
Abstract
Ion channels are ubiquitously expressed in almost all living cells, and are the third-largest category of drug targets, following enzymes and receptors. The transient receptor potential melastatin (TRPM) subfamily of ion channels are important to cell function and survival. Studies have shown upregulation of the TRPM family of ion channels in various brain tumours. Gliomas are the most prevalent form of primary malignant brain tumours with no effective treatment; thus, drug development is eagerly needed. TRPM2 is an essential ion channel for cell function and has important roles in oxidative stress and inflammation. In response to oxidative stress, ADP-ribose (ADPR) is produced, and in turn activates TRPM2 by binding to the NUDT9-H domain on the C-terminal. TRPM2 has been implicated in various cancers and is significantly upregulated in brain tumours. This article reviews the current understanding of TRPM2 in the context of brain tumours and overviews the effects of potential drug therapies targeting TRPM2 including hydrogen peroxide (H2O2), curcumin, docetaxel and selenium, paclitaxel and resveratrol, and botulinum toxin. It is long withstanding knowledge that gliomas are difficult to treat effectively, therefore investigating TRPM2 as a potential therapeutic target for brain tumours may be of considerable interest in the fields of ion channels and pharmacology.
Similar content being viewed by others


Introduction
Ion channels are eminent therapeutic targets in the treatment of various pathophysiological conditions and currently represent the third-largest targets for drug research [1], following enzymes and receptors. Pharmacological research has shown that these proteins modulate a dynamic range of physiological processes and the dysfunction of which results in various pathologies [2]. Drugs have widespread potential as they can regulate ion channels in their ability to open or block the channels, act as receptor agonists or antagonists, or modify enzyme activities [3]. Ion channels have established their extensive capabilities in regulating cellular functions and processes and have gained potential therapeutic relevance in various pathological conditions.
Primary brain tumours are a heterogeneous group of tumorigenic cells within the central nervous system (CNS), and can be malignant or benign [4]. There are histologically over 100 distinct types of primary CNS tumours. Further classification is based on the cell type, location, and degree of malignancy [5]. Despite developments in research and medicine, interventions remain challenging [6]. The most common primary brain tumour is glioma, derived from glial cells and includes astrocytoma, ependymomas, oligodendrogliomas, and glioblastoma [7]. WHO classification of gliomas is categorized into four grades, each increasing in malignancy. Grades I–II represent low-grade and Grades III–IV represent high-grade gliomas. In adults diagnosed with glioblastoma, grade IV type by WHO classification, and one of the most aggressive forms of cancer, only 18% will survive 2 years after the initial diagnosis [8, 9]. As mentioned, the difficulty in the treatment of primary brain tumours is due to the development of resistance to current therapies such as chemoradiation and chemotherapy. Targeting ion channels in drug development may aid in enhancing therapeutic outcomes.
TRPM2 (transient receptor potential melastatin), the second member of the TRPM subfamily of channels is essential to cell survival and proliferation [10]. It is permeable to Ca2+, Na+, and K+ and is especially important to the cellular response to oxidative stress. This article will review the many roles of TRPM2 and discuss its importance and therapeutic potential as a drug target in brain tumour pathologies.
TRP family
While most major ion channel families had been identified by the late 20th century, there were clusters of sensory transduction channels, calcium-activated non-selective cation channels, and second messenger channels that had yet to be molecularly identified. With such diversity in channel properties, mechanisms of action, and regulatory function, it is quite remarkable that one major family encompasses most of said ion channels. This major ion channel family is the transient receptor potential (TRP), a polymodal superfamily of non-selective cation channels involved in widespread physiological and pathological functions [11].
Initially described in the trp-mutant fruit fly due to its transient potential to light [12], the Drosophila TRP protein was the first in the TRP superfamily to be sequenced [13]. Since the initial discovery, a multitude of TRP channel subfamilies within the TRP superfamily have been recognized: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPML (mucolipin), and TRPP (polycystic). All TRP proteins share a characteristic of six transmembrane segments (S1–S6) with a pore loop between S5 and S6 [14]. The N- and C-termini domains in the cytosol contain different functional and structural elements varying between each TRP subfamily. In the TRPC family, there are seven members, unique in structure with ankyrin repeat domains and a coiled-coil domain on the N-terminus. The C-terminal domain contains a TRP motif and a calmodulin/inositol-1,4,5-trisphosphate (Ins(1,4,5)P3) receptor-binding domain [15]. In literature, TRPC channels have been implicated in PLC pathways, and internal Ca2+ store regulation [16]. In glioblastoma patients, gene expression of TRPC6, and TRPC1 were found to be upregulated [17], with both contributing to increased migration and invasion in the progression of gliomas [18].
TRPV, family to six channels and originally sourced from capsaicin, also has N-terminal ankyrin repeat domains and a TRP motif, but differs at its pore, where there are two gate regions, unique to TRPV1, TRPV2, TRPV6, TRPA1, and TRPP2 [19]. It is widely expressed in sensory nerves, smooth muscle, and vascular endothelial cells. TRPV channels are found to have an important role in the digestive, respiratory, and cardiovascular systems [20], as well as pain perception, temperature sensation, and calcium homoeostasis [21]. Further, TRPV1 and TRPV2 have been found to be significantly upregulated in glioblastoma patients exhibiting a 79- and 31.5-fold change, respectively [17]. The inhibition of both channels resulted in a corresponding decrease in proliferation and oncogenic activity in glioblastoma [22, 23].
TRPA, which has only one member, TRPA1, contains 14–17 ankyrin repeats on the N-terminal domain, and no TRP motif on its C-terminal domain [15]. TRPA1 is found on epithelial cells, mast cells, and fibroblasts [24].
TRPML, consisting of three members, is unique in its lack of ankyrin repeats and TRP motif, with only an endoplasmic reticulum (ER) retention domain on its C-terminus [15]. The TRPML subfamily is found in the adrenal gland, bladder, placenta, lung, various immune cells, cochlear sensory cells, and melanocytes [25]. Accordingly, TRPML plays a critical role in the lysosome systems, but also in immune and inflammatory responses [25].
Finally, the TRPP subfamily has five members, and like TRPML, has an ER retention domain, and no ankyrin repeats nor a TRP motif. TRPP channels are ubiquitously expressed in vertebrates and are best known for the association of TRPP2 and autosomal dominant polycystic kidney disease [26]. However, other functions of the TRPP channels include signalling in primary cilia and tube morphogenesis [27].
Beyond the discussion of TRPM2 presented in this review, there are several other subfamilies of TRP channels that are relevant in the discussion of brain tumours. In literature, many other TRP channels have been employed as therapeutic targets for various pathological conditions and are of great relevance in the context of drug development [28].
TRPM subfamily
The TRPM (melastatin) subfamily of proteins is the most widespread, diverse, and largest group in the TRP family. There are eight members, TRPM1-8, and the majority (TRPM1-3, TRPM6-8) are Ca2+ permeable, with all members playing a critical role in the regulation of apoptosis, magnesium homoeostasis, vascular tone, and more [29]. Structurally, in comparison to the other TRP proteins, TRPM channels are the largest, spanning between 732 to 1611 amino acids in length [29]. Characteristic to most members of the TRPM subfamily are four TRPM homology region (MHR) domains on the N-terminus, six transmembrane domains, a TRP domain, and a coiled-coil domain on the C-terminus.
TRPM1, the founding member of the TRPM subfamily is found on the retinal ON-bipolar cells, and in melanocytes of the skin. It has been implicated in melanoma [30], and is regulated by the mGluR6 cascade and Gβ3 [31]. Physiological functions include the development of ON rod bipolar dendrites, melanocytogenesis, and pigmentation.
TRPM3 is primarily found in beta cells of the pancreas, the kidney, and is used for nociception. Its structure remains highly conserved to other members of the TRPM subfamily, however, there are different isoforms of TRPM3 that can alter the channel permeability to Ca2+ [32, 33].
TRPM4 and TRPM5 both exhibit similar structures to other TRPM channels and are activated by intracellular Ca2+ but differ in their heightened sensitivity to Ca2+ [34]. Both TRPM4 and TRPM5 are voltage-gated and regulated by PLC and PI(4,5)P2 [35]. Strong evidence suggests that the channel plays a role in cytokine secretion in T lymphocytes and in cerebral arterial myogenic constriction [36].
