
Abstract
Furmonertinib was designed for the treatment of non-small cell lung cancer (NSCLC) patients with EGFR T790M mutation. In this study, we investigated the metabolic disposition and mass balance in humans and tissue distribution in rats. After a single oral administration of 97.9 μCi/81.5 mg [14C]-furmonertinib mesylate to six healthy male volunteers, the absorption process of furmonertinib was fast with a tmax of total plasma radioactivity at 0.75 h. Afterward, furmonertinib was extensively metabolized, with the parent drug and active metabolite AST5902 accounting for 1.68% and 0.97% of total radioactivity in plasma. The terminal t1/2 of total radioactivity in plasma was as long as 333 h, suggesting that the covalent binding of drug-related substances to plasma proteins was irreversible to a great extent. The most abundant metabolites identified in feces were desmethyl metabolite (AST5902), cysteine conjugate (M19), and parent drug (M0), which accounted for 6.28%, 5.52%, and 1.38% of the dose, respectively. After intragastric administration of 124 μCi/9.93 mg/kg [14C]-furmonertinib to rats, drug-related substances were widely and rapidly distributed in tissues within 4 h. The concentration of total radioactivity in the lung was 100-fold higher than that in rat plasma, which could be beneficial to the treatment of lung cancer. Mass balance in humans was achieved with 77.8% of the administered dose recovered in excretions within 35 days after administration, including 6.63% and 71.2% in urine and feces, respectively. In conclusion, [14C]-furmonertinib is completely absorbed and rapidly distributed into lung tissue, extensively metabolized in humans, presented mostly as covalent conjugates in plasma, and slowly eliminated mostly via fecal route.
Similar content being viewed by others
Introduction
Acquired drug resistance caused by epidermal growth factor receptor (EGFR) mutation usually occurs 9–13 months after the usage of first- and second-generation EGFR tyrosine kinase inhibitors (EGFR-TKIs) in patients with non-small-cell lung cancer (NSCLC) [1]. The EGFR T790M mutation is the most widespread, accounting for ~50%–60% of drug-resistant cases [2, 3]. Research and development of third-generation EGFR-TKIs have been placed on the agenda, and this class of drugs has been proven to be effective in the treatment of NSCLC with the EGFR T790M mutation [4]. Osimertinib (Fig. 1) was the first third-generation EGFR-TKI approved by the FDA. In phase III clinical trials of osimertinib, the median duration of progression-free survival was remarkably longer than that of other treatments [5]. Olmutinib and almonertinib have also been marketed in South Korea and China, respectively, as third-generation EGFR inhibitors for the treatment of locally advanced or metastatic EGFR T790M mutation-positive NSCLC [6].

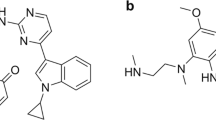
Structures of furmonertinib and osimertinib. *denotes 14C.
Furmonertinib (alflutinib, AST2818, Fig. 1) is a new third-generation irreversible and selective EGFR-TKI developed by Shanghai Allist Pharmaceutical Technology Co., Ltd. [7]. It is a structural analog of osimertinib, with a trifluoroethoxypyridine moiety in place of the anisole of osimertinib. Preclinical studies have shown enhanced safety and tolerability of furmonertinib in rats and dogs. Furmonertinib also exhibited antitumor activity comparable to that of osimertinib [7]. In the clinical phase I dose-escalation human trial of furmonertinib (20–240 mg), no dose-limiting toxicity was observed. Plasma exposure to the parent drug (M0) and its major metabolite AST5902 was similar at the steady-state after multiple oral administrations of furmonertinib to patients with NSCLC in all dose groups [7]. A new drug application for furmonertinib has been submitted to the National Medical Products Administration for the treatment of patients with EGFR T790M mutation-positive NSCLC.
In this study, the metabolic disposition of furmonertinib in humans was investigated, and the main metabolic pathways of furmonertinib were reported for the first time. Mass balance was achieved following oral administration of 97.9 μCi/81.5 mg [14C]-furmonertinib mesylate to healthy male volunteers and the major elimination route in humans was illustrated. The tissue distribution of furmonertinib in rats was also investigated, and an in vivo comparison of the metabolic disposition between furmonertinib and osimertinib was conducted. The results could provide data support for the safety and efficacy of furmonertinib.
