
Abstract
Ischemia/reperfusion (I/R) injury is a major cause of acute kidney injury (AKI) in clinic. The activation of NLRP3 inflammasome is associated with inflammation and renal injury in I/R-induced AKI. In the current study we explored the molecular and cellular mechanisms for NLRP3 inflammasome activation following renal I/R. Mice were subjected to I/R renal injury by clamping bilateral renal pedicles. We showed that I/R injury markedly increased caspase-11 expression and the cleavage of pannexin 1 (panx1) in the kidneys accompanied by NLRP3 inflammasome activation evidenced by the activation of caspase-1 and interlukin-1β (IL-1β) maturation. In Casp-11−/− mice, I/R-induced panx1 cleavage, NLRP3 inflammasome activation as well as renal functional deterioration and tubular morphological changes were significantly attenuated. In cultured primary tubular cells (PTCs) and NRK-52E cells, hypoxia/reoxygenation (H/R) markedly increased caspase-11 expression, NLRP3 inflammasome activation, IL-1β maturation and panx1 cleavage. Knockdown of caspase-11 attenuated all those changes; similar effects were observed in PTCs isolated from Casp-11−/− mice. In NRK-52E cells, overexpression of caspase-11 promoted panx1 cleavage; pretreatment with panx1 inhibitor carbenoxolone or knockdown of panx1 significantly attenuated H/R-induced intracellular ATP reduction, extracellular ATP elevation and NLRP3 inflammasome activation without apparent influence on H/R-induced caspase-11 increase; pretreatment with P2X7 receptor inhibitor AZD9056 also attenuated NLRP3 inflammasome activation. The above results demonstrate that the cleavage of panx1 by upregulated caspase-11 is involved in facilitating ATP release and then NLRP3 inflammasome activation in I/R-induced AKI. This study provides new insight into the molecular mechanism of NLRP3 inflammasome activation in AKI.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI) is a syndrome characterized by the rapid loss of kidney excretory function [1]. Ischemia/reperfusion (I/R) is the major cause of AKI in native kidneys and kidney allografts [2]. Renal tubules, a major component of the kidney, are vulnerable to a variety of injuries, including hypoxia and toxins [3]. For decades, numerous studies have investigated the molecular and cellular mechanisms of I/R-induced renal injury; however, the mechanistic understanding is incomplete.
The nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) inflammasome is activated in response to pathogens and endogenous danger signals. Activation of the NLRP3 inflammasome has been implicated in AKI induced by different stimuli [4, 5]. Activation of the NLRP3 inflammasome is mediated by two steps. The first step, referred to as priming, includes the upregulation of NLRP3 and pro-IL-1β expression via the activation of NF-κB; the second step is the assembly of the NLRP3 inflammasome in response to various stimuli, including ATP, bacterial pore-forming toxins, or particulate matter. ATP, the major biological energy molecule of the cell, acts as a signaling mediator and modulates multiple cellular functions. By activating and opening the purinergic receptor P2X (P2X7), a ligand-gated ion channel, ATP induces cations in the intracellular fluid and extracellular fluid to traverse the plasma membrane, resulting in K+ efflux and Na+ influx. It has been demonstrated that the efflux of intracellular K+ is a trigger for NLRP3 inflammasome activation [6] and thereby activates caspase-1 and leads to the proteolytic cleavage of proinflammatory substrates such as pro-IL-1β into active mediators [7]. Previous studies have demonstrated that the activation of the NLRP3 inflammasome plays an important role in renal damage [8,9,10,11]. However, the exact mechanism of NLRP3 inflammasome activation during AKI remains poorly understood.
Cysteine-aspartic protease (caspase)-11 is a member of the inflammatory caspase subfamily. Caspase-11 and its human orthologs caspase-4/caspase-5 were first identified as sensors of intracellular lipopolysaccharide (LPS) derived from gram-negative bacteria in macrophage-mediated inflammatory responses [12, 13]. In the noncanonical inflammasome pathway, activation of caspase-11 by direct binding of LPS promotes proinflammatory cytokine maturation independent of NLRP3 activation. Later studies showed that caspase-11 could also be activated by noninfectious factors [14, 15], especially in kidney diseases, such as unilateral ureteral obstruction [16], and in I/R-induced AKI mouse models [17, 18]. Recently, an interesting phenomenon was noticed: the noncanonical caspase-11 inflammasome pathway can also promote IL-1β maturation and secretion by triggering the activation of the canonical NLRP3 inflammasome [13]; however, the link between caspase-11 and the NLRP3 inflammasome remains unclear.
The pannexin1 (panx1) channel is a nonselective, large-pore channel that is responsible for the release of nucleotides and ATP [19]. In response to apoptotic stimuli, the panx1 channel can be functionally activated by caspase-3-mediated cleavage of the intracellular autoinhibitory distal domain [20, 21]. It was recently noticed that the panx1 channel can also be activated by caspase-11-dependent cleavage following LPS stimulation [22]. Panx1 is present in polarized epithelial cells in the lung and kidney [23, 24]. In the kidney, panx1 expression was found in the apical membranes of proximal tubules, thick descending limbs, collecting ducts and renal vasculature [24]. Although recent studies have revealed that pharmacologic inhibition of panx1 protects mouse kidneys from I/R-induced injury [25], the mechanism by which panx1 activation mediates tubular epithelial cell injury has not been explored.