Unique to TRPM6 and TRPM7 lies a serine/threonine kinase domain, functionally allowing the channels to act as “chanzymes,” due to their dual function as both a channel and an enzyme [37]. TRPM7 is ubiquitously expressed all over the body, and is highly implicated in numerous CNS pathologies [38]. Both TRPM6 and TRPM7 are preferentially permeable to Zn2+, Mg2+, and Ca2+, but are perhaps most essential in their maintenance of physiological Mg2+ homoeostasis [39].
Finally, TRPM8 is primarily expressed in sensory neurons and in the prostate, serving a role in the regulation of Ca2+ homoeostasis, apoptosis, thermosensation, inflammation, and pain. Various disorders such as irritable bowel syndrome, migraines, cancer, and chronic cough have been linked with TRPM8 in their pathologies [40]. TRPM2, 3, 7, and 8 have all been found to be upregulated in patients with glioblastoma. TRPM8 had an 1850-fold increase in gene expression in patients with glioblastoma and has thus been heavily studied in the literature in the context of brain tumours [17]. TRPM2 gene expression increased 57.5-fold, while TRPM7 showed a 17.5-fold increase [17]. However, while TRPM7 has recently gained traction as a potential therapeutic target for brain tumours [41,42,43,44], the literature surrounding TRPM2 in the same context remains limited. As reviewed here, the TRPM family holds great significance and has gained recent recognition as promising therapeutic targets due to their extensive involvement in physiological function and pathologies.
TRPM2 channel
The second member of the TRPM subfamily to be cloned, TRPM2 (Fig. 1), is a non-selective cation channel permeable to Na+, K+, and Ca2+. First described by Nagamine et al. [45], it has been previously known as TRPC7 and LTRPC and is widely distributed in the CNS. It is highly expressed in neurons, dendritic cells [46], astrocytes, and microglia [45, 47,48,49], as well as immune cells including macrophages [50], neutrophils [51], lymphocytes [52], and monocytes [53]. TRPM2 has also been detected in peripheral tissues such as the liver, pancreas, heart, and lung [49]. This widespread expression reflects the broad involvement of TRPM2 in a multitude of physiological processes and pathologies. Furthermore, its defining characteristic lies in its NUDT9-H domain [48]. TRPM6 and TRPM7, as mentioned earlier, and in addition, TRPM2, are the only TRP channels that contain an enzyme domain in the C-terminus. The NUDT9-H domain in TRPM2 holds a similar homology to human nucleoside diphosphate linked moiety X type (Nudix) hydrolase motif 9 (NUDT9). ADPR, a substrate of NUDT9, is also an agonist for TRPM2, which suggests that the channel has a critical role in the modulation of oxidative stress [54].

The structure includes 4 N-terminal MHR domains, six transmembrane helices labelled S1–S6, a TRP domain, a C-terminal coiled-coil domain, and a ~270 residue NUDT9-H domain on the C-terminus which interacts with ADP-ribose (ADPR).
The human TRPM2 gene is situated on chromosome 21q22.3, consisting of 32 exons, spanning 90 kb. There are multiple alternative splicing isoforms of TRPM2 expressed in specific regions within the body [55]. Structurally, like the other members of the TRPM subfamily, TRPM2 shares the characteristic homology of an N-terminal region (MHR1-4) [48], a coiled-coil domain, and a TRP motif on the C-terminus. The TRPM2 channel protein also includes six transmembrane domains (S1–S6), with the pore loop located between domains S5 and S6, and an ion channel located at residues 762–1048, out of a total of 1503 [56]. The Glu-960, Gln-981, Asp-987, and Glu-1022, contribute to the channel’s divalent cation permeable properties, with the first three residues especially critical to Ca2+ permeability [57]. The regulation of Ca2+ in TRPM2 is also based in the calmodulin (CaM) binding IQ-like motif found in the 700 amino acid length N-terminus [56]. Conserved among the members of the TRPM subfamily are the Cys-996 and Cys-1008 residues, which are located around the pore region of TRPM2. Findings from site-directed mutagenesis have revealed the necessity of these cysteine residues in channel activation by ADPR, and thus, proper channel functioning [58].
TRPM2 modulation and signalling
TRPM2 signalling (Fig. 2) under oxidative stress primarily centres around poly (ADP-ribose) polymerase (PARP), especially PARP1. Activation of PARP/poly (ADP-ribose) glycohydrolase (PARG) pathways in response to ROS leads to the production of NAD+ metabolite, ADPR. Accumulated ADPR move freely in the cytosol and act on TRPM2’s NUDT9-H domain, triggering Ca2+ influx into the cell. Specifically, the NUDT9-H domain senses ADPR and upon binding, rotates 27° with the MHR1/2 domains priming the channel to open. TRPM2 then undergoes a 15° rotation in the cytosolic domains, causing the TRP motif to tilt, and twisting open the S6 gating domain to result in channel opening [59]. While primarily regulated by ADPR, some researchers suggest that NUDT9-H may be activated by ROS independently of ADPR [60, 61], although the pathways are not postulated to be mutually exclusive. Finally, TRPM2 currents are also regulated by intracellular Ca2+ which is dependent on the CaM-binding IQ-like motif [62].

a Increased reactive oxygen species due to tumour necrosis factor-α (TNF-α), induces caspase 8-dependent mitochondrial ROS generation. b ROS generation in the mitochondria induces poly (ADPR) polymerase-1 (PARP1) activation, ADPR production, and Ca2+ entry across the plasma membrane through the TRPM2 channel. In a similar fashion, H2O2 and oxidative stress result in the activation of PARP1, which in turn produces ADPR. ADP-ribose binds to the NUDT9-H domain allowing for Ca2+ entry across the plasma membrane, which overactivates the caspase 3 and 9 cascades and ultimately triggers apoptosis.
Activation
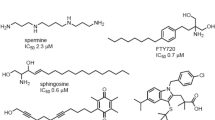
Due to the broad distribution profile and varied physiological roles of TRPM2, there are several external stimuli involved in its activation. Some controversy is presented in the literature regarding the activation of TRPM2, especially regarding nucleotide activation [63]. Generally, ADPR is considered the TRPM2 agonist with the highest affinity (EC50 of 10–80 μM) [48]. Other external activators include tumour necrosis factor-α (TNFα) [64], ROS [65], Zn2+ [66], concanavalin A [67], amyloid β peptide [68], paclitaxel and resveratrol [69], docetaxel and selenium [70], and hydrogen peroxide (H2O2) [71]. However, most of these act through ADPR as their mechanism of activation. Other more controversial activators presented in the literature include, cyclic ADPR (cADPR) [63], nicotinic acid adenine dinucleotide (NAAD), NAAD-phosphate (NAADP) [72], and nicotinamide-adenine dinucleotide (NAD) [61]. As some of these activators function at a much lower affinity, their direct role in TRPM2 activation has been disputed. Finally, other factors of activation depend on intracellular Ca2+ to sensitize TRPM2 to ADPR through the interaction with CaM, and are also able to gate TRPM2 without ADPR [62] in its spliced isoforms [73].
Inhibition
In a similar fashion, there are a plethora of agents that inhibit and target TRPM2. Predominant inhibitors that have been discussed in the literature include: N-(p-amylcinnamoyl) anthranilic acid (ACA), clotrimazole, econazole, flufenamic acid (FFA), compounds 7i and 8a, scalaradial, 2-aminoethoxydiphenyl borate (2-APB), AG490/555/556, curcumin, tat-M2NX, low extracellular pH and glutathione (GSH). KO mice models of depleted TRPM2 are viable, but exhibit significant changes in oxidative stress signalling [55]. ACA has previously been used in the context of phospholipase A2 (PLA2), but has been found to completely block the ADPR- and H2O2-induced TRPM2 currents (IC50 = 1.7 μM) in HEK293 cells [74]. Similarly, 2-aminoethoxydiphenyl borate (2-APB) induced rapid and reversible inhibition of the channel and could be modulated by heat [75]. AG490/555/556 were found by Shimizu et al., to inhibit the H2O2-induced TRPM2 Ca2+ entry, which was later confirmed by Li et al. using AG490 in mouse models in vivo [76, 77]. Closely related compounds, like AG555 and 556, were also shown to inhibit TRPM2 [76,77,78]. Clotrimazole and econazole are both antifungal agents that inhibit TRPM2 channels in concentrations ranging from 3 to 30 μM, with the latter completely inhibiting the ADPR-activated currents in HEK293 cells [79]. FFA is a non-steroid anti-inflammatory agent, found to inhibit TRPM2 currents at 50 to 1000 μM, and was shown to be influenced by the surrounding pH [80]. Furthermore, a low extracellular and intracellular pH was found to inhibit TRPM2 at an IC50 of pHo = 5.3, and IC50 of pHi = 6.7, respectively [81]. Antioxidants like glutathione and curcumin were also demonstrated to inhibit TRPM2 and regulate the oxidative stress within a cell [82]. Recently, Luo et al. were able to synthesize two new analogues to inhibit TRPM2 selectively: compounds 7i and 8a with an IC50 of 5.7 and 5.4 μM, respectively. The compounds are specific to TRPM2, selectively inhibiting TRPM2 over close homologues like TRPV1, TRPV3, TRPM7, and TRPM8 [83]. Scalaradial, a scalarane sesterterpenoid extracted from Cacospongia mollior, has an anti-inflammatory profile and was also found to inhibit TRPM2 Ca2+ influx [84]. Most recently, tat-M2NX had been discovered to inhibit 90% of TRPM2 currents at 2 μM, acting through interactions with the NUDT9-H domain. Although still in its early stages of characterization, tat-M2NX has been proven to be an incredibly potent TRPM2 antagonist, with great potential as a novel instrument to study TRPM2 mechanisms within the cell, and an opportunity to be utilized in numerous pathologies [85].
