDAC1-mediated regulation of autophagy. J Mol Cell Cardiol. 2018;119:40–50.
Ren X, Chen L, Xie J, Zhang Z, Dong G, Liang J, et al. Resveratrol ameliorates mitochondrial elongation via Drp1/Parkin/PINK1 signaling in senescent-like cardiomyocytes. Oxid Med Cell Longev. 2017;2017:4175353.
Manzella N, Santin Y, Maggiorani D, Martini H, Douin-Echinard V, Passos JF, et al. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of Parkin-mediated mitophagy. Aging Cell. 2018;17:e12811.
Woodall BP, Orogo AM, Najor RH, Cortez MQ, Moreno ER, Wang H, et al. Parkin does not prevent accelerated cardiac aging in mitochondrial DNA mutator mice. JCI Insight 2019;5:e127713.
Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, et al. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 2011;30:2853–67.
Aguirre JD, Dunkerley KM, Mercier P, Shaw GS. Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc Natl Acad Sci USA. 2017;114:298–303.
Beasley SA, Safadi SS, Barber KR, Shaw GS. Solution structure of the E3 ligase HOIL-1 Ubl domain. Protein Sci. 2012;21:1085–92.
Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, et al. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI3K-Akt signalling. Nat Cell Biol. 2006;8:834–42.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81773735, 81903610, 91949130, 81961138018, and 81730012) and the National Key R&D Program of China (2017YFC1702003).
Author information
Authors and Affiliations
Contributions
BFY and YZ conceived and designed the study and wrote the paper. HML, XL and ZYM performed major experiments and analyzed the data. LW, LMZ, HC, ZXW, and XQT performed Western blotting, immunochemistry and qPCR experiments. HC, XHL, and XB performed primary cell culture and transfection experiments. YL, WNH, and HL performed animal experiments. All authors contributed to and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, Hm., Liu, X., Meng, Zy. et al. Kanglexin delays heart aging by promoting mitophagy. Acta Pharmacol Sin 43, 613–623 (2022). https://doi.org/10.1038/s41401-021-00686-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00686-5
Keywords
This article is cited by
-
Nitidine chloride induces cardiac hypertrophy in mice by targeting autophagy-related 4B cysteine peptidase
Acta Pharmacologica Sinica (2023)
-
Tetrahydroberberrubine retards heart aging in mice by promoting PHB2-mediated mitophagy
Acta Pharmacologica Sinica (2023)