In this study, we investigated how caspase-11 contributes to AKI. Our data from I/R-induced AKI mice, hypoxia/reoxygenation (H/R)-treated NRK-52E cells and cultured primary tubular cells (PTCs) showed that the expression of caspase-11 was increased after I/R or H/R treatment and associated with NLRP3 inflammasome activation. Caspase-11 activated panx1 by catalytic cleavage and then facilitated the release of ATP, which in turn stimulated NLRP3 inflammasome activation and promoted kidney injury.
Materials and methods
Animals and surgical protocol
Mice were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). Eight- to 10-week-old C57BL/6J mice (20–25 g) were used for all experiments. Casp-11−/− C57BL/6J mice were obtained from The Jackson laboratory (ME, USA). Tail DNA was isolated to analyze the genotype at the age of 2–3 weeks. The PCR primers used for genotyping were as follows: a, 5′-CGCTTCCTCGTGCTTTACGGTAT-3′; b, 5′-ACAATTGCCACTGTCCAGGT-3′; and c, 5′- CATTGCTGACCTTATTTCTGTATGG-3′. The PCR product formutant (Casp-11−/−) mice was 650 bp, and the PCR product for wild-type (WT) mice was 495 bp. Mice were maintained in standard vivarium housing on a chow diet with freely available water. All animal experiments were performed according to the Criteria of the Medical Laboratory Animal Administrative Committee of Shanghai and the Guide for Care and Use of Laboratory Animals of Fudan University. The protocols were approved by the Ethics Committee for Experimental Research of Shanghai Medical College, Fudan University (20160225–087).
Bilateral ischemia-reperfusion was performed according to a procedure described previously [26]. Briefly, both renal pedicles were clamped for 45 min followed by reperfusion. After reperfusion, the kidneys were inspected to ensure blood flow (the color returned from dark red to their original color within 1 min). Kidney cortices were harvested after 24 h. Littermates with sham operations were used as control subjects.
Evaluation of renal function
The urine creatinine level was assessed using the QuantiChromCreatinine Assay Kit (DICT-500, BioAssay Systems, CA, USA). A standard curve was created from the stock 50 mg/dL creatinine standard. Concentrations of 6, 2, 1, 0.5, and 0 mg/dL were used to create the standard curve. Creatinine concentrations were determined by measuring absorbance per the manufacturer’s instructions. The level was measured by an automatic chemistry analyzer (Cobas C 501, Roche, Basel, Switzerland). The BUN level was measured using a QuantiChrom assay kit (DIUR-500).
Cell culture and exposure to H/R
Primary renal tubular cells were freshly isolated after harvesting the kidneys. The renal cortices were digested with 0.75 mg/mL collagenase at 37 °C for 30 min. The digestion was stopped by DMEM (HyClone) supplemented with 10% FBS (Gibco). The suspension was filtered through an 80 μm strainer and collected. Finally, the freshly isolated tubule fragments were plated into 60 mm dishes and cultured in DMEM supplemented with 10% FBS at 37 °C. After 4 days, the cultured cells were identified as renal tubular epithelial cells with AQP1-positive and α-SMA-negative staining.
NRK-52E (rat renal tubular epithelial cell line) cells were purchased from the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin in 5% CO2 and 95% air at 37 °C.
For cell exposure to H/R, cells were cultured under hypoxic conditions (1% O2, 94% N2, and 5% CO2) at 37 °C for 24 h to induce hypoxic injury and then returned to 5% CO2 and 95% air for reoxygenation. Cells and supernatants were harvested at 24 h after reoxygenation.
A recombinant adenovirus vector carrying caspase-11 (Ad-caspase-11) and an empty adenovirus vector (Ad-null) were obtained from ObioTechnology Company (Shanghai, China).
ATP measurement
ATP levels were measured in cells using an ATP Assay Kit per the manufacturer’s recommendations (Beyotime Institute of Biotechnology, Shanghai, China). Briefly, extracellular ATP was measured in the culture medium, which was mixed at 1:1 with the luciferase reagent. Cultured cells were harvested and lysed with lysis buffer, followed by centrifugation at 12,000 × g for 2 min at 4 °C. Finally, the level of ATP was determined by mixing the supernatant with the luciferase reagent, which catalyzes the production of light by ATP and luciferin. The emitted light was linearly related to the ATP concentration and was measured using a microplate luminometer.