TRPM2 in CNS diseases
TRPM2 is primarily localized and expressed in the peripheral immune system where it is critical to the activation and recruitment involved in the inflammatory response. Previous experiments have indicated differing levels of cytokines, IFNγ and IL-12, and a compromised immune system in TRPM2 KO mice [86]. Further, as TRPM2 has essential functions in mediating oxidative stress within a cell, it has been found to be highly expressed in various cancers all over the body, including melanoma, leukaemia, breast, prostate, and pancreatic cancers [87]. Within the CNS, TRPM2 mRNA is amongst the most expressed out of all the TRP channels [49]. As a result, a rising amount of evidence supports the importance of TRPM2 in the physiology and pathology of the CNS [88]. TRPM2 is expressed in various regions of the CNS, including the spinal cord, cortex, striatum, hippocampus, and substantia nigra [88, 89].
Stroke is an extremely complex condition wherein ischemia and/or hypoxia triggers the activation of various excitotoxic cascades, causing lasting and varying levels of impairment in patients [90]. Repair and recovery from periods of ischemia and hypoxia are heavily dependent on the regulation of inflammatory cascades and apoptosis [91]. Given the postulated role of TRPM2 in these two processes, along with Ca2+ and Zn2+ regulation, and oxidative stress, TRPM2 represents a potential drug target in the treatment of stroke. Accordingly, various inhibitors of TRPM2, such as ACA, have been shown to promote protective effects against ischemia [92].
Neuronal excitotoxicity in neurodegenerative diseases has also classically been studied using models focused on the glutamate N-methyl-D-aspartate receptors (NMDARs), to very little avail. The search for therapeutic targets has since moved to focus on mediating the downstream effects of NMDAR activation [93] as well as Ca2+ signalling, relevant to TRPM2 channels. Dysregulation of Ca2+ homoeostasis contributes to AD, PD, and Huntington’s disease (HD) [94], as well as the physiological process of aging [95]. With age, GSH [96], a TRPM2 inhibitor, decreases and in turn, increases [Ca2+]i and oxidative stress, causing the internal environment of the cell to become toxic [97, 98]. Inflammation has also been implicated in the development and severity of AD [99], PD [100], and HD [101]. As TRPM2 is expressed in astrocytes and microglia, responds to oxidative stress, and activates downstream cascades such as JNK, NF-κB, and p38 MAPK, it has been suggested to play a role in the progression of these neurodegenerative pathologies [102].
TRPM2 in brain tumours
Existing literature has shown that TRPM2 is found to be upregulated in gliomas [17, 103]. Experiments measuring TRP gene expression levels with qRT-PCR, showed a significant increase in TRPM2 gene expression, especially in older patients with higher-grade gliomas (Grades III-IV) [17]. In U251 and U87 cell lines, models of human primary glioblastoma, the overexpression of TRPM2-AS is met with increased proliferation, invasion, and migration, hallmark characteristics of cancer malignancy [103]. The same characteristics were suppressed when TRPM2-AS was downregulated using siRNA [103]. Further, TRPM2-AS expression directly corresponded with levels of regulator of G protein signaling 4, a tumour promoter [103]. Another experiment studying the gene expression levels of the TRP ion channel proteins in GBM patients found that TRPM2 gene expression was significantly higher, showing a 57.5-fold change compared to controls [17]. Together, both studies establish TRPM2 as, not only a significant contributor to the hallmark characteristics seen in cancer cells, but also a significantly upregulated ion channel in glioma patients [17]. With TRPM2’s role in ROS regulation, antioxidants, oxidants, and various chemotherapeutic agents have been investigated as potential therapies. The following sections review various pharmacological agents and their interactions through TRPM2 in the context of brain tumours.
Hydrogen peroxide and ROS
Various studies have begun the process of exploiting the characteristics and mechanisms of TRPM2 as a potential therapeutic target for brain tumours. It had been previously established that H2O2 induces a robust increase in Ca2+ influx via the TRPM2 channel of microglia [47]. In addition, activation of TRPM2 via ROS eventually causes the cell to progress to necrotic cell death. Early experiments used this characteristic of TRPM2 to selectively induce cell death with the insertion of TRPM2 in A172 human GBM cells [47, 71]. Ishii et al. found that TRPM2 transfected cells treated with 0.1–5 mM H2O2, could enhance cell death [71]. This was accomplished by creating a plasmid construct with human TRPM2 cDNA mixed into the solution where the A172 cells were electroporated. Transfection of the TRPM2 plasmid construct into the A172 cells was confirmed through RT-PCR, while cell death was assessed through a Trypan blue exclusion assay. The results showed that cell death was mediated by the TRPM2 channels that had been transfected into A172 cells, while cell proliferation, invasion, and migration were surprisingly unaffected. This experiment confirmed that TRPM2 was activated by ROS and that inducing cell death with H2O2 may provide an alternative treatment method without exacerbating the malignancy in GBM cells. H2O2 is a model system for experimental oxidative stress, and was confirmed as an activator for TRPM2 in this experiment. These experiments collectively provided evidence that the evoked Ca2+ overload was derived from TRPM2 activation [60]. Thus, targeting TRPM2 activation in order to mediate cell death in pathological cells is a promising therapeutic direction for cancer that other labs have since begun to explore.
Curcumin
Turmeric is a household common spice that had been traditionally used for its medicinal properties. The natural polyphenol sourced from turmeric and where it derives its medicinal capabilities is curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione). Curcumin has been more recently investigated in its use as an anti-inflammatory [104], antioxidant [105], and chemoprotective agent [106]. Curcumin is sourced from rhizomes of Curcuma longa L. (turmeric) and similar species [107]. Previously, curcumin has demonstrated protective properties in various neurodegenerative diseases, and neuropsychiatric disorders [108]. Tumour suppressive effects of curcumin had previously been identified to show oncoprotection on A1207 human glioma cells [109]. The mechanism of which was shown to be through the NEDD4 E3 ubiquitin ligase [109], transcriptional activation of which is modulated through ROS [110]. While it had been established that the TRPM2 channel was independently activated by oxidative stress, leading to cell death, less literature had studied if an antioxidant could regulate or inhibit TRPM2 activity. However, curcumin’s ability to modulate ion channels and transporters has been established [111], with literature demonstrating the effects of curcumin and its analogues on other TRP proteins, including activation of TRPA1, and inhibition of TRPM8 [112] and TRPV1 [113]. Öz and Çelik tested the effects of curcumin on neuroblastoma cell lines, measuring TRPM2 inhibition/activation with electrophysiology, as well as the effects on PARP1, caspase 3, and 9 levels via Western blotting [114]. Neuroblastoma cells were transfected to overexpress TRPM2 [114, 115]. Experimental groups were separated into controls and those incubated in media containing concentrations of curcumin. Since curcumin had previously been known to have antioxidant effects, action through the TRPM2 channel was confirmed through whole-cell recordings, and compared to a known TRPM2 antagonist ACA [74]. Experimenters were able to show that curcumin inhibited the TRPM2 currents; when triggered by H2O2, TRPM2 current density was significantly reduced in the curcumin-incubated TRPM2-transfected groups and further, curcumin was found to attenuate the H2O2-induced currents more than ACA (Fig. 3). By extension, intracellular free calcium concentrations were also significantly reduced by curcumin treatment. In various assays, the curcumin-incubated TRPM2-transfected group also showed reduced levels of procaspase 9 and 3, as well as reduced ROS production and mitochondrial membrane potential (MtMP) levels compared to the curcumin-free TRPM2-transfected groups. Previous literature regarding curcumin in rat hepatocytes showed that ADPR-induced TRPM2 cationic currents were inhibited by curcumin [116], which suggests that ADPR is implicated in this pathway as well. In the end, it was concluded that curcumin was able to partially rescue the severity of ROS and oxidative stress through inhibition of the TRPM2 channel.