Materials and methods
Chemicals and reference compounds
[14C]-Furmonertinib mesylate (99% purity) and [14C]-furmonertinib (99% purity) were synthesized by Jiangsu Wuxi Pharmaceutical Co., Ltd. (Shanghai, China). Nonradiolabeled furmonertinib mesylate tablets (40 mg), furmonertinib (98.8% purify), AST5902 (98.2% purity), and d3-furmonertinib (96.0% purity) were supplied by Shanghai Allist Pharmaceutical Co., Ltd. (Shanghai, China). [14C]-Furmonertinib was used to investigate the tissue distribution in rats, while [14C]-furmonertinib mesylate and nonradiolabeled furmonertinib mesylate tablets were used to investigate the metabolic disposition and mass balance in healthy male volunteers. Furmonertinib, AST5902, and d3-furmonertinib were used for quantitative determination in biological samples using liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Tissue distribution in rats
The tissue distribution of the total radioactivity was determined using a liquid scintillation counter (PerkinElmer, Waltham, MA, USA) after a single intragastric administration of 124 μCi/9.93 mg/kg [14C]-furmonertinib to Sprague Dawley (SD) rats. After [14C]-furmonertinib dosing, six rats (three males and three females) from each group were euthanized at 4, 8, 48, and 168 h. Blood, plasma, and selected tissues (or organs), including the heart, liver, spleen, lung, kidney, brain, bladder, pancreas, testis, ovary, uterus, stomach, and duodenum, were collected. All collected samples were stored at −80°C until analysis. SD rats were provided by Shanghai Sippe-Bk Lab Animal Co., Ltd. (production license number SCXK 2013–0016). All animal study procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
Study design
This study adopted a single-center, single-dose, nonrandom open design. It was conducted at The First Affiliated Hospital of Soochow University (Suzhou, China). Six healthy Chinese male volunteers aged 18–45 years with a body mass index between 19 and 26 kg/m2 were recruited for this study. The use of prescribed or nonprescribed concomitant medications was not permitted 14 days before the start of the study and during the study. In addition, the use of medications with CYP450 enzyme-inducing properties was not permitted 30 days before the administration of furmonertinib. Volunteers were also excluded if they had any intake of products containing grapefruit, irregular bowel movements, and abnormal values for clinical laboratory tests (hematology, serum chemistry, urinalysis, and stool analysis), or 12-lead electrocardiography. An independent ethics committee of The First Affiliated Hospital of Soochow University approved this study, which was conducted in accordance with the ethical principles derived from the Helsinki Declaration and met the good clinical practice and applicable regulatory requirements. All patients provided written informed consent before enrollment.
After 97.9 μCi/81.5 mg of [14C]-furmonertinib mesylate with unlabeled furmonertinib mesylate was orally administered to the volunteers, plasma, urine, and feces were collected for 84 days after dosing, and the volunteers were discharged from the clinic when one of the following criteria was satisfied: the accumulated excreted radioactivity was more than 80% of the administered radioactivity, the excreted radioactivity over 2 consecutive days was less than 1% of the administered radioactivity, and the measured radioactivity for 2 consecutive days in the plasma collected was below two times that of the background level.
Sample collection
Blood samples (~4 mL) were collected pre-dose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, 48, 72, 96, 120, 144, 168, 192, 216, 240, 264, 288, 312, 336, 360, 384, 408, 432, 456, 480, 504, 672, 840, 1008, 1176, 1344, 1512, 1680, 1848, and 2016 h postdose into tubes containing the anticoagulant K2-EDTA. The tubes were centrifuged to obtain plasma samples for determination of the concentrations of total radioactivity, furmonertinib, and AST5902. In addition, 12 mL blood samples were collected at 4, 10, 24, 48, 96, and 168 h postdose for metabolite identification and determination of the total radioactivity in whole blood. Urinary and fecal samples of each volunteer were collected at a fixed interval of 0–2016 h after drug administration. All collected samples were stored at −80°C until analysis.
Quantitation of total radioactivity, furmonertinib, and AST5902 in the biological samples
The total radioactivity in rat plasma, human plasma, and urine was determined using a Packard Tri-Carb 3100 TR liquid scintillation counter (PerkinElmer, USA). Rat tissues, human blood, and fecal samples were combusted using an OX-501 Biological Oxidizer (R. J. Harvey, USA) prior to liquid scintillation counting. Data were acquired using a QuantaSmart data system (PerkinElmer, USA). The concentrations of furmonertinib and AST5902 in human plasma were simultaneously determined using a validated LC-MS/MS method [8]. The linearity ranges of the calibration curves for furmonertinib and AST5902 were 0.200–100 and 0.0500–25.0 ng/mL, respectively.