RNA interference
Knockdown of the target gene was performed by transfection of specific siRNA facilitated by Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, 13778030). Caspase-11 siRNA (5′-GCAAUGUAUUGAAAUUAAAdTdT-3′ sense, 3′-UUUAAUUUCAAUACAUUGCdTdT-5′ antisense) and panx1 siRNA (5′-AGCAGUACUUGAAGACGAAdTdT-3′ sense, 3′-UUCGUCUUCAAGUACUGCUdTdT-5′ antisense) were purchased from Biotend Company (Guangzhou, China). A nonsilencing siRNA (NC-siRNA) oligonucleotide that does not recognize any known homolog of mammalian genes was used as a negative control.
Western blot
For Western blot analyses, total tissue or cellular lysate was subjected to 5%–15% SDS-PAGE. The fractionated proteins were transferred onto PVDF membranes that were then incubated with primary antibodies. Primary antibodies against the following proteins were used: caspase-11 (ab180673, 1:1000), NLRP3 (ab214185, 1:1000) and IL-1β (ab9722, 1:1000) (all purchased from Abcam Biotechnology, MA, USA). The anti-panx1 (91137S,1:1000) antibody was purchased from Cell Signaling Technology (MA, USA). The anti-caspase-1 (sc-514, 1:500) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-α-tubulin (66031–1-lg, 1:5000) antibody was purchased from Proteintech Group (MA, USA). After overnight incubation, the membranes were immersed in a solution containing individual secondary antibodies (A0216 and A0208, 1:1000, Beyotime Institute of Biotechnology, Shanghai, China). The bands were visualized by an ECL system (Amersham Biosciences).
Assessment of kidney histology
The kidneys were fixed in 10% formalin and embedded in paraffin. Sections were cut at a thickness of 4 μm. The sections were subjected to HE, periodic acid-Schiff (PAS) and immunohistochemical staining. In the HE-stained sections, the extent of tubular detachment, foamy degeneration, leukocyte infiltration and necrosis were evaluated and scored as follows: normal, 0; mild injury, 1; moderate injury, 2; and severe injury, 3.
Immunohistochemistry and immunofluorescence staining
The renal sections were deparaffinized and rehydrated and then subjected to antigen retrieval in 0.01 M citrate buffer (pH 6.0) by microwaving. After blocking with 5% bovine serum albumin, the sections were incubated with anti-caspase-11 (sc-374615, 1:500, Santa Cruz Biotechnology), anti-AQP1 (ab15080, 1:500, Abcam), anti-MPO (ab208670, 1:1000, Abcam) or anti-F4/80 (70076,1:1000, Cell Signaling Technology) antibody overnight at 4 °C, followed by secondary antibody (Jackson ImmunoResearch Laboratories, PA, USA). Sections were viewed under a fluorescence microscope (Leica, Wetzlar, Germany).
Statistical analysis
The data are shown as the mean ± SEM (standard error of the mean) and were analyzed by GraphPad Prism 7.0 (CA, USA).
Statistical differences among groups were analyzed using the t-test (2 groups) and one-way ANOVA plus Tukey’s posttest (≥3 groups). Statistical significance was set at P < 0.05, P < 0.01, and P < 0.001.
Results
Caspase-11 expression was increased in I/R mouse kidneys
The expression of caspase-11 was examined in I/R mouse kidneys. As shown in Fig. 1a, caspase-11 expression was significantly increased in I/R mice compared with sham animals. Immunohistological analyses confirmed that caspase-11 was mainly expressed in renal tubules and was markedly upregulated in I/R mice (Fig. 1b). Immunofluorescence imaging also showed that caspase-11 expression in the renal tubules was increased in I/R mice compared with sham mice, and caspase-11 colocalized with the proximal tubular epithelial cell marker AQP1 (Fig. 1c). To elucidate the role of caspase-11 in I/R-induced AKI, Casp-11−/− mice were obtained for further experiments. The genotyping PCR results showed that the expected 495 bp band was observed in WT mice, while the 650 bp band was observed in Casp-11−/− mice. Western blot results also showed that, as expected, caspase-11 was expressed in WT mice but was undetectable in Casp-11−/− mice (Fig. 1d, e).

Kidney and blood samples were harvested from mice after bilateral ischemia or sham operation followed by 24 h of reperfusion (n = 6). a Representative Western blot and quantitative data showing the expression of caspase-11 in the sham and I/R mice. ***P < 0.001 compared with the sham group. b The expression of caspase-11 was detected by immunohistological analysis in the sham and I/R mice. ***P < 0.001 compared with the sham group. c Coimmunofluorescence staining for caspase-11 (red) and AQP1 (green) in the sham and I/R-treated mice. d The identification of Casp-11−/− mice. Mice exhibiting only the 650-base pair (bp) polymerase chain reaction product were assessed as Casp-11−/− mice. e Western blot of caspase-11 in WT and Casp-11−/− mouse kidneys. Data are presented as the mean ± SEM. AQP1 aquaporin-1, I/R ischemia/reperfusion.
Knockout of caspase-11 ameliorated renal morphological changes and dysfunction after I/R treatment
Histological analysis showed that renal tubular injury, including tubular dilation, loss of brush border, and cast formation, was observed in I/R mice, while the morphological changes were significantly ameliorated in Casp-11−/− mice (Fig. 2a, b). The levels of serum BUN and serum creatinine were significantly increased in I/R mice compared with sham mice, and the increases were significantly suppressed in Casp-11−/− mice (Fig. 2c, d). These data suggested that the elevation in caspase-11 contributed to renal tubular injury and dysfunction in I/R-induced AKI mice.

