
Abstract
Endoplasmic reticulum stress (ER stress) plays a key role in the development of cardiac hypertrophy and diabetic cardiomyopathy (DCM). Zonisamide (ZNS) was originally developed as an antiepileptic drug. Studies have shown that ZNS suppresses ER stress-induced neuronal cell damage in the experimental models of Parkinson’s disease. Herein, we investigated whether ZNS improved DCM by attenuating ER stress-induced apoptosis. C57BL/6J mice were fed with high-fat diet (HFD) and intraperitoneally injected with low-dose streptozotocin (STZ) to induce type 2 diabetes mellitus (T2DM), and then treated with ZNS (40 mg·kg−1·d−1, i.g.) for 16 weeks. We showed that ZNS administration slightly ameliorated the blood glucose levels, but significantly alleviated diabetes-induced cardiac dysfunction and hypertrophy. Furthermore, ZNS administration significantly inhibited the Bax and caspase-3 activity, upregulated Bcl-2 activity, and decreased the proportion of TUNEL-positive cells in heart tissues. We analyzed the hallmarks of ER stress in heart tissues, and revealed that ZNS administration significantly decreased the protein levels of GRP78, XBP-1s, ATF6, PERK, ATF4, and CHOP, and elevated Hrd1 protein. In high glucose (HG)-treated primary cardiomyocytes, application of ZNS (3 μM) significantly alleviated HG-induced cardiomyocyte hypertrophy and apoptosis. ZNS application also suppressed activated ER stress in HG-treated cardiomyocytes. Moreover, preapplication of the specific ER stress inducer tunicamycin (10 ng/mL) eliminated the protective effects of ZNS against HG-induced cardiac hypertrophy and ER stress-mediated apoptosis. Our findings suggest that ZNS improves the cardiac diastolic function in diabetic mice and prevents T2DM-induced cardiac hypertrophy by attenuating ER stress-mediated apoptosis.
Similar content being viewed by others
Introduction
Type 2 diabetes mellitus (T2DM) is a heterogeneous, multifactorial disorder characterized by hyperglycemia and a gradual decline in insulin action (insulin resistance), followed by the inability of β cells to compensate for insulin resistance (pancreatic β cell dysfunction) [1]. Diabetic cardiomyopathy (DCM) is a major cardiovascular complication of T2DM, which causes increased mortality and morbidity associated with heart failure in patients with T2DM [2]. The cardinal features of DCM include cardiac stiffness, myocardial fibrosis, hypertrophy, and cardiac diastolic dysfunction; later, systolic dysfunction and eventually clinical heart failure occur [3]. In particular, cardiomyocyte apoptosis is a major characteristic of DCM [4,5,6,7,8]. Therefore, antiapoptotic therapy for DCM has received increasing attention.
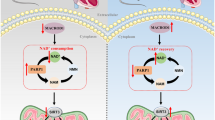
Recently, accumulating evidence has indicated that endoplasmic reticulum (ER) stress plays a key role in the onset and progression of DCM. Inhibition of ER stress can attenuate diabetes-induced cardiac hypertrophy [9,10,11,12,13,14,15]. Various factors can perturb the normal function of the ER and cause ER stress, including hypoxia, hyperglycemia, oxidative stress, lipotoxicity, and inflammation. The accumulation of misfolded and unfolded proteins elicits the unfolded protein response (UPR). The UPR maintains ER homeostasis when ER stress is mild. ER-associated degradation (ERAD) is also activated by the UPR to clear irreparably misfolded proteins. ER stress is initiated by three effector proteins: dsRNA-activated protein kinase-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). During ER stress or the UPR, immunoglobulin-binding protein (Bip)/glucose-regulated protein 78 (GRP78) dissociates from the effector proteins, which causes activation of downstream proteins. HMG-CoA reductase degradation 1 (Hrd1), an important component of ERAD, encodes an E3 ubiquitin ligase and contributes to protection against ER stress-induced cell death by ubiquitinating the nonfunctional proteins that accumulate in the ER lumen for proteasomal degradation [16].