Curcumin decreased PARP1 expression and altered caspase 3 and 9 activities. TRPM2 inhibition was confirmed through measuring TRPM2 currents in response to H2O2 in curcumin-transfected cells. The effect of curcumin was compared to N-(p-amylcinnamoyl) anthranilic acid (ACA), a known TRPM2 inhibitor. Curcumin was proved to block more H2O2-induced currents compared to ACA.
Most recently, curcumin was used in combination with cisplatin, a chemotherapeutic agent well established in the literature and used in a variety of cancer treatments. Cisplatin is a taxane that had previously been used to treat neuron metastasis, breast, and prostate cancers [117]. The combination of curcumin and cisplatin showed that curcumin enhanced cisplatin-induced cancer cell death via TRPM2 in human laryngeal squamous cancer (HEP2) cells [118]. Curcumin could enhance the effects of cisplatin even in drug-resistant conditions, thus proving a worthy combination drug therapy for brain tumours as well. In SH-SY5Y cells, pairing the compounds prevented one of the most common side effects of cisplatin, severe visual loss [119]. TRPM2 was confirmed in experiments measuring the fluorescence intensity of Ca2+, where the intensity was significantly lower in the cisplatin and curcumin group compared to an increase in Ca2+ fluorescence intensity in the cisplatin-only group. Vision loss occurred through the excessive production of ROS, implicating the activation of TRPM2. However, experimenters found that curcumin was able to help reduce the detrimental effects of cisplatin on the optic nerve via prevention of oxidative stress-induced cell death [120]. The combination was proved to be beneficial in reducing the potential negative side effects of chemotherapeutic agents. Similar dysregulation of redox status is highly characteristic of brain tumours like GBM. Redox therapeutics have been proposed to reduce oxidative stress and to prevent further damage and mutation to genomic and mitochondrial DNA, eventually leading to gliomagenesis [121]. Therapeutic agents that are able to control the redox status would be a favourable treatment.
Docetaxel and selenium
While maintenance of redox status would stand to be advantageous in some scenarios, there is a delicate balance between oxidants and antioxidants in brain tumours. In the context of cancer, where activation of TRPM2 can increase oxidative stress, inhibition of TRPM2 to rescue the redox status, as exemplified with curcumin, may also be applicable. While it may be important to have antioxidants in the case of prevention and treatment of healthy cells, drug treatments that increase ROS may be advantageous in inducing cell death in pathological cells [122]. Antioxidants and oxidants may act as inhibitors or inducers of tumorigenesis, respectively. Since TRPM2 has a critical role in ROS status management, both antioxidants and oxidants have been investigated as potential therapeutic treatments in the context of brain cancer. Docetaxel, a semisynthetic taxane, is a well-established anti-cancer agent within the literature that is most often used to promote apoptosis and cell cycle arrest by binding to β-tubulin and promoting aggregation of intracellular microtubules [123]. Docetaxel and other taxanes are often combined and used as chemotherapeutic agents, like in the example of curcumin and cisplatin. One commonly paired with docetaxel is selenium, an essential micronutrient, and has anticarcinogenic and chemoprotective properties. Selenium has been found to have an inverse relationship with cancer mortality in women with breast cancer [124]. Similarly, it had been previously found that there were lower than average levels of selenium in brain tumour patients [125]. Further, it was found that selenium could reduce the growth and viability of human GBM cells in vitro [126]. The combination of selenium and docetaxel has been used to treat human breast cancer cell lines where it was reported to inhibit proliferation and induce apoptosis in vitro [127]. While docetaxel alone has been used to treat prostate cancer and GBM, it occasionally can have adverse side effects such as neurotoxicity and nephrotoxicity. Since taxanes are lipophilic compounds, adipose tissue is much more sensitive [128]. Selenium and its co-factor glutathione peroxidase as an antioxidant supplement, have previously been used to attenuate the hypothesized mechanism of toxicity, excessive mitochondrial ROS [129]. The combination of selenium and docetaxel had previously been investigated in the TRPV1 channel and its role in mediating neuropathic pain. Like TRPM2, TRPV1 is also activated by ROS [130] and is upregulated in brain tumour patients. In addition, it had also been implicated in the pathway of cisplatin-induced neuropathic pain [131]. Experiments using both selenium and docetaxel, found that selenium acted through TRPV1 to attenuate the docetaxel-induced damage to neurons.
Ertilav et al. were able to investigate the mechanisms of selenium and docetaxel in the context of human GBM cells [70]. Docetaxel had previously been tested and shown to regulate other members of the TRP family of proteins in the context of neuropathic pain [132]. TRPM2 is likely involved in proliferation, as it is critical to Ca2+ regulation. In a similar fashion to that in the curcumin cancer study, it had yet to be investigated if selenium could potentially attenuate the adverse neurotoxic effects of docetaxel and thus provide a more efficient chemotherapeutic treatment (Fig. 4). Ertilav et al. split the study groups into separate selenium and docetaxel concentrations, a control, and one with a combination of both docetaxel and selenium. Experimenters measured various characteristics such as viability, cell number, apoptosis via phosphatidylserine residue detection, ROS, MtMP (JC1), PARP activity, and TRPM2 activation. Cell numbers and viability decreased in the docetaxel and selenium combined treatment groups, whereas apoptotic indicators showed the inverse tendency, presenting significantly higher levels in the combination group. To confirm the mechanism of action was through TRPM2, experimenters measured the fluorescence intensity of Ca2+. Intracellular Ca2+ fluorescence intensity was increased by selenium and docetaxel following TRPM2 activation. TRPM2 activation increased ROS, mitochondrial depolarization, and PARP activity compared to the ACA treatment. Further, the side effects of docetaxel were alleviated by the addition of selenium. As mentioned, adjuvant therapies are useful in the case where patients build resistance to individual chemotherapeutic agents [133]. This experiment supported that notion while also providing much needed insight into the synergistic mechanisms of action of the combination of docetaxel and selenium in cancer therapeutics. Changes in PARP levels, NADH, ADPR, and TRPM2 activation corresponded with the current understanding of the TRPM2 signalling pathway. In summary, this experiment confirmed the benefits of the combination of docetaxel and selenium, but also demonstrated their promise as chemotherapeutic agents, while validating TRPM2 as an effective target in the treatment of brain tumours.

Together, acting primarily through the generation of excessive mitochondrial ROS, docetaxel and selenium inhibited cell proliferation in glioblastoma cell lines. This combination also induced a significant increase of PARP activity.
Paclitaxel and resveratrol
Resveratrol (trans-3,4′,5-trihydroxystilbene) is a well-known polyphenol that has gained recognition in the literature over the past few decades for its benefits against cancer and cardiovascular diseases [134]. It is a naturally occurring stilbene polyphenol and like curcumin, also possesses various anticarcinogenic, and antioxidant properties. One of the most popularized main sources of resveratrol is within red wine [135]. Due to its poor solubility and potential for adverse side effects, it stands as one of the more controversial popular antioxidants [136]. In literature, resveratrol has been investigated to modulate certain TRP family channels, such as TRPA1 in neurons [137]. Paclitaxel is a potent anti-cancer drug discovered in the late 1970s and is another taxane similar to docetaxel [138]. The mechanisms of action are alike as well, disrupting microtubule dynamics and effectively inducing cell cycle arrest and apoptosis, and has been used in the contexts of ovarian, lung, and breast cancers [139,140,141]. Paclitaxel had been previously found to modulate TRPA1, TRPV4, TRPM8, TRPV1, and TRPV2 channels [74, 142]. The combination of paclitaxel and resveratrol as chemotherapeutic agents has shown mixed results in forms of cancer. While it had been previously established in the literature that resveratrol could sensitize cancer to other drugs, Fukui et al. had found that in contrast, resveratrol had strongly attenuated the effects of paclitaxel in the treatment of human breast cancer [143]. More recently, however, studies have shown the benefit of combining resveratrol and paclitaxel treatments, and in the same human breast cancer line, it was found that resveratrol augments the effects of paclitaxel, making it especially relevant for paclitaxel-resistant breast cancer cells [144]. In the context of brain cancers, mixed findings have also been reported. Nicolini et al. showed that trans-resveratrol had attenuated the apoptotic effects of paclitaxel in neuroblastoma cells [145].