Kidney and blood samples were harvested from mice after bilateral ischemia or sham operation followed by 24 h of reperfusion (n = 6). a Histological damage in the HE-stained kidney cortex sections.The renal tissue injury score was used to grade renal tubular damage in the HE-stained kidney sections. **P < 0.01 compared with the sham group. ##P < 0.01 compared with the WT + I/R group. b Histological damage in the PAS-stained kidney cortex sections. The renal tissue injury score was used to grade renal tubular damage in the PAS-stained kidney sections. **P < 0.01 compared with the sham group. ##P < 0.01 compared with the WT + I/R group. c, d Serum samples were collected for measurements of BUN and serum creatinine levels. **P < 0.01 compared with the sham group. ##P < 0.01 compared with the WT + I/R group. Data are presented as the mean ± SEM. WT wild-type, I/R ischemia/reperfusion, PAS periodic acid-Schiff, BUN blood urea nitrogen.
Knockout of caspase-11 suppressed NLRP3 inflammasome activation and neutrophil infiltration in I/R mice
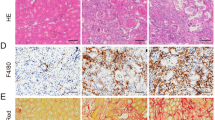
NLRP3 inflammasome activation is essential to initiate renal inflammation and exacerbates I/R-induced kidney injury [27]. NLRP3 inflammasome activation was evaluated by measuring the cleavage of caspase-1 (p20 subunit of caspase-1) and the maturation of IL-1β (p17 subunit of IL-1β) by Western blotting. The expression of NLRP3 and the cleavage of caspase-1 and IL-1β were significantly increased in I/R mice. All these changes were significant and were attenuated in Casp-11−/− mice (Fig. 3a). The activation of renal inflammation stimulates the infiltration of immune cells and then promotes the progression of AKI [28]. Immunohistochemistry results indicated that the numbers of infiltrated MPO-positive neutrophils and F4/80-positive macrophages were significantly increased in the kidneys of I/R mice and were reduced in Casp-11−/− mice (Fig. 3b). These results suggested that knocking out caspase-11 attenuated NLRP3 inflammasome activation and renal infiltration of immune cells in I/R mice.

Mouse kidneys were harvested after bilateral ischemia or sham operation followed by 24 h of reperfusion (n = 6). a Representative Western blot and quantitative data showing the expression of NLRP3, caspase-1 and IL-1β in sham or I/R-treated kidneys from WT and Casp-11−/− mice. **P < 0.01 compared with the sham group. #P < 0.05 compared with the WT + I/R group. b Renal neutrophil infiltration was evaluated by immunohistochemical detection of MPO. Renal macrophage infiltration was evaluated by immunohistochemical detection of F4/80. Scale bar = 100 μm. Data are presented as the mean ± SEM. I/R ischemia/reperfusion.
Knockdown of caspase-11 reduced NLRP3 inflammasome activation in tubular epithelial cells after H/R treatment
To identify the relationship between caspase-11 elevation and NLRP3 inflammasome activation, an in vitro H/R approach was used. In both NRK-52E cells and PTCs, caspase-11 expression was increased significantly after H/R treatment (Fig. 4a, b). Caspase-1 activation and IL-1β maturation were also increased following H/R treatment in NRK-52E cells (Fig. 4c). Small interfering RNA (siRNA) targeting caspase-11 (si-Casp-11) was used to knockdown caspase-11 expression in NRK-52E cells. As shown in Fig. 4d, caspase-11 expression in NRK-52E cells was decreased by ~80% after transfection with si-Casp-11. Knockdown of caspase-11 inhibited NLRP3 inflammasome activation, as indicated by the suppression of caspase-1 activation and IL-1β maturation in NRK-52E cells after H/R treatment (Fig. 4e). In PTCs, caspase-1 activation and IL-1β maturation were increased after H/R treatment, while these changes were attenuated in PTCs isolated from Casp-11−/− mice (Fig. 4f, g). These results indicated that the increase in caspase-11 expression was associated with H/R-induced NLRP3 inflammasome activation.