ER stress causes cell dysfunction and apoptosis if the UPR fails to decrease ER stress and restore homeostasis. Signaling switches from prosurvival (adaptive response) to proapoptosis (maladaptive response) by transcriptional induction of C/EBP homologous protein (CHOP) or by activation of the c-Jun N-terminal kinase (JNK)- and caspase-12-dependent pathways [17,18,19,20,21]. CHOP is known to play a key role in ER stress-induced apoptosis, which is regulated by three factors, including PERK, ATF6, and IRE1. It has been reported that ER stress-induced apoptosis contributes to the pathogenesis of DCM [14]. Thus, ER stress has become an important therapeutic target for DCM.
Zonisamide (ZNS) was developed as an antiepileptic drug and has been widely used for the treatment of seizures. The most common adverse effects of ZNS are sleepiness, anorexia and drowsiness [22]. It has been reported that ZNS inhibits neuronal cell death in patients with Parkinson’s disease by increasing Hrd1 protein levels [23]. ZNS also suppresses ER stress-induced neuronal cell damage by inhibiting caspase-3 activity in vitro and in vivo [24]. In addition, our previous study showed that ZNS attenuated abdominal aortic constriction-induced cardiac hypertrophy in rats and angiotensin II-induced myocardial hypertrophy in primary cardiomyocytes from neonatal rats (unpublished). However, the effect of ZNS on DCM remains unknown.
Therefore, we hypothesized that ZNS has a protective effect on diabetic myocardial damage by inhibiting ER stress. In this study, we evaluated this hypothesis in both high glucose (HG)-treated cardiomyocytes and T2DM mice.
Materials and methods
Reagents and antibodies
ZNS was purchased from Target Molecule Corp (Boston, MA, USA). Streptozotocin (STZ) and 4-phenylbutyrate (4-PBA) were purchased from Sigma Chemicals (St. Louis, MO, USA). Tunicamycin (Tm) was purchased from Aladdin (Shanghai, China). The primary antibodies used in this study are listed in the “Materials and methods” section. Peroxidase-conjugated anti-mouse and anti-rabbit IgG were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The enhanced chemiluminescence (ECL) kit was obtained from Pierce (Rockford, IL, USA). The ABC staining system was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Animals and experimental protocols
Four-week-old male C57BL/6J mice were purchased from Guangdong Medical Laboratory Animal Center (Guangzhou, China). The mice were maintained with a standard laboratory diet and free access to tap water in a controlled environment with room temperature (22 °C ± 1 °C), humidity (65% ± 5%) and a 12:12 h light/dark cycle. All animal studies were performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health and were approved by the Committee on the Ethics of Animal Experiments of Guangzhou Medical University.
According to a previous report [25, 26], 24 mice were fed a high-fat diet (HFD) (D12492, 60 kcal% fat, Guangzhou, China) for 3 weeks to induce insulin resistance, and 12 mice were fed a normal diet as the control group. After 3 weeks of HFD feeding, the mice were intraperitoneally injected with 85 mg·kg−1 freshly prepared STZ (in 0.1 M citrate buffer, pH 4.5) twice within 72 h to cause mild insulin secretion deficiency and hyperglycemia. After the STZ injections, 20 mice with hyperglycemia (6 h fasting blood glucose levels >11.6 mmol/L) were defined as the diabetic mice and were selected for subsequent studies (Fig. 1a), and four mice were excluded from the study.

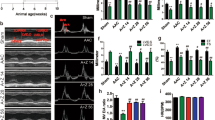
a Experimental design for high-fat diet (HFD)/streptozotocin (STZ)-induced type 2 diabetic mice. Male 4-week-old C57BL/6J mice were fed either the normal diet (ND) or a HFD for 3 weeks. The HFD-fed mice were then intraperitoneally (i.p.) injected with freshly prepared STZ (85 mg·kg−1) twice within 72 h, followed by continued HFD feeding for an additional 5 weeks. Beginning at the 9th week, the HFD-fed/STZ-treated mice were administered ZNS at 40 mg·kg−1·d−1 or vehicle by oral gavage (i.g.) for 16 weeks. Control mice were fed a normal diet and received vehicle administration. b Representative images of M-mode and pulsed-wave Doppler across the mitral valve in the Con, T2DM, and T2DM + ZNS groups. c Evaluation of the ejection fraction (EF%). d Fraction shortening (FS%). e Peak E to peak A ratio (MV E/A). f Left ventricular end-diastolic anterior wall thickness (LVAWd). g Left ventricular end-diastolic posterior wall thickness (LVPWd). h Left ventricular internal dimension at diastole (LVIDd). i Left ventricular end-systolic anterior wall thickness (LVAWs). j Left ventricular end-systolic posterior wall thickness (LVPWs). k Left ventricular internal dimension at systole (LVIDs). The data are presented as the mean ± SEM (n = 10). *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. T2DM. Con, control group; T2DM, type 2 diabetes mellitus; T2DM + ZNS, diabetic mice treated with ZNS.