The combination of resveratrol and paclitaxel was studied in DBTRG glioblastoma cells by Öztürk et al. who were able to show that in a mechanism involving TRPM2, resveratrol enhances the effects of paclitaxel (Fig. 5) [69]. Via increasing the intracellular ROS levels and inducing dysfunction in the mitochondria, resveratrol and paclitaxel further increased apoptosis, which suggests the potential of TRPM2 in regulating GBM apoptosis. Study groups were divided into control, one without ACA, ADPR, paclitaxel only, resveratrol only, and one group with both paclitaxel and resveratrol. TRPM2 function was measured using whole-cell patch-clamp recordings and Ca2+ fluorescence intensity. Using whole-cell patch-clamp recordings, experimenters found that TRPM2 current density was highest in the paclitaxel, resveratrol, and ADPR group and was significantly higher than the control and ADPR groups. The current density was severely reduced with the addition of ACA, which confirms paclitaxel and resveratrol can induce TRPM2 currents. Caspase 3 and 9, and the MtMP were also assessed. TRPM2 currents increased in a stepwise manner with the addition of paclitaxel and then in combination with resveratrol. Similarly, the levels of apoptosis, caspase 3 and 9 activities, and mitochondrial depolarization increased progressively with the addition of paclitaxel alone but were further increased in the presence of both [69]. Adjuvant therapy with resveratrol therefore proved to be a promising potential treatment especially in cases of chemotherapy resistance. In addition, this study revealed that paclitaxel and resveratrol can work synergistically to increase apoptosis and oxidative stress via activation of the TRPM2 channel. One limitation, however, lies in the fact that resveratrol has low bioavailable forms for clinical use [146], a drawback that can be addressed in future developments.

Paclitaxel, a chemotherapeutic agent, induces mitochondrial ROS generation, increasing the production of ADPR and resulting in TRPM2 activation. Resveratrol, a well-known antioxidant, is proposed to act through proliferator-activated receptor-gamma coactivator-1 (PGC-α) to increase mitochondrial membrane depolarization. Together, the effects of paclitaxel and resveratrol in cancer cells are apoptotic and prooxidant.
Clostridium botulinum neurotoxin A
Botulinium neurotoxins (BTXs) are produced by strains of bacteria of the genus Clostridium (Clostridium botulinum, Clostridium butyrricum, Clostridium barati, and Clostridium argentinensis) [147]. C. botulinum has seven serotypes (A–G). There are a wide variety of applications of Botulinium neurotoxins type A (BTX-A) and B (BTX-B) in the clinical context, where it has been used to treat the spasticity of multiple sclerosis, stroke, cerebral palsy, and spinal cord injuries [148]. BTX-A have recently gained traction as potential treatments used in cancer-related pain [149], and in vitro cultures of cancer cell lines have shown BTXs to promote apoptosis, and slow the mitotic activity of cells [150]. Only within the past few years has BTX-A shown anti-cancer potential. In prostate cancer cells (DU145), BTX-A elevated the expression levels of p53, suggesting anticarcinogenic properties to trigger apoptosis in cancer cells [151]. Botulinum neurotoxin type C (BTX-C) has been studied in brain tumours, where BTX-C proteases coupled to neuroblastoma antibodies could selectively induce apoptosis in differentiated neuroblastoma cells [152].
Akpinar et al. tested the therapeutic potential of BTX-A in DBTRG glioblastoma (Fig. 6), and SH-SY5Y cells. The experimenters suggested that BTX and its substrate (RAC1) induced excessive ROS production, triggering excessive Ca2+ influx via TRPM2 activation, and ultimately leading to mitochondrial breakdown and cell death. First, inhibition of the TRPM2 channels with ACA and 2-APB increased cell viability, whereas the BTX group showed significantly increased cell death. Next, measurements of Ca2+ fluorescence intensity were used to confirm the involvement of TRPM2. The BTX-treated group showed the highest fluorescence intensity compared to both the control and H2O2-treated groups, while the addition of ACA significantly reduced the fluorescence intensity. In addition, BTX induced increases of caspase 3 and 9 activities that were attenuated with ACA and 2-APB. Laser confocal microscopy measuring ROS generation showed that BTX induced increases in mitochondrial membrane depolarization, and ROS in both the mitochondria and cytosol [153]. These results are more controversial as this would be the first experiment showing that BTX directly elevated cytosolic ROS. In contrast, previous literature suggests that BTX-A exerts protective effects after ischemia in mouse models [154] via prevention of ROS accumulation in vascular endothelial cells [154]. While the results of this study are unique compared to the surrounding literature of BTXs, it implicates TRPM2 in BTX-mediated mitochondrial and cytosolic ROS generation, and neuronal death.

BTX acts through increasing mitochondrial ROS and inducing a corresponding increase in caspase 3 and 9 activities. The mechanism where this is achieved was not identified in the experiments, however, it likely follows the PARP pathway in TRPM2 function.
The studies discussed above have tested the potential of certain drugs acting through TRPM2 to regulate activity in brain tumour cells. However, only a few of the known TRPM2 inhibitors have been tested in cancer contexts, and even fewer in the context of brain tumours. As mentioned earlier, there are several non-specific TRPM2 inhibitors such as clotrimazole, PLC inhibitor FFA, 2-APB, and ACA [155]. While current literature holds promise for the profile of TRPM2 as a therapeutic target, future development into drug pharmacokinetic/dynamic properties is required for more clinical significance in targeting TRPM2.
Conclusion and future directions
Ion channels are the third-largest group in drug development. Although medicine has significantly progressed over the past few decades in the treatment of brain tumours, employed treatments still lack the efficacy and specificity required for optimal expected patient outcomes. While it has been known that TRPM2 is widely spread in its involvement in physiological processes, there is increasing evidence suggesting TRPM2 to be a potential target in a wide variety of CNS diseases, including brain tumours. TRPM2 has been found to be upregulated in various gliomas and is broadly expressed in the CNS. Thus, TRPM2 may provide more specificity as a target for drug development depending on the cell types. Further, TRPM2 has been implicated in the mechanisms of well-established pre-existing chemotherapeutics like taxanes and naturally derived anti-cancer agents, thus proving the requisite for more extensive prospective studies exploiting TRPM2 in a therapeutic context and investigating the processes that modulate such effects. Studies reviewed in this article suggest that TRPM2 has a multitude of functions in cancer cells, ranging from preserving viability, increasing the sensitivities to chemotherapies, or inducing cell cycle arrest and apoptosis, all of which represent potential targets for novel therapeutic approaches. The dual nature of TRPM2 in its essential ability to regulate or exacerbate the redox status of a cell also highlights its potential in controlling the health of a cell. TRPM2 inhibitors and activators alike therefore offer promising results in future pre-clinical evaluations of brain tumour treatment.
References
Dabrowski MA, Dekermendjian K, Lund PE, Krupp JJ, Sinclair J, Larsson O. Ion channel screening technology. CNS Neurol Disord Drug Targets. 2008;7:122–8.
Ashcroft FM. Ion channels and disease. Oxford: Academic Press; 1999.
Kaczorowski GJ, McManus OB, Priest BT, Garcia ML. Ion channels as drug targets: the next GPCRs. J Gen Physiol. 2008;131:399–405.
Lapointe S, Perry A, Butowski NA. Primary brain tumours in adults. Lancet. 2018;392:432–46.
Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro-Oncol. 2019;21:v1–v100.
Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol. 2019;16:509–20.
Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117.
Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–22.
Poon MTC, Sudlow CLM, Figueroa JD, Brennan PM. Longer-term (≥2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: a systematic review and meta-analysis. Sci Rep. 2020;10:11622.
Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217.
Akopian A. Role of TRP ion channels in physiology and pathology. Semin Immunopathol. 2016;38:275–6.
Cosens DJ, Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature. 1969;224:285–7.
Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–23.
Phillips AM, Bull A, Kelly LE. Identification of a Drosophila gene encoding a calmodulin-binding protein with homology to the trp phototransduction gene. Neuron. 1992;8:631–42.
Moran MM, McAlexander MA, Bíró T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10:601–20.
Bavencoffe A, Zhu MX, Tian J-b. New aspects of the contribution of ER to SOCE regulation: TRPC proteins as a link between plasma membrane ion transport and intracellular Ca2+ stores. In: Groschner K, Graier WF,Romanin C, editors. Store-operated Ca²+ Entry (SOCE) pathways: emerging signaling concepts in human (Patho)physiology. Cham: Springer International Publishing; 2017. p. 239–55.