NRK-52E cells and isolated mouse primary tubular cells were exposed to hypoxia for 24 h followed by 24 h of reoxygenation. a Representative Western blot and quantitative data showing the expression of caspase-11 in control and H/R-treated NRK-52E cells. ***P < 0.001 compared with the control group. b Representative Western blot and quantitative data showing the expression of caspase-11 in control and H/R-treated primary tubular epithelial cells. ***P < 0.001 compared with the control group. c Western blotting was performed to visualize NLRP3, caspase-1 and IL-1β expression in control and H/R-treated NRK-52E cells. **P < 0.01 compared with the control group. d Western blotting was performed to examine the silencing efficiency oftransfected si-Casp-11. **P < 0.01 compared with the NC-siRNA group. e After transfection with caspase-11 siRNA or NC-siRNA and treatment with H/R, cells were harvested to evaluate the expression of caspase-11, NLRP3, caspase-1 and IL-1β. Representative Western blot analyses of the expression of NLRP3, caspase-1 and IL-1β in H/R-treated NRK-52E cells are shown. **P < 0.01, ***P < 0.001 compared with the Con+NC-siRNA group. #P < 0.05 compared with the H/R + NC-siRNA group. f Western blotting was performed to examine the silencing efficiency of caspase-11 knockout. ***P < 0.001 compared with the WT group. g Representative Western blot analyses of the expression of NLRP3, caspase-1 and IL-1β in H/R-treated mouse primary tubular cells. **P < 0.01, ***P < 0.001 compared with the control group. #P < 0.05 compared with the Con+H/R group. Data are presented as the mean ± SEM. Con control, H/R hypoxia/reperfusion, NC-siRNA negative control siRNA, si-Casp-11 caspase-11 small interfering RNA, WT wild-type.
Knockout of caspase-11 prevented the cleavage of panx1 in vivo and in vitro
Panx1 can be activated by cleavage of the COOH-terminal autoinhibitory domain [20, 21]. The caspase-11-mediated noncanonical inflammasome pathway is engaged in the activation of the panx1 channel [22]. To elucidate the relationship between caspase-11 and panx1 cleavage after H/R treatment, the expression and cleavage of panx1 were detected by Western blot. The expression and cleavage of panx1 was significantly increased in NRK-52E cells after H/R treatment (Fig. 5a). To determine whether caspase-11 is able to induce the cleavage of panx1, the expression of caspase-11 in NRK-52E cells was knocked down by transfection with si-Casp-11. The cleavage of panx1 induced by H/R was significantly suppressed by si-Casp-11 (Fig. 5b). This result was confirmed in PTCs in which the expression levels of both caspase-11 and panx1 were increased after H/R, while these changes were significantly inhibited in PTCs isolated from Casp-11−/− mice (Fig. 5c). To further determine the effect of caspase-11 on the cleavage of panx1, Ad-casp-11 was transfected into NRK-52E cells to overexpress caspase-11. As shown in Fig. 5d, overexpression of caspase-11 significantly promoted the cleavage of panx1 in Ad-casp-11 NRK-52E cells compared with that in Ad-null cells. As shown in Fig. 5e, the expression and cleavage of panx1 were also increased in I/R mice. The increase in panx1 cleavage was attenuated in Casp-11−/− mice. These results suggested that the increase in caspase-11 was responsible for the I/R-induced cleavage of panx1.

NRK-52E cells were exposed to hypoxia for 24 h followed by 24 h of reoxygenation. a Representative Western blot and quantitative data showing the expression and cleavage of panx1 in control and H/R-treated NRK-52E cells. **P < 0.01 compared with the control group. b After transfection with caspase-11 siRNA or NC-siRNA and treatment with H/R, cells were harvested to evaluate the expression of caspase-11 and panx1. Representative Western blot and quantitative data showing the expression of caspase-11 and panx1 in NRK-52E cells. **P < 0.01 compared with the Con+NC-siRNA group. #P < 0.05 compared with the H/R + NC-siRNA group. c Representative Western blot and quantitative data showing the expression of caspase-11 and panx1 in H/R-treated primary tubular epithelial cells from WT and Casp-11−/− mice. **P < 0.01 compared with the control group. #P < 0.05, ##P < 0.01 compared with the H/R + Con group. d NRK-52E cells were treated with Ad-Casp-11 or Ad-null. Western blotting was performed to examine the expression of caspase-11 and panx1. **P < 0.01, ***P < 0.001 compared with the Ad-nullgroup. e Representative Western blot and quantitative data showing the expression of caspase-11 and panx1 in I/R-treated kidneys from WT and Casp-11−/−mice. **P < 0.01 compared with the sham mice. #P < 0.05, ##P < 0.01 compared with the WT + I/R group. Data are presented as the mean ± SEM. Con control, H/R hypoxia/reperfusion, Ad-null empty adenovirus vector, Ad-Casp-11 adenovirus containing the caspase-11 gene, NC-siRNA negative control siRNA, si-Casp-11 caspase-11 small interfering RNA.