Pharmacokinetic analysis
The pharmacokinetic parameters of total radioactivity, furmonertinib, and AST5902 in the plasma were derived using standard noncompartmental analysis methods with Phoenix WinNonlin Professional version 7.0 (Pharsight Corporation, Mountain View, CA, USA). The following pharmacokinetic parameters were assessed: maximum concentration (Cmax); time to reach Cmax (tmax); area under the concentration-time curve (AUC) from time 0 to 24 h (AUC0–24 h); AUC from time 0 to time of the last quantifiable concentration (AUC0-last); AUC from time 0 to infinite time (AUC0-∞), calculated as the sum of AUC0-last and Clast/λz, where Clast is the last observed quantifiable concentration and λz is the elimination half-life time to the last quantifiable concentration; the apparent volume of distribution (Vd/F) based on the terminal phase, calculated as D/(λz × AUC0-∞); and total drug clearance (CL/F) after extravascular administration, calculated as D/AUC0-∞.
Cumulative excretion from 0 to 504 h (Ae0–504) was calculated by the summation of the percent recovery at each collection interval, while the cumulative excretion from 504 to 2016 h was calculated by the trapezoidal method (as the area under the excretion rate versus time curve through day 2016) according to Ae0–504 [9].
Sample pretreatment for metabolite profiling
The plasma samples of each subject were pooled at equal volumes at 4, 10, 24, 48, 96, and 168 h to obtain pooled plasma samples (6 mL). A 1 mL aliquot of each of the pooled plasma samples was used to determine the total radioactivity, while a 5 mL aliquot of the pooled plasma samples was pretreated with 10 mL of acetonitrile. This mixture was vortexed for 1 min and centrifuged for 5 min at 3500 r/min. A 1 mL aliquot of the supernatant was used to determine the total radioactivity after extraction, while the rest of the supernatant was moved to another container and evaporated under a nitrogen stream at 40°C. Reconstitution was performed for the remaining fraction in 500 μL of an acetonitrile/water (1:1, v/v) mixture, and 90 and 7 µL of the mixture were analyzed via high-performance liquid chromatography (HPLC) with off-line radiodetection and via HPLC/Triple TOF MS, respectively.
The urine samples of each subject between 0 and 72 h were pooled in accordance with the volumes excreted during each time period to obtain pooled urine samples (10 mL). A 1 mL aliquot of the pooled urine samples was used to determine the total radioactivity, while a 5 mL aliquot of the pooled urine samples was evaporated under a nitrogen stream at 40°C. Reconstitution was performed for the remaining fraction in 600 μL of an acetonitrile/water (1:1, v/v) mixture. In addition, 100 μL of the mixture was used to determine the total radioactivity after extraction, and 90 and 7 µL of the mixture were analyzed via HPLC with off-line radiodetection and via HPLC/Triple TOF MS, respectively.
The fecal homogenates of each subject between 0 and 240 h were pooled in accordance with the weight from each time period to obtain pooled fecal samples (3 g). Then, 0.3 g of these samples were used to determine the total radioactivity, while 2.7 g was pretreated using 0.9 mL of acetonitrile/methanol (1:1, v/v) three times before centrifugation for 5 min at 3500 r/min. All of the supernatants were combined, 1 mL of which was used to determine the total radioactivity after extraction, and the rest was evaporated under a nitrogen stream at 40°C. Reconstitution was performed for the remaining fraction in 600 μL of an acetonitrile/water (1:1, v/v) mixture; then, 100 μL of the mixture was used to determine the total radioactivity after extraction, while 90 and 7 µL of the mixture were analyzed via HPLC with off-line radiodetection and via HPLC/Triple TOF MS, respectively.
HPLC conditions
Metabolite profiling was performed on an 1100 HPLC system (Agilent Technologies, Inc., Santa Clara, CA, USA). Furmonertinib and its metabolites were separated using a Shim-pack CLCl C8 column (150 mm × 4.6 mm, 5 μm) with a C18 guard column (4.0 mm × 3.0 mm, 5 μm) equilibrated at room temperature. The mobile phase consisted of 5 mM aqueous ammonium acetate containing 0.02% formic acid (A) as the aqueous phase and acetonitrile (B) as the organic phase. Subsequently, gradient elution was performed linearly from 30% B to 55% B from 0 to 45 min and to 95% B for the next 5 min and maintained at this ratio for another 5 min. The flow rate was maintained at 0.6 mL/min throughout this process.
Metabolite identification and profiling using radiodetection and MS
Radiodetection was conducted with an off-line TopCount NXT microplate scintillation and luminescence counter (PerkinElmer, Waltham, MA, USA). HPLC eluents collected in a LumaPlate 96-well plate were dried and counted using the TopCount NXT counter. Data were applied to construct radiochromatograms by using an ARC data system (AIM Research Company, Hockessin, DE, USA).