Alptekin M, Eroglu S, Tutar E, Sencan S, Geyik MA, Ulasli M, et al. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumor Biol. 2015;36:9209–13.
Li S, Ding X. TRPC channels and glioma. In: Wang Y, editor Transient receptor potential canonical channels and brain diseases. Dordrecht: Springer Netherlands; 2017. p 157–65.
Samanta A, Hughes TET, Moiseenkova-Bell VY. Transient receptor potential (TRP) channels. Subcell Biochem. 2018;87:141–65.
Du Q, Liao Q, Chen C, Yang X, Xie R, Xu J. The role of transient receptor potential vanilloid 1 in common diseases of the digestive tract and the cardiovascular and respiratory system. Front Physiol. 2019;10:1064.
Niemeyer BA. Structure-function analysis of TRPV channels. Naunyn-Schmiedeberg’s Arch Pharmacol. 2005;371:285–94.
Santoni G, Amantini C, Maggi F, Marinelli O, Santoni M, Nabissi M, et al. The TRPV2 cation channels: from urothelial cancer invasiveness to glioblastoma multiforme interactome signature. Lab Investig. 2020;100:186–98.
Nersesyan Y, Asuthkar S, Velpula K, Sun X, Demirkhanyan L, Zakharian E. Role of TRPV1 channels in glioma cell viability and survival. Biophys J. 2015;108:124a.
Zygmunt PM, Högestätt ED. TRPA1. Handb Exp Pharmacol. 2014;222:583–630.
Santoni G, Santoni M, Maggi F, Marinelli O, Morelli MB. Emerging role of mucolipins TRPML channels in cancer. Front Oncol. 2020;10:659.
Semmo M, Köttgen M, Hofherr A. The TRPP subfamily and polycystin-1 proteins. Handb Exp Pharmacol. 2014;222:675–711.
Zhou J. Polycystins and primary cilia: primers for cell cycle progression. Annu Rev Physiol. 2009;71:83–113.
Nishida M, Kuwahara K, Kozai D, Sakaguchi R, Mori Y. TRP channels: their function and potentiality as drug targets. In: Nakao K, Minato N, Uemoto S, editors. Innovative medicine: basic research and development. Tokyo: Springer; 2015. p. 195–218.
Huang Y, Fliegert R, Guse AH, Lü W, Du J. A structural overview of the ion channels of the TRPM family. Cell Calcium. 2020;85:102111.
Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, et al. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 1998;58:1515–20.
Koike C, Obara T, Uriu Y, Numata T, Sanuki R, Miyata K, et al. TRPM1 is a component of the retinal ON bipolar cell transduction channel in the mGluR6 cascade. Proc Natl Acad Sci USA. 2010;107:332–7.
Lee N, Chen J, Sun L, Wu S, Gray KR, Rich A, et al. Expression and characterization of human transient receptor potential melastatin 3 (hTRPM3). J Biol Chem. 2003;278:20890–7.
Frühwald J, Camacho Londoño J, Dembla S, Mannebach S, Lis A, Drews A, et al. Alternative splicing of a protein domain indispensable for function of transient receptor potential melastatin 3 (TRPM3) ion channels. J Biol Chem. 2012;287:36663–72.
Ullrich ND, Voets T, Prenen J, Vennekens R, Talavera K, Droogmans G, et al. Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium. 2005;37:267–78.
Rohacs T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv Biol Regul. 2013;53:341–55.
Liman E. The Ca2+-activated TRP channels: TRPM4 and TRPM5. TRP Ion Channel function in sensory transduction and cellular signaling cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007. p. 203–11.
Ferioli S, Zierler S, Zaißerer J, Schredelseker J, Gudermann T, Chubanov V. TRPM6 and TRPM7 differentially contribute to the relief of heteromeric TRPM6/7 channels from inhibition by cytosolic Mg2+ and Mg·ATP. Sci Rep. 2017;7:8806.
Abumaria N, Li W, Clarkson AN. Role of the chanzyme TRPM7 in the nervous system in health and disease. Cell Mol Life Sci. 2019;76:3301–10.
Cabezas-Bratesco D, Brauchi S, González-Teuber V, Steinberg X, Valencia I, Colenso C. The different roles of the channel-kinases TRPM6 and TRPM7. Curr Med Chem. 2015;22:2943–53.
Liu Y, Mikrani R, He Y, Faran Ashraf Baig MM, Abbas M, Naveed M, et al. TRPM8 channels: a review of distribution and clinical role. Eur J Pharmacol. 2020;882:173312.
Wong R, Gong H, Alanazi R, Bondoc A, Luck A, Sabha N, et al. Inhibition of TRPM7 with waixenicin A reduces glioblastoma cellular functions. Cell Calcium. 2020;92:102307.
Wong R, Turlova E, Feng Z-P, Rutka JT, Sun H-S. Activation of TRPM7 by naltriben enhances migration and invasion of glioblastoma cells. Oncotarget. 2017;8:11239–48.
Chen W-L, Barszczyk A, Turlova E, Deurloo M, Liu B, Yang BB, et al. Inhibition of TRPM7 by carvacrol suppresses glioblastoma cell proliferation, migration and invasion. Oncotarget. 2015;6:16321–40.
Chen W-L, Turlova E, Sun CLF, Kim J-S, Huang S, Zhong X, et al. Xyloketal B suppresses glioblastoma cell proliferation and migration in vitro through inhibiting TRPM7-regulated PI3K/Akt and MEK/ERK signaling pathways. Mar Drugs. 2015;13:2505–25.
Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F, et al. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54:124–31.
Sumoza-Toledo A, Lange I, Cortado H, Bhagat H, Mori Y, Fleig A, et al. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011;25:3529–42.
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, et al. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–37.
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–9.
Fonfria E, Murdock PR, Cusdin FS, Benham CD, Kelsell RE, McNulty S. Tissue distribution profiles of the human TRPM cation channel family. J Recept Signal Transduct Res. 2006;26:159–78.
Di A, Kiya T, Gong H, Gao X, Malik AB. Role of the phagosomal redox-sensitive TRP channel TRPM2 in regulating bactericidal activity of macrophages. J Cell Sci. 2017;130:735–44.
Wang G, Cao L, Liu X, Sieracki NA, Di A, Wen X, et al. Oxidant Sensing by TRPM2 Inhibits Neutrophil Migration and Mitigates Inflammation. Dev Cell. 2016;38:453–62.
Roedding AS, Gao AF, Au-Yeung W, Scarcelli T, Li PP, Warsh JJ. Effect of oxidative stress on TRPM2 and TRPC3 channels in B lymphoblast cells in bipolar disorder. Bipolar Disord. 2012;14:151–61.
Wehrhahn J, Kraft R, Harteneck C, Hauschildt S. Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J Immunol. 2010;184:2386–93.
Kühn FJP. Structure-function relationship of TRPM2: recent advances, contradictions, and open questions. Int J Mol Sci. 2020;21:6481.
Jiang L-H, Yang W, Zou J, Beech DJ. TRPM2 channel properties, functions and therapeutic potentials. Expert Opin Therapeutic Targets. 2010;14:973–88.
Li J, Gao Y, Bao X, Li F, Yao W, Feng Z, et al. TRPM2: a potential drug target to retard oxidative stress. Front Biosci. 2017;22:1427–38.
Xia R, Mei ZZ, Mao HJ, Yang W, Dong L, Bradley H, et al. Identification of pore residues engaged in determining divalent cationic permeation in transient receptor potential melastatin subtype channel 2. J Biol Chem. 2008;283:27426–32.
Mei Z-Z, Mao H-J, Jiang L-H. Conserved cysteine residues in the pore region are obligatory for human TRPM2 channel function. Am J Physiol-Cell Physiol. 2006;291:C1022–C8.
Wang L, Fu T-M, Zhou Y, Xia S, Greka A, Wu H. Structures and gating mechanism of human TRPM2. Science. 2018;362:eaav4809.
Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–73.
Naziroğlu M, Lückhoff A. A calcium influx pathway regulated separately by oxidative stress and ADP-Ribose in TRPM2 channels: single channel events. Neurochem Res. 2008;33:1256–62.
Tong Q, Zhang W, Conrad K, Mostoller K, Cheung JY, Peterson BZ, et al. Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J Biol Chem. 2006;281:9076–85.
Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–9.
Roberge S, Roussel J, Andersson D, Meli A, Vidal B, Blandel F, et al. TNF-mediated caspase-8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovasc Res. 2014;103:90–9.