Inhibiting panx1 activation and ATP release reduced NLRP3 inflammasome activation in NRK-52E cells after H/R
An increase in extracellular ATP released by panx1 stimulates renal inflammation [25]. To determine whether the activation of panx1 by caspase-11-induced cleavage was related to H/R-induced NLRP3 inflammasome activation, panx1 was inhibited by the inhibitor CBX or siRNA knockdown. As shown in Fig. 6a, CBX pretreatment significantly suppressed H/R-induced caspase-1 cleavage and IL-1β maturation, which indicated that the activation of the NLRP3 inflammasome was suppressed by the panx1 inhibitor, although the expression of caspase-11 and NLRP3 and the cleavage of panx1 were not obviously affected. Intracellular ATP was significantly reduced in cells exposed to H/R compared with control cells. CBX significantly attenuated the decrease in intracellular ATP after H/R (Fig. 6b). In contrast, extracellular ATP was significantly increased in the cells exposed to H/R compared with the control cells, and CBX significantly attenuated the increase in extracellular ATP after H/R (Fig. 6c). All these results indicated that the release of ATP was increased after H/R and that panx1 was involved in facilitating the release of ATP. Similarly, knockdown of panx1 by siRNA attenuated H/R-induced NLRP3 inflammasome activation, which was indicated by the increase in caspase-1 cleavage and IL-1β maturation. The decrease in intracellular ATP and increase in extracellular ATP after H/R treatment were attenuated using panx1 siRNA (Fig. 6d, e). Pharmacological blockade of the ATP P2X7 receptor by AZD9056 also significantly attenuated H/R-induced caspase-1 cleavage and IL-1β maturation (Fig. 6h). These results demonstrated that the panx1-mediated release of ATP was involved in H/R-induced NLRP3 inflammasome activation.

NRK-52E cells were exposed to hypoxia for 24 h followed by 24 h of reoxygenation. a Representative Western blot and quantitative data showing the expression of caspase-11, panx1, NLRP3, caspase-1 and IL-1β in H/R-treated NRK-52E cells with or without 50 μM CBX pretreatment. **P < 0.01, ***P < 0.001 compared with the control group. #P < 0.05 compared with the H/R group. b The intracellular ATP levels in cultured cells were measured. **P < 0.01 compared with the control group. #P < 0.05 compared with the H/R group. c The extracellular ATP levels in the culture mediawere measured. *P < 0.05, **P < 0.01 compared with the control group. #P < 0.05 compared with the H/R group. d Western blotting was performed to examine the silencing efficiency of the transfected panx1 siRNA. e After transfection with panx1 siRNA or NC-siRNA and treatment with H/R, cells were harvested to evaluate the expression of caspase-1 and IL-1β. The expression of caspase-1 and IL-1β was assayed using Western blotting. **P < 0.01 compared with the control group. #P < 0.05 compared with the H/R group. f The intracellular ATP levels in cultured cells were measured. **P < 0.01 compared with the control group. ##P < 0.01 compared with the H/R group. g The extracellular ATP levels in the culture mediawere measured. *P < 0.05, **P < 0.01 compared with the control group. #P < 0.05 compared with the H/R group. h Representative Western blot and quantitative data showing the expression of caspase-1 and IL-1β in H/R-treated NRK-52E cells with or without pretreatment with 12.5 μM the P2X7 inhibitor AZD9056. **P < 0.01 compared with the control group. ##P < 0.01 compared with the H/R group. Data are presented as the mean ± SEM. Con control, H/R hypoxia/reperfusion, CBX carbenoxolone(a panx1 inhibitor), NC-siRNA negative control siRNA, si-panx1 panx1 small interfering RNA.
Discussion
In addition to rapid loss of kidney function, another characteristic of AKI is severe inflammation. The activation of the NLRP3 inflammasome plays an essential role in inflammation in AKI induced by different stimuli. However, the exact mechanism of NLRP3 inflammasome activation remains unclear.
Caspase-11 belongs to the inflammatory caspase subfamily and was originally identified to play a crucial role in innate immune defense against gram-negative bacteria. However, later studies demonstrated that in addition to LPS, a series of noninfectious factors, including age-related macular degeneration [15], can also promote the transcription and activation of caspase-11. Previous studies have shown that caspase-11 is upregulated in noninfectious renal injury models, such as unilateral ureteral obstruction-induced renal fibrosis and cisplatin- or I/R-induced AKI [16,17,18]. In this study, caspase-11 was upregulated in the kidneys of I/R-induced AKI mice and was mainly distributed in tubular epithelial cells, which was consistent with previous observations. Knockout of caspase-11 significantly reduced tubular injury, renal functional deterioration, immune cell infiltration and inflammatory cytokine maturation after I/R. These results indicated that the elevation of caspase-11 was related to renal injury as well as inflammation in I/R-induced AKI.
Panx1 belongs to the pannexin family of plasma membrane proteins (panx1, panx2, and panx3). Panx1 is widely expressed in almost all tissues, whereas panx2 is predominantly expressed in the central nervous system, and panx3 is expressed in the skin, bone, and cartilage. In the kidney, panx1 was detected in the proximal tubule, the thin descending limb of the loop of Henle, and the collecting duct system. Similar to other members of the pannexin family, the activation of panx1 is regulated by posttranslational modifications. Panx1 is a channel formation protein that is activated by proteolytic cleavage at the distal end of its intracellular domain. After cleavage, panx1 is observed as a 42 kDa fragment lacking 46 amino acids on its COOH-terminal segment. The cleavage of panx1 and opening of the channel were first identified to be initiated by caspase-3 [20, 21, 29]. Pharmacological inhibition or genetic ablation of the panx1 channel protected kidneys from ischemic injury [25]. PeptideCutter (web.expasy.org/peptide_cutter/) analysis showed that panx1 exhibits a predicted caspase-11 cleavage site. In 2015, Yang D et al. identified that the cleavage of panx1 can also be triggered by caspase-11 [22]. In this study, our results showed that overexpression of caspase-11 significantly promoted the cleavage of panx1 in NRK-52E cells . Knockdown of caspase-11 suppressed the cleavage of panx1 in NRK-52E cells and PTCs after H/R treatment as well as in I/R mice. All these results indicated that the upregulation of caspase-11 after I/R or H/R was involved in the cleavage and activation of panx1.