Metabolite identification was conducted on a Triple TOF 5600+ MS system (AB SCIEX, Concord, Ontario, CA, USA) with a DuoSpray ion source in positive ion mode. The mass range was set at m/z 80–1000. The following parameter settings were used: ion spray voltage, 5500 V; declustering potential, 80 eV; ion source heater, 500°C; curtain gas, 40 psi; ion source gas 1, 55 psi; and ion source gas 2, 50 psi. The collision energy was set to 10 eV during the TOF MS scans. Moreover, the collision energy of the product ion scans was fixed at 45 eV, with a spread of 15 eV (MS/MS). Data were acquired using Analyst TF 1.6 software (AB SCIEX) and processed using MetabolitePilot 1.5 software (AB SCIEX).
Results
Tissue distribution in rats
The tissue distribution in 18 selected rat tissues was investigated after a single intragastric administration of 124 μCi/9.93 mg/kg [14C]-furmonertinib, and the results are shown in Fig. 2. Drug-related substances were mainly distributed in the therapeutic target organ, the lung. These substances were distributed in the lung within 4 h after administration, as the concentration of total radioactivity in the lung was 100-fold than that in the plasma, followed by the digestive tract, spleen, kidney, bladder, and ovary. The tissue-to-plasma ratios of the radioactivity in all tissues decreased from 4 h to 48 h, and the total radioactivity in tissues was almost completely eliminated at 168 h after administration. Therefore, there was no potential tissue accumulation of drug-related substances.

Radioactivity of selected tissues after intragastric administration of 124 μCi/9.93 mg/kg [14C]-furmonertinib to SD rats.
Pharmacokinetics
The concentration-time curves of total radioactivity, furmonertinib, and AST5902 in the plasma are shown in Fig. 3, and the main pharmacokinetic parameters are summarized in Table 1. After a single oral administration of 97.9 μCi/81.5 mg [14C]-furmonertinib mesylate to healthy male volunteers, the concentration of total radioactivity in the plasma peaked at 0.75 h, indicating rapid absorption, while the tmax values of furmonertinib and AST5902 in the plasma were 6.0 and 10.0 h, respectively. The exposure of total radioactivity (AUC0-∞) was 42400 h × ng eq/mL, while furmonertinib and AST5902 accounted for only 1.68% and 0.972%, respectively, of the total radioactivity according to the geometric mean AUC0-∞ ratios. The terminal phase t1/2 values for total radioactivity, furmonertinib, and AST5902 were 333, 37.2, and 62.1 h, respectively, suggesting slow elimination of the drug-related materials from the plasma. In addition, the ratio of total radioactivity in the blood to the plasma increased from 0.959 to 1.86 from 4 to 336 h (data are shown in Supplementary Table S1), indicating that the drug-related substances did not prefer to distribute into red blood cells but were eliminated more slowly from the blood than from plasma.

Mean plasma concentration-time curves of total radioactivity, furmonertinib, and AST5902 following a single oral administration of 97.9 μCi/81.5 mg [14C]-furmonertinib mesylate to male volunteers (n = 6).
Mass balance
As shown in Fig. 4, the mean cumulative recovery of radioactivity in excretions up to 840 h (35 days) after dosing was 77.8% (range of 67.4%–84.8%). Fecal excretion was the predominant route of elimination, accounting for 71.2% (range of 59.6%–79.5%) of the administered dose, while the value of urinary excretion accounted for 6.63% (range of 5.35%–7.78%). The excretion of total radioactivity was slow, and the majority of radioactivity in excretions was recovered in the first 504 h (21 days), accounting for 75.2% (range 64.1%–83.3%). Twenty-one days after administration, small amounts of total radioactivity excreted through urine and feces remained, but the mean weekly excretion was less than 2% of the dose (data shown in Supplementary Table S2).

Cumulative recovery of radioactivity in excretions following a single oral administration of 97.9 μCi/81.5 mg [14C]-furmonertinib mesylate to male volunteers (n = 6).
Metabolite identification and profiling
Under the described experimental conditions, radiochromatograms, retention times (tR), and mass fragmentation pathways of the furmonertinib and AST5902 reference compounds were obtained using HPLC/Triple TOF MS and HPLC radiodetection. As shown in Figs. 5 and 6, the tR of furmonertinib and AST5902 in the MS chromatograms were 2.1 and 1.9 min later, respectively than those in the radiochromatograms. Therefore, the correlation between MS chromatograms and radiochromatograms was established in terms of the relations in tR, achieving metabolite identification.