Twelve mice were fed a normal diet and were used as the control group (Con), and diabetic mice were randomly divided into two other groups (n = 10 in each group): T2DM and diabetic mice were treated with ZNS (T2DM + ZNS). The dose of 40 mg·kg−1 ZNS was chosen from three doses tested in the preliminary experiment that was performed according to our previous study (unpublished) and based on previous reports [22, 27]. ZNS was suspended in 1% DMSO diluted with ultrapure water. Animals in the T2DM + ZNS group were administered ZNS via oral gavage daily for an additional 16 weeks. Animals in the control group and T2DM group received the same volume of vehicle (1% DMSO diluted with ultrapure water). During the experiment, mice in the control group were fed a normal diet, while all diabetic mice continued to be fed an HFD (Fig. 1a). Blood glucose was measured every 2 weeks.
Echocardiography
Transthoracic echocardiography was performed to measure cardiac function with a Visual-Sonics instrument (Vevo 2100; VisualSonics Inc., ON, Canada) equipped with a 30 MHz imaging transducer. Briefly, after 16 weeks of ZNS treatment, the hair was removed from the chest of each mouse (n = 10 for each group) with a topical depilatory agent under inhaled isoflurane (1%) anesthesia. Transthoracic M-mode echocardiographs at the level of the papillary muscles were used to assess changes in the wall thickness of the heart, such as left ventricular anterior wall and left ventricular posterior wall at the diastolic and systolic stage (LVAWd, LVAWs, LVPWd, and LVPWs), the left ventricle internal diameter at systole (LVIDs) and left ventricle internal diameter at diastolic (LVIDd), the ejection fraction (EF%) and fraction shortening (FS%), to assess systolic function. To evaluate diastolic function, we used pulsed-wave Doppler across the mitral valve from the apical four chamber views to measure the peak velocity of early (E) and late (A) ventricular filling velocity and the ratio of early-to-late mitral inflow velocity (E/A). Throughout the procedure, heart rate (HR) was monitored. The HR of each mouse was maintained in a consistent range.
Oral glucose tolerance test (OGTT)
The OGTT was performed as described to assess insulin sensitivity. Briefly, after 16 weeks of ZNS treatment, 12 h-fasted mice in all groups were administered a glucose solution (2 g/kg) via oral gavage, and blood glucose levels were measured at 0, 30, 60, 90, and 120 min in tail vein blood using a glucometer (SD Biosensor, Inc., Chungcheongbuk-do, Korea). Total glycemic responses were calculated from respective areas under the curve (AUCs) during the 120 min observation period.
Histological examination of the heart
At the end of the experiment, the mice were anaesthetized with inhaled isoflurane (1%), and once the mice were determined to be fully anaesthetized, the mice were sacrificed. Then, the hearts were quickly removed and rinsed with PBS. Cardiac tissue (n = 3 per group) was fixed with 4% buffered paraformaldehyde solution for 24 h at room temperature before being embedded in paraffin. Then, the tissues were cut into 5-μm-thick sections and stained with hematoxylin and eosin (HE) for histological examination. The sections were examined by light microscopy (Leica, Germany). The percentage of the cell diameter was calculated using ImageJ software (NIH, Bethesda, USA).