Aminzadeh M, Roghani M, Sarfallah A, Riazi GH. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int Immunopharmacol. 2018;54:78–85.
Mortadza SS, Sim JA, Stacey M, Jiang L-H. Signalling mechanisms mediating Zn2+-induced TRPM2 channel activation and cell death in microglial cells. Sci Rep. 2017;7:45032.
Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, et al. Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem. 2006;281:2489–96.
Fonfria E, Marshall IC, Boyfield I, Skaper SD, Hughes JP, Owen DE, et al. Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J Neurochem. 2005;95:715–23.
Öztürk Y, Günaydın C, Yalçın F, Nazıroğlu M, Braidy N. Resveratrol enhances apoptotic and oxidant effects of paclitaxel through TRPM2 channel activation in DBTRG glioblastoma cells. Oxid Med Cell Longev. 2019;2019:4619865.
Ertilav K, Nazıroğlu M, Ataizi ZS, Braidy N. Selenium enhances the apoptotic efficacy of docetaxel through activation of TRPM2 channel in DBTRG glioblastoma cells. Neurotox Res. 2019;35:797–808.
Ishii M, Oyama A, Hagiwara T, Miyazaki A, Mori Y, Kiuchi Y, et al. Facilitation of H2O2-induced A172 human glioblastoma cell death by insertion of oxidative stress-sensitive TRPM2 channels. Anticancer Res. 2007;27:3987–92.
Tóth B, Csanády L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J Biol Chem. 2010;285:30091–102.
Du J, Xie J, Yue L. Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc Natl Acad Sci USA. 2009;106:7239–44.
Kraft R, Grimm C, Frenzel H, Harteneck C. Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol. 2006;148:264–73.
Togashi K, Inada H, Tominaga M. Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol. 2008;153:1324–30.
Shimizu S, Yonezawa R, Hagiwara T, Yoshida T, Takahashi N, Hamano S, et al. Inhibitory effects of AG490 on H2O2-induced TRPM2-mediated Ca2+ entry. Eur J Pharmacol. 2014;742:22–30.
Li F, Wong R, Luo Z, Du L, Turlova E, Britto LRG, et al. Neuroprotective effects of AG490 in neonatal hypoxic-ischemic brain injury. Mol Neurobiol. 2019;56:8109–23.
Yamamoto S, Toda T, Yonezawa R, Negoro T, Shimizu S. Tyrphostin AG-related compounds attenuate H2O2-induced TRPM2-dependent and -independent cellular responses. J Pharm Sci. 2017;134:68–74.
Hill K, McNulty S, Randall AD. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:227–37.
Hill K, Benham CD, McNulty S, Randall AD. Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology. 2004;47:450–60.
Du J, Xie J, Yue L. Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J Gen Physiol. 2009;134:471–88.
Nazıroğlu M, Özgül C, Çiğ B, Doğan S, Uğuz AC. Glutathione modulates Ca2+ influx and oxidative toxicity through TRPM2 channel in rat dorsal root ganglion neurons. J Membr Biol. 2011;242:109–18.
Luo X, Li M, Zhan K, Yang W, Zhang L, Wang K, et al. Selective inhibition of TRPM2 channel by two novel synthesized ADPR analogues. Chem Biol Drug Des. 2018;91:552–66.
Starkus JG, Poerzgen P, Layugan K, Kawabata KG, Goto JI, Suzuki S, et al. ScalaradiaL is a potent inhibitor of transient receptor potential melastatin 2 (TRPM2) ion channels. J Nat Prod. 2017;80:2741–50.
Cruz-Torres I, Backos DS, Herson PS. Characterization and optimization of the novel TRPM2 antagonist tatM2NX. Mol Pharmacol. 2019:mol.119.117549.
Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, et al. Transient Receptor Potential Melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc Natl Acad Sci USA. 2011;108:11578–83.
Miller BA. TRPM2 in cancer. Cell Calcium. 2019;80:8–17.
Belrose JC. TRPM2 in the central nervous system: physiological role and critical regulatory pathways. [dissertation]. London (ON): Western Univ; 2012.
Emir TLR. Neurobiology of TRP channels. Boca Raton (FL): CRC Press; 2017.
Khaku AS, Tadi P. Cerebrovascular disease/stroke. J Clin Hypertens. 2007;9:A27–A31.
Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci. 2012;13:267–78.
Gelderblom M, Melzer N, Schattling B, Göb E, Hicking G, Arunachalam P, et al. Transient receptor potential melastatin subfamily member 2 cation channel regulates detrimental immune cell invasion in ischemic stroke. Stroke. 2014;45:3395–402.
Xie Y-F, Macdonald JF, Jackson MF. TRPM2 calcium and neurodegenerative diseases. Int J Physiol Pathophysiol Pharmacol. 2010;2:95–103.
Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener. 2009;4:20.
Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–50.
Parihar MS, Kunz EA, Brewer GJ. Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. J Neurosci Res. 2008;86:2339–52.
Jurma OP, Hom DG, Andersen JK. Decreased glutathione results in calcium-mediated cell death in PC12. Free Radic Biol Med. 1997;23:1055–66.
Belrose JC, Xie YF, Gierszewski LJ, MacDonald JF, Jackson MF. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol Brain. 2012;5:11.
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018;4:575–90.
Tufekci KU, Meuwissen R, Genc S, Genc K. Inflammation in Parkinson’s disease. Adv Protein Chem Struct Biol. 2012;88:69–132.
Crotti A, Glass CK. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015;36:364–73.
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev. 2016;2016:4350965.
Bao MH, Lv QL, Szeto V, Wong R, Zhu SZ, Zhang YY, et al. TRPM2‐AS inhibits the growth, migration, and invasion of gliomas through JNK, c‐Jun, and RGS4. J Cell Physiol. 2020;235:4594–604.
Mukhopadhyay A, Basu N, Ghatak N, Gujral PK. Anti-inflammatory and irritant activities of curcumin analogues in rats. Agents Actions. 1982;12:508–15.
Weber WM, Hunsaker LA, Abcouwer SF, Deck LM, Vander Jagt DL. Anti-oxidant activities of curcumin and related enones. Bioorg Med Chem. 2005;13:3811–20.
Kawamori T, Lubet R, Steele VE, Kelloff GJ, Kaskey RB, Rao CV, et al. Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res. 1999;59:597.
Ohshiro M, Kuroyanagi M, Ueno A. Structures of sesquiterpenes from Curcuma longa. Phytochemistry. 1990;29:2201–5.
Kulkarni SK, Dhir A. An overview of curcumin in neurological disorders. Indian J Pharm Sci. 2010;72:149–54.
Wang X, Deng J, Yuan J, Tang X, Wang Y, Chen H, et al. Curcumin exerts its tumor suppressive function via inhibition of NEDD4 oncoprotein in glioma cancer cells. Int J Oncol. 2017;51:467–77.
Kwak YD, Wang B, Li JJ, Wang R, Deng Q, Diao S, et al. Upregulation of the E3 ligase NEDD4-1 by oxidative stress degrades IGF-1 receptor protein in neurodegeneration. J Neurosci. 2012;32:10971–81.
Zhang X, Chen Q, Wang Y, Peng W, Cai H. Effects of curcumin on ion channels and transporters. Front Physiol. 2014;5:94.
Nalli M, Ortar G, Schiano Moriello A, Di Marzo V, De, Petrocellis L. Effects of curcumin and curcumin analogues on TRP channels. Fitoterapia. 2017;122:126–31.
Yeon KY, Kim SA, Kim YH, Lee MK, Ahn DK, Kim HJ, et al. Curcumin produces an antihyperalgesic effect via antagonism of TRPV1. J Dent Res. 2009;89:170–4.
Öz A, Çelik Ö. Curcumin inhibits oxidative stress-induced TRPM2 channel activation, calcium ion entry and apoptosis values in SH-SY5Y neuroblastoma cells: involvement of transfection procedure. Mol Membr Biol. 2016;33:76–88.
Hirschler-Laszkiewicz I, Chen SJ, Bao L, Wang J, Zhang XQ, Shanmughapriya S, et al. The human ion channel TRPM2 modulates neuroblastoma cell survival and mitochondrial function through Pyk2, CREB, and MCU activation. Am J Physiol Cell Physiol. 2018;315:C571–c86.
Kheradpezhouh E, Barritt GJ, Rychkov GY. Curcumin inhibits activation of TRPM2 channels in rat hepatocytes. Redox Biol. 2016;7:1–7.
Shah N, Mohammad AS, Saralkar P, Sprowls SA, Vickers SD, John D, et al. Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol Res. 2018;132:47–68.