Chromatograms of furmonertinib and AST5902 from HPLC/Triple TOF MS (a), respectively. Proposed fragmentation pathways of furmonertinib (b) and AST5902 (c).

Chromatograms of reference standards in solution (a) and metabolites observed from pooled urine samples (b) and pooled fecal samples (c) via HPLC–radiodetection.
Furmonertinib and AST5902 produced protonated molecular ions [M + H]+ at m/z 569.256 and 555.240, respectively, and shared similar MS2 fragment ions, including m/z 524.199, 441.189, 386.171, 302.103, and 225.113. However, the unique MS2 fragment ions were m/z 370.175 and 355.165 for furmonertinib and m/z 498.181, 480.173, 415.173, 360.154, 344.159, and 225.113 for AST5902. The proposed fragmentation pathways of furmonertinib and AST5902 are displayed in Fig. 5b, c.
Plasma, urine, and fecal data were obtained using HPLC radiodetection and HPLC/Triple TOF MS after a single oral administration of 97.9 μCi/81.5 mg [14C]-furmonertinib mesylate to healthy male volunteers. A total of 11 metabolites were observed in humans. Tables 2 and 3 list the specific information of each metabolite detected by HPLC-radiodetection and HPLC/Triple TOF MS, respectively. The identification process in feces has been introduced in detail as an example, given that the majority of the metabolites were found in feces. The MS2 fragmentation spectra of the following metabolites are displayed in Supplementary Fig. S1.
M0 (parent drug)
The chromatographic peak of M0 was found at 32.5 min, and it displayed a protonated molecule at m/z 569.255, indicating that the elemental composition was C28H31F3N8O2. The tR and mass spectral fragmentation arrangements of M0 were the same as those of furmonertinib, indicating the unchanged drug.
M2
M2 demonstrated a protonated molecular mass of m/z 487.254, and it was eluted at 13.4 min. M2’s elemental composition was C26H30N8O2, indicating the loss of C2HF3 (82 Da) in comparison to M0. The fragment ion of M2 at m/z 442.197 also had a loss of 82 Da compared with that of furmonertinib. Therefore, M2 was confirmed as the O-detrifluoroethyl metabolite of furmonertinib.
M5
M5 was eluted at 22.5 min and displayed an [M + H]+ ion at m/z 541.225, indicating that the chemical formula was C26H27F3N8O2, with a loss of C2H4 compared to M0. The major fragment ions were at m/z 484.169, 401.159, 372.156, 346.139, 330.145, and 211.098, which all had a loss of CH2 compared to those of AST5902. Thus, M5 has accordingly identified as the N,N-demethylated metabolite of furmonertinib.
M6
M6-1, M6-2, and M6-3 were eluted at 22.7, 25.0, and 30.0 min, respectively, with an [M + H]+ ion at m/z 555.239. They demonstrated an elemental composition of C27H29F3N8O2 with the loss of CH2 from M0. The fragment ions of M6-1 at m/z 510.183, 427.174, and 372.155 and M6-2 at m/z 441.189 and 386.171 were 14 Da lower than those of M0, indicating that furmonertinib demethylation occurred. By contrast, the fragment ions of M6-3 at m/z 498.183, 441.188, 415.173, 386.170, 360.155, 344.160, 302.102, and 225.113 were the same as those of the AST5902 reference compound, with the same retention time. Thus, M6-1, M6-2, and M6-3 were all N-demethylated metabolites of furmonertinib.
M8
M8-1 and M8-2 were eluted at 16.8 and 20.1 min, respectively. Both metabolites displayed the same [M + H]+ ions at m/z 571.234. Their elemental compositions were C27H29F3N8O3, with the addition of an oxygen atom and the loss of CH2 compared to M0. The fragment ions of M8-1 at m/z 514.180, 376.151, and 241.109 were 16 Da larger than those of AST5902, and they shared a similar fragment ion at m/z 360.157. Thus, M8-1 was a single oxidation metabolite of AST5902. The fragment ion of M8-2 at m/z 496.170 was 32 Da lower than that of M0 at m/z 524.198, and the ions at m/z 484.170, 427.177, and 330.146 were 14 Da lower than those of AST5902, suggesting that M8-2 was the N-demethylated single oxidation metabolite of furmonertinib.
M9
M9 was eluted at 32.3 min, and it exhibited an [M + H]+ ion at m/z 585.249. Its chemical formula was C28H31F3N8O3, with the addition of an oxygen atom compared to M0. The fragment ions of M9 were at m/z 524.205, 441.188, and 370.175, which were all the same as those of M0. Thus, M9 was a single oxidation metabolite of furmonertinib.