Under physiological conditions, activated panx1 conducts molecules up to 1 kDa in size across the plasma membrane, which includes releasing intracellular ATP. This phenomenon was also observed in the present study. H/R induced a decrease in intracellular ATP in cultured epithelial cells with a simultaneous increase in ATP concentration in the culture medium, while pharmacological blockade or knockdown of panx1 attenuated the decrease in intracellular ATP and increase in culture medium ATP. These results suggested that the activation of panx1 induced the release of ATP. Although the significance has not been fully elucidated, it is possible that the release of ATP facilitates cell accommodation to pathological conditions, such as ischemia and inflammation [30]. In addition, the accumulation of extracellular ATP is also considered an important danger-associated molecular pattern. Extracellular ATP can activate all P2X receptors and some P2Y receptors [31]. By acting on P2 purinergic receptors, extracellular ATP activates NLRP3 inflammasome-dependent signaling [32] and is involved in different renal pathological conditions. In the present study, inhibition of panx1 by CBX or knockdown of panx1 by siRNA attenuated ATP release induced by H/R treatment. Pretreatment of the cells with the P2X7 inhibitor AZD9056 attenuated H/R-induced NLRP3 inflammasome activation. These results suggested that ATP release from panx1 channels contributed to H/R-induced NLRP3 inflammasome activation and the inflammatory response.
Renal tubular epithelial cells are extremely susceptible to intrinsic oxidative stress, particularly during the reperfusion phase of I/R [33]. During AKI, the tubule is not only the key site of injury but also an important source of inflammatory cytokines. The expression of inflammatory cytokines by tubular cells is an important determinant of the injury and repair phases. Renal inflammation is a universal response to infectious and noninfectious triggers [34]. Increasing data now show that NLRP3 inflammasome activation and IL-1β maturation are essential in many kidney diseases, including diabetes, amyloidosis, malaria, crystal-related diseases, and autoinflammatory disorders. Thus, the innate immune pathway is considered an attractive therapeutic target for renal diseases [34]. In this study, I/R-induced activation of the NLRP3 inflammasome was diminished in Casp-11−/− mice. Similar results were also observed in NRK-52E cells after transfection with caspase-11 siRNA and H/R treatment. Pharmacological inhibition or siRNA knockdown of panx1 and blockade of the ATP P2X7 receptor suppressed ATP release and NLRP3 inflammasome activation after H/R treatment. All these observations implicated the cleavage of panx1 by caspase-11 in facilitating NLRP3 inflammasome activation during H/R.
Our study provides new insight into the mechanism of NLRP3 inflammasome activation and IL-1β maturation. These results suggest that caspase-11-mediated panx1 cleavage is implicated in inflammation related to NLRP3 activation. However, NLRP3 inflammasome activation includes two steps, priming and activation, and our study focuses only on the mechanism of the second step. The initial priming phase involves NF-κB-dependent transcription of NLRP3 and pro-IL-1β. The relationship between caspase-11 elevation and NF-κB needs to be clarified in further studies.
In conclusion, the data obtained in this study suggest that the cleavage of panx1 by caspase-11 is implicated in NLRP3 inflammasome activation and subsequent inflammation in IR-induced AKI. Upregulation of caspase-11 promoted the cleavage of panx1. Activation of the panx1 channel mediated ATP release and promoted NLRP3 inflammasome activation, inflammatory cytokine release and kidney injury. Based on these observations, suppressing caspase-11-induced NLRP3 inflammasome activation might have beneficial effects in alleviating AKI (Fig. 7).

The expression of caspase-11 was increased under I/R-induced acute kidney injury, which induced renal functional deterioration and neutrophil infiltration. Caspase-11-mediated cleavage of panx1 caused panx1 channel opening and thus ATP release. Extracellular ATP released by the renal tubular epithelial cells activated P2X7 to trigger K+ efflux and NLRP3 inflammasome activation under H/R conditions. The activated NLRP3 inflammasome catalyzed pro-IL-1β cleavage into IL-1β. IL-1β has been implicated in the progression of acute kidney disease. I/R ischemia/reperfusion.
References
Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380:756–66.
Bonventre JV, Weinberg JM. Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol. 2003;14:2199–210.
Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93:568–79.
Kim SM, Kim YG, Kim DJ, Park SH, Jeong KH, Lee YH, et al. Inflammasome-independent role of NLRP3 mediates mitochondrial regulation in renal injury. Front Immunol. 2018;9:2563.