Gökçe Kütük S, Gökçe G, Kütük M, Gürses Cila HE, Nazıroğlu M. Curcumin enhances cisplatin-induced human laryngeal squamous cancer cell death through activation of TRPM2 channel and mitochondrial oxidative stress. Sci Rep. 2019;9:17784.
Omoti AE, Omoti CE. Ocular toxicity of systemic anticancer chemotherapy. Pharm Pract. 2006;4:55–9.
Özkaya D, Nazıroğlu M. Curcumin diminishes cisplatin-induced apoptosis and mitochondrial oxidative stress through inhibition of TRPM2 channel signaling pathway in mouse optic nerve. J Recept Signal Transduct Res. 2020;40:97–108.
Salazar-Ramiro A, Ramírez-Ortega D, Pérez de la Cruz V, Hérnandez-Pedro NY, González-Esquivel DF, Sotelo J, et al. Role of redox status in development of glioblastoma. Front Immunol. 2016;7:156.
Ramírez-Expósito MJ, Martínez-Martos JM. The delicate equilibrium between oxidants and antioxidants in brain glioma. Curr Neuropharmacol. 2019;17:342–51.
Montero A, Fossella F, Hortobagyi G, Valero V. Docetaxel for treatment of solid tumours: a systematic review of clinical data. Lancet Oncol. 2005;6:229–39.
Harris HR, Bergkvist L, Wolk A. Selenium intake and breast cancer mortality in a cohort of Swedish women. Breast Cancer Res Treat. 2012;134:1269–77.
Yakubov E, Buchfelder M, Eyüpoglu IY, Savaskan NE. Selenium action in neuro-oncology. Biol Trace Elem Res. 2014;161:246–54.
Hazane-Puch F, Arnaud J, Trocmé C, Faure P, Laporte F, Champelovier P. Sodium selenite decreased HDAC activity, cell proliferation and induced apoptosis in three human glioblastoma cells. Anticancer Agents Med Chem. 2016;16:490–500.
Park SO, Yoo YB, Kim YH, Baek KJ, Yang J-H, Choi PC, et al. Effects of combination therapy of docetaxel with selenium on the human breast cancer cell lines MDA-MB-231 and MCF-7. Ann Surg Treat Res. 2015;88:55–62.
Desmedt C, Fornili M, Clatot F, Demicheli R, De Bortoli D, Di, et al. Differential benefit of adjuvant docetaxel-based chemotherapy in patients with early breast cancer according to baseline body mass index. J Clin Oncol. 2020;38:2883–91.
Baş E, Naziroğlu M. Selenium attenuates docetaxel-induced apoptosis and mitochondrial oxidative stress in kidney cells. Anticancer Drugs. 2019;30:339–46.
Ibi M, Matsuno K, Shiba D, Katsuyama M, Iwata K, Kakehi T, et al. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci. 2008;28:9486.
Ta LE, Bieber AJ, Carlton SM, Loprinzi CL, Low PA, Windebank AJ. Transient Receptor Potential Vanilloid 1 is essential for cisplatin-induced heat hyperalgesia in mice. Mol Pain. 2010;6:15.
Huang K, Bian D, Jiang B, Zhai Q, Gao N, Wang R. TRPA1 contributed to the neuropathic pain induced by docetaxel treatment. Cell Biochem Funct. 2017;35:141–3.
Spengler G, Gajdács M, Marć MA, Domínguez-Álvarez E, Sanmartín C. Organoselenium compounds as novel adjuvants of chemotherapy drugs—a promising approach to fight cancer drug resistance. Molecules. 2019;24:336.
Tangney CC, Rasmussen HE. Polyphenols, inflammation, and cardiovascular disease. Curr Atheroscler Rep. 2013;15:324.
Snopek L, Mlcek J, Sochorova L, Baron M, Hlavacova I, Jurikova T, et al. Contribution of red wine consumption to human health protection. Molecules. 2018;23:1684.
Salehi B, Mishra AP, Nigam M, Sener B, Kilic M, Sharifi-Rad M, et al. Resveratrol: a double-edged sword in health benefits. Biomedicines. 2018;6:91.
Yu L, Wang S, Kogure Y, Yamamoto S, Noguchi K, Dai Y. Modulation of TRP channels by resveratrol and other stilbenoids. Mol Pain. 2013;9:3.
Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–7.
Johnston SRD. Ovarian cancer: review of the National Institute for Clinical Excellence (NICE) guidance recommendations. Cancer Invest. 2004;22:730–42.
Harper E, Dang W, Lapidus RG, Garver RI. Enhanced efficacy of a novel controlled release paclitaxel formulation (PACLIMER Delivery System) for local-regional therapy of lung cancer tumor nodules in mice. Clin Cancer Res. 1999;5:4242.
Watatani M, Ueda K, Daito K, Azumi T, Hirai T, Yamato M, et al. Clinical experience of weekly paclitaxel-based treatment as preoperative chemotherapy for patients with primary breast cancer. Breast Cancer. 2004;11:187–93.
Nazıroğlu M, Braidy N. Thermo-sensitive TRP channels: novel targets for treating chemotherapy-induced peripheral pain. Front Physiol. 2017;8:1040. -
Fukui M, Yamabe N, Zhu BT. Resveratrol attenuates the anticancer efficacy of paclitaxel in human breast cancer cells in vitro and in vivo. Eur J Cancer. 2010;46:1882–91.
Sprouse AA, Herbert B-S. Resveratrol augments paclitaxel treatment in MDA-MB-231 and paclitaxel-resistant MDA-MB-231 breast cancer cells. Anticancer Res. 2014;34:5363.
Nicolini G, Rigolio R, Miloso M, Bertelli AAE, Tredici G. Anti-apoptotic effect of trans-resveratrol on paclitaxel-induced apoptosis in the human neuroblastoma SH-SY5Y cell line. Neurosci Lett. 2001;302:41–4.
Walle T. Bioavailability of resveratrol. Ann N Y Acad Sci. 2011;1215:9–15.
Pirazzini M, Rossetto O, Eleopra R, Montecucco C. Botulinum neurotoxins: biology, pharmacology, and toxicology. Pharmacol Rev. 2017;69:200–35.
Jabbari B. Botulinum toxin treatment in clinical medicine. Springer International Publishing; 2018.
Shaw L, Bazzell AF, Dains JE. Botulinum toxin for side-effect management and prevention of surgical complications in patients treated for head and neck cancers and esophageal cancer. J Adv Pract Oncol. 2019;10:40–52.
Mittal SO, Jabbari B. Botulinum neurotoxins and cancer-a review of the literature. Toxins. 2020;12:32.
Shebl RI. Anti-cancer potential of captopril and botulinum toxin type-a and associated p53 gene apototic stimulating activity. Iran J Pharm Res. 2019;18:1967–77.
Rust A, Leese C, Binz T, Davletov B. Botulinum neurotoxin type C protease induces apoptosis in differentiated human neuroblastoma cells. Oncotarget. 2016;7:33220–8.
Akpınar O, Özşimşek A, Güzel M, Nazıroğlu M. Clostridium botulinum neurotoxin A induces apoptosis and mitochondrial oxidative stress via activation of TRPM2 channel signaling pathway in neuroblastoma and glioblastoma tumor cells. J Recept Signal Transduct. 2020;40:620–32.
Uchiyama A, Yamada K, Perera B, Ogino S, Yokoyama Y, Takeuchi Y, et al. Protective effect of botulinum toxin A after cutaneous ischemia-reperfusion injury. Sci Rep. 2015;5:9072.
Nazıroğlu M. TRPM2 cation channels, oxidative stress and neurological diseases: where are we now? Neurochem Res. 2011;36:355–66.
Acknowledgements
This work was supported by grants to ZPF from the Canadian Institutes of Health Research (CIHR PJT-153155) and HSS from the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2016-04574).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Ji, D., Luo, Zw., Ovcjak, A. et al. Role of TRPM2 in brain tumours and potential as a drug target. Acta Pharmacol Sin 43, 759–770 (2022). https://doi.org/10.1038/s41401-021-00679-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00679-4
Keywords
This article is cited by
-
TRPM2-mediated Ca2+ signaling as a potential therapeutic target in cancer treatment: an updated review of its role in survival and proliferation of cancer cells
Cell Communication and Signaling (2023)
-
Paclitaxel Promotes Oxidative Stress–Mediated Human Laryngeal Squamous Tumor Cell Death through the Stimulation of Calcium and Zinc Signaling Pathways: No Synergic Action of Melatonin
Biological Trace Element Research (2022)