M10
M10 was eluted at 14.3 min, and it displayed an [M + H]+ ion at m/z 601.246. The elemental composition of M10 was C28H31F3N8O4 with the addition of two oxygen atoms (32 Da) compared to M0. The major fragment ions of M10 were at m/z 556.193, 473.182, and 418.165, which were also all 32 Da larger than those of M0, indicating that M10 was a double oxidation metabolite of furmonertinib.
M19
M19 was eluted at 17.6 min with an [M + H]+ ion at m/z 690.274, and its elemental composition was C31H38F3N9O4S. The fragment ion of M19 at m/z 562.207 was 121 Da larger than that of M0, indicating that cysteine conjugation occurred on M0, whereas the fragment ion at m/z 386.180 was identical to that of M0. Thus, M19 was proposed to be a cysteine conjugate of furmonertinib.
M20
M20 was eluted at 23.0 min, and its parent ion was displayed at m/z 732.285. The elemental composition of M20 was C33H40F3N9O5S, which suggested the addition of acetylcysteine to M0. The fragment ions at m/z 687.226, 604.217, and 386.171 were identical to those of M0. Therefore, M20 was speculated to be an N-acetylcysteine conjugate of furmonertinib.
The extraction recovery was investigated by comparing the total radioactivity in biological samples before and after the extraction process. The radioactivity values of the pooled plasma samples were too low to detect metabolites by radiodetection, which indicated the covalent binding of drug-related substances to plasma proteins. Therefore, the metabolites in the plasma were mostly identified using HPLC/Triple TOF MS. M0 and AST5902 were the main forms of furmonertinib found in the plasma, and small amounts of M19, M6-2, and M10 were found in the pooled plasma at 4, 10, 24, and 48 h after administration (Supplementary Fig. S2).
The extraction and reconstitution procedure was evaluated with [14C]-furmonertinib spiked into the control urine sample and control fecal homogenate. The use of acetonitrile/methanol (50/50, v/v) extraction ensured that greater than 96% of the total radioactivity was recovered from the control homogenate. Reconstitution with acetonitrile/water (50/50, v/v) ensured greater than 95% and 80% total radioactivity recovery for the control urine sample and the control homogenate, respectively. Consequently, this extraction procedure was used to prepare urine and feces. The recovery of the total radioactivity was 85.4% and 67.3% for the urine and fecal samples, respectively. In the urine, only M0 and AST5902 were detected at the administered dose, accounting for 1.99% and 1.16%, respectively. The radioactivity recoveries in feces were 1.38% and 6.28% for furmonertinib and AST5902, respectively, while the value of M19 reached 5.52%. Another eight metabolites were observed in the feces, including M2, M5, M20, M9, M8-1, M6-2, M10, and M8-2, sorted by excretion recovery. Each of these was present at concentrations less than 1.5% of the dose.
Fig. 6 and 7 display the metabolites observed from the pooled urine and fecal samples via HPLC–radiodetection and HPLC/Triple TOF MS, respectively. In the urine, only M0 and AST5902 were detected at the administered dose, accounting for 1.99% and 1.16%, respectively. The radioactivity recoveries in feces were 1.38% and 6.28% for furmonertinib and AST5902, respectively, while the value of M19 reached 5.52%. Another eight metabolites were observed in the feces, including M2, M5, M20, M9, M8-1, M6-2, M10, and M8-2, sorted by excretion recovery. Each of these compounds was present at concentrations less than 1.5% of the dose. The proposed metabolic pathways of furmonertinib in humans are shown in Fig. 8.

Metabolites observed from pooled urine samples (a) and pooled fecal samples (b) via HPLC/Triple TOF MS.

Proposed metabolic pathways of furmonertinib in humans.
Discussion
In this study, the metabolic disposition and mass balance of furmonertinib in humans were investigated after a single oral administration of 97.9 μCi/81.5 mg [14C]-furmonertinib mesylate to healthy male volunteers. The absorption process of furmonertinib was fast, with the tmax of total radioactivity occurred at 0.5 h. The parent drug recovered in the feces accounted for only 1.38% of the dose, indicating nearly complete absorption of furmonertinib in the gastrointestinal tract. Furmonertinib is widely metabolized in humans, while the parent drug and AST5902 accounted for 1.68% and 0.97% of the total radioactivity in the plasma, respectively. The tmax values of furmonertinib and AST5902 were comparable to those obtained in the pharmacokinetic study following a single 80 mg oral dose of furmonertinib mesylate in patients with NSCLC [7].