Wen Y, Liu YR, Tang TT, Pan MM, Xu SC, Ma KL, et al. mROS-TXNIP axis activates NLRP3 inflammasome to mediate renal injury during ischemic AKI. Int J Biochem Cell Biol. 2018;98:43–53.
Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010;21:1732–44.
Ludwig-Portugall I, Bartok E, Dhana E, Evers BD, Primiano MJ, Hall JP, et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016;90:525–39.
Kim SM, Lee SH, Kim YG, Kim SY, Seo JW, Choi YW, et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am J Physiol Ren Physiol. 2015;308:F993–F1003.
Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87:74–84.
Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. 2015;32:78–83.
Kayagaki N, Warming S, Lamkanfi M, Vande WL, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21.
Wang J, Sahoo M, Lantier L, Warawa J, Cordero H, Deobald K, et al. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018;14:e1007105.
Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2018;24:50–61.
Miao NJ, Xie HY, Xu D, Yin JY, Wang YZ, Wang B, et al. Caspase-11 promotes renal fibrosis by stimulating IL-1beta maturation via activating caspase-1. Acta Pharmacol Sin. 2019;40:790–800.
Yang JR, Yao FH, Zhang JG, Ji ZY, Li KL, Zhan J, et al. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Ren Physiol. 2014;306:F75–F84.
Miao N, Yin F, Xie H, Wang Y, Xu Y, Shen Y, et al. The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int. 2019;96:1105–20.
D’Hondt C, Ponsaerts R, De Smedt H, Bultynck G, Himpens B. Pannexins, distant relatives of the connexin family with specific cellular functions? Bioessays. 2009;31:953–74.
Sandilos JK, Chiu YH, Chekeni FB, Armstrong AJ, Walk SF, Ravichandran KS, et al. Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J Biol Chem. 2012;287:11303–11.
Boyd-Tressler A, Penuela S, Laird DW, Dubyak GR. Chemotherapeutic drugs induce ATP release via caspase-gated pannexin-1 channels and a caspase/pannexin-1-independent mechanism. J Biol Chem. 2014;289:27246–63.
Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43:923–32.
Ransford GA, Fregien N, Qiu F, Dahl G, Conner GE, Salathe M. Pannexin 1 contributes to ATP release in airway epithelia. Am J Respir Cell Mol Biol. 2009;41:525–34.
Hanner F, Lam L, Nguyen MT, Yu A, Peti-Peterdi J. Intrarenal localization of the plasma membrane ATP channel pannexin1. Am J Physiol Ren Physiol. 2012;303:F1454–F1459.
Jankowski J, Perry HM, Medina CB, Huang L, Yao J, Bajwa A, et al. Epithelial and endothelial pannexin1 channels mediate AKI. J Am Soc Nephrol. 2018;29:1887–99.
Gao S, Zhu Y, Li H, Xia Z, Wu Q, Yao S, et al. Remote ischemic postconditioning protects against renal ischemia/reperfusion injury by activation of T-LAK-cell-originated protein kinase (TOPK)/PTEN/Akt signaling pathway mediated anti-oxidation and anti-inflammation. Int Immunopharmacol. 2016;38:395–401.
Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, et al. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol. 2010;185:6277–85.
Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. 2015;11:88–101.
Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature. 2010;467:863–7.
Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med. 2010;16:1434–8.
Vallon V. P2 receptors in the regulation of renal transport mechanisms. Am J Physiol Ren Physiol. 2008;294:F10–F27.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8.
Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–6.
Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22:1007–18.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 82070712, 81873603, 81670664) to LML, and (No. 81873603) to XXW. The authors would like to thank the research participants for volunteering their time in the study.
Author information
Authors and Affiliations
Contributions
FY performed experiments, data collection and analysis, and wrote the paper. PQZ and LQZ participated in experiments in vitro and animal model establishment. NJM provided some knockout mice. QC, ZLZ, PPC, HYX, JYL and JYN contributed to the material preparation and data collection.YZW, LZ and WZ contributed to the study conception and design. XXW, JL and LML conducted study design and supervised the findings.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Yin, F., Zheng, Pq., Zhao, Lq. et al. Caspase-11 promotes NLRP3 inflammasome activation via the cleavage of pannexin1 in acute kidney disease. Acta Pharmacol Sin 43, 86–95 (2022). https://doi.org/10.1038/s41401-021-00619-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00619-2
Keywords
This article is cited by
-
P2X7 receptors: a bibliometric review from 2002 to 2023
Purinergic Signalling (2024)
-
Caffeine and neonatal acute kidney injury
Pediatric Nephrology (2024)
-
TLR2/caspase-5/Panx1 pathway mediates necrosis-induced NLRP3 inflammasome activation in macrophages during acute kidney injury
Cell Death Discovery (2022)
-
Co-treatment with Esculin and erythropoietin protects against renal ischemia–reperfusion injury via P2X7 receptor inhibition and PI3K/Akt activation
Scientific Reports (2022)
-
Novel insights into NOD-like receptors in renal diseases
Acta Pharmacologica Sinica (2022)