The delayed absorption of the parent drug (tmax: 6 h) in comparison with total radioactivity (tmax: 0.5 h) may be caused by the strong lipid solubility of furmonertinib (log P > 4). At 0.5 h after administration, most of the radioactivity in plasma was present as covalent conjugates between furmonertinib and plasma proteins. In addition, most furmonertinib was distributed in tissues (Vd/F: 937 L). The subsequent release of free furmonertinib from the tissues to the plasma increased the amount of free and noncovalently bound forms of furmonertinib in the plasma. Under these circumstances, furmonertinib could reach its Cmax at 6 h. In fact, the tmax of total radioactivity was too early to rule out the influence caused by an error during radioanalysis.
The terminal t1/2 of the total radioactivity in the plasma was as long as 333 h, indicating that the covalent binding of drug-related substances to plasma proteins was irreversible to a great extent. Subsequent research in the researchers’ lab confirmed this point. The extraction efficiency during the metabolite identification process was investigated, and the values in the pooled plasma samples were too low for radiodetection. However, no such phenomenon occurred in the pooled urinary and fecal samples. Furthermore, the covalent adduct between furmonertinib and lysine-190 of human serum albumin (HSA) was confirmed using LC-MS [10]. Therefore, covalent binding to HSA caused the slow elimination of furmonertinib from the plasma, as the half-life of HSA is 19–20 days. EGFR-TKIs were designed to combine a nuclear structure with electrophilic groups, the former of which could competitively bind with the target and the latter of which covalently binds with a cysteine residue of the target [11]. Therefore, HSA covalent adducts have been found after the metabolism of various EGFR-TKIs, including neratinib, pyrotinib, osimertinib, and ibrutinib [12,13,14,15].
Furmonertinib is widely metabolized in humans after fast absorption. The main metabolic pathways of furmonertinib were N-demethylation (AST5902) and cysteine conjugation (M19) of the acrylamide group, considering that the total radioactivity values of AST5902 and M19 recovered in excretions were 6.28% and 5.52%, respectively, with respect to the dose. In addition, small amounts of M2, M5, M20, M9, M8-1, M6-2, M10, and M8-2 were found in feces, and each of them accounted for less than 1.5% of the dose. Considering that the metabolites of furmonertinib detected in feces were mainly oxidative metabolites, it is unlikely that the intestinal flora participates in the metabolism of furmonertinib. The metabolic enzyme CYP3A4 mostly contributes to the biotransformation from the parent drug to AST5902 [16]. Moreover, furmonertinib and AST5902 are potent inducers of CYP3A4, and the parent drug is also a CYP3A4 substrate. The induction degree of furmonertinib is similar to that of rifampicin, while the induction ability of AST5902 is relatively weak [16]. Therefore, attention should be paid to clinical drug–drug interactions when using furmonertinib for treatment, and plasma exposure to AST5902 may increase significantly when CYP3A4 is induced in vivo. Furthermore, the terminal t1/2 of AST5902 (62.1 h) in plasma is much longer than that of furmonertinib (37.2 h), and they possess similar pharmacological activity [7]. Thus, AST5902 may substantially contribute to the in vivo efficacy.
Furmonertinib is a structural analog of the marketed drug osimertinib, which is a third-generation tyrosine kinase inhibitor designed for the treatment of NSCLC. Furmonertinib and osimertinib have similar metabolic pathways, such as N-dealkylation, in humans. In addition to oxidative metabolism, cysteine and acetylcysteine conjugates were found after in vivo metabolism of furmonertinib and osimertinib [14]. The terminal t1/2 of the total radioactivity of osimertinib in human plasma was 474 h, which is slightly longer than that of furmonertinib (333 h). This finding was attributed to the reversible binding of furmonertinib with HSA, with a significantly lower binding rate than that of osimertinib [10]. Osimertinib was reported to be distributed mostly in the liver and lung, and the concentration of osimertinib-related radioactivity reached the highest at 6 h after administration to rats. However, furmonertinib was found to be primarily distributed in the lung within 4 h after administration. In addition, in vitro experiments showed that the t1/2 of the reaction between furmonertinib and plasma proteins was approximately 10.5 h, and this value for AST5902 was 12.7 h, thus demonstrating that furmonertinib was distributed to tissues faster than plasma protein binding occurred [10]. Therefore, furmonertinib may increase the lung cancer treatment efficacy and decrease hepatotoxicity compared with osimertinib.
After furmonertinib was orally administered, the total radioactivity recovered in excretions accounted for 77.8% of the dose within 840 h (35 days). Among the six volunteers, fecal excretion (71.2% of the dose) was the main excretion route of furmonertinib, with minor excretion via urine (6.63% of the dose).
In conclusion, [14C]-furmonertinib was completely absorbed, and the radioactivity rapidly distributed to tissues after its administration, with the highest concentration in the lung, the therapeutic target organ. In human plasma, more than 95% of furmonertinib and its metabolites covalently bound to plasma proteins, and the main forms of free drugs were the parent drug and AST5902, which possess similar pharmacological activity in vivo. Furmonertinib is metabolized primarily by CYP3A4 and subsequently eliminated via fecal excretion [16].
References
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34.
Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–7.
Han W, Du Y. Recent development of the second and third generation irreversible epidermal growth factor receptor inhibitors. Chem Biodivers. 2017; 14. https://doi.org/10.1002/cbdv.201600372.
Murtuza A, Bulbul A, Shen JP, Keshavarzian P, Woodward BD, Lopez-Diaz FJ, et al. Novel third-generation EGFR tyrosine kinase inhibitors and strategies to overcome therapeutic resistance in lung cancer. Cancer Res. 2019;79:689–98.
Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376:629–40.
Kim ES. Olmutinib: first global approval. Drugs. 2016;76:1153–7.
Shi Y, Zhang S, Hu X, Feng J, Ma Z, Zhou J, et al. Safety, clinical activity, and pharmacokinetics of alflutinib (AST2818) in patients with advanced NSCLC with EGFR T790M mutation. J Thorac Oncol. 2020;15:1015–26.
Liu X, Li W, Zhang Y, Jiang Y, Zhao Q, Zhong D. Simultaneous determination of alflutinib and its active metabolite in human plasma using liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2019;176:112735.
Graham RA, Lum BL, Morrison G, Chang I, Jorga K, Dean B, et al. A single dose mass balance study of the Hedgehog pathway inhibitor vismodegib (GDC-0449) in humans using accelerator mass spectrometry. Drug Metab Dispos. 2011;39:1460–7.
Liu X, Feng D, Zheng M, Zhong D. Characterization of covalent binding to human plasma proteins by selected covalent tyrisine kinase inhibitors. Drug Metab Pharmacokinet. 2020;35:456–65.
Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Chem Biol. 2011;10:307–17.
Meng J, Liu XY, Ma S, Zhang H, Yu SD, Zhang YF, et al. Metabolism and disposition of pyrotinib in healthy male volunteers: covalent binding with human plasma protein. Acta Pharmacol Sin. 2019;40:980–8.
Wang J, Li-Chan XX, Atherton J, Deng L, Espina R, Yu L, et al. Characterization of HKI-272 covalent binding to human serum albumin. Drug Metab Dispos. 2010;38:1083–93.
Dickinson PA, Cantarini MV, Collier J, Frewer P, Martin S, Pickup K, et al. Metabolic disposition of osimertinib in rats, dogs, and humans: insights into a drug designed to bind covalently to a cysteine residue of epidermal growth factor receptor. Drug Metab Dispos. 2016;44:1201–12.
Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, et al. Absorption, metabolism and excretion of oral 14C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 2014;43:289–97.
Liu X, Guo Z, Chen Z, Zhang Y, Zhou J, Jiang Y, et al. Alflutinib (AST2818), primarily metabolized by CYP3A4, is a potent CYP3A4 inducer. Acta Pharmacol Sin. 2020;41:1366–76.
Acknowledgements
This article was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA12050306) and the National Natural Science Foundation of China (No. 81521005).
Author information
Authors and Affiliations
Contributions
JM, ZDC, and DFZ contributed to the writing of this paper. JM, XYL, and DFZ participated in the drug metabolism study. HZ and LYM participated in clinical trials. JJB and YJ conducted pharmacological experiments. ZDC and YFZ performed pharmaceutical analysis and data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Meng, J., Zhang, H., Bao, Jj. et al. Metabolic disposition of the EGFR covalent inhibitor furmonertinib in humans. Acta Pharmacol Sin 43, 494–503 (2022). https://doi.org/10.1038/s41401-021-00667-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00667-8
Keywords
This article is cited by
-
Recent advances in targeting the “undruggable” proteins: from drug discovery to clinical trials
Signal Transduction and Targeted Therapy (2023)
-
Effect of autoinduction and food on the pharmacokinetics of furmonertinib and its active metabolite characterized by a population pharmacokinetic model
Acta Pharmacologica Sinica (2022)
-
Furmonertinib: First Approval
Drugs (2021)