Isolation and culture of neonatal rat cardiomyocytes (NRCMs)
NRCMs were isolated from newborn Sprague–Dawley rats (1–3-day old) by trypsin (0.05%) and collagenase II (0.1%) (Life Technologies, Grand Island, NY, USA) digestion according to previous studies [28]. Briefly, NRCMs were cultured in high‐glucose Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific, Inc., Waltham, MA, USA), 1% penicillin–streptomycin and 0.1 mM bromodeoxyuridine in an incubator with 5% CO2 at 37 °C. After 24 h, the culture medium was changed to fresh low-glucose DMEM containing 1% FBS, 1% penicillin–streptomycin, and 0.1 mM bromodeoxyuridine. After 24 h, upon reaching 80%–90% confluence, the cells were treated with glucose (5.5 and 33 mM) or ZNS (3 µM, dissolved in ddH2O and 0.1% dimethyl sulfoxide), alone or in combination. Moreover, a specific ERS inhibitor (4-PBA) and an inducer (Tm) were used to identify the role of ZNS in HG-induced cardiac hypertrophy and ER stress-induced apoptosis. The cells were incubated with 4-PBA (5 mM) and Tm (10 ng/mL) for 2 h before glucose (33 µM) administration. After 24 h of treatment, the cells were harvested for analysis.
Cell surface area measurement
The cell surface area was measured using phalloidin–tetramethylrhodamine isothiocyanate (TRITC) to stain F-actin as described previously [29]. Briefly, after HG and ZNS exposure for 24 h, NRCMs were fixed with 4% paraformaldehyde for 10 min at room temperature. Then, the cells were incubated with 1% Triton X-100 in PBS for 20 min. After being blocked with 10% normal goat serum for 1 h, the slides were incubated with 50 μg/mL phalloidin–TRITC (Sigma-Aldrich, MO, USA) for 40 min at room temperature to stain F-actin. Cell nuclei were counterstained with DAPI (Sigma-Aldrich, MO, USA). After the cells were washed in PBS three times, images were captured by laser scanning confocal microscopy (A1 si, Nikon, Japan) and the cell surface area was semiquantitatively analyzed with ImageJ software (NIH, Bethesda, USA).
TUNEL staining
A TUNEL assay was performed to determine myocardial apoptosis. We used the in situ cell death detection kit TMR red (No. 12156792910; Roche, IN, USA) and the in situ cell death detection kit POD (No. 11684817910; Roche, IN, USA), which were used to label apoptotic cells according to the manufacturer’s instructions. Briefly, cells were fixed for 1 h with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then stained. Mouse heart tissue sections were deparaffinized and hydrated. The samples were treated with 20 μg/mL proteinase K for 15 min and were then washed with PBS. Then, the samples were incubated with 3% H2O2 for 15 min. After adding the equilibration buffer, the samples were stained with the TUNEL reaction mixture for 1 h in a humidified chamber in the dark at 37 °C. DAPI staining was used to label myocardial cell nuclei. Apoptotic cells were examined using a laser scanning confocal microscope (A1si, Nikon, Japan). TUNEL-positive cells containing red fluorescent nuclei were quantitatively assessed with ImageJ software (NIH, Bethesda, USA).
Real-time RT-PCR
Total RNA from cultured NRCMs was extracted using TRIzol reagent (Life Technologies, Grand Island, NY, USA). All procedures were performed as previously described [30]. Briefly, first-strand cDNA was prepared from total RNA by using the Prime Script RT reagent kit with gDNA Eraser (Takara Bio Inc., Kusatsu, Shiga, Japan), Real-time PCR amplification reactions were performed using a SYBR Premix Ex Taq kit with ROX (Takara Bio Inc., Kusatsu, Shiga, Japan) in triplicate using an ABI StepOne Plus Real-Time PCR system (Thermo Fisher Scientific Inc., Waltham, MA, USA). The expression scores were obtained by the 2−ΔΔCt calculation method and were relatively quantified by normalization to GAPDH expression levels. The data are presented as the fold change in the expression of the gene of interest relative to that of the control groups. The primer sequences (Life Technologies) are shown in Table 1.
Western blotting
Frozen heart tissues (n = 7 per group) and NRCM lysates were prepared by homogenization in RIPA buffer containing a protease inhibitor and phosphatase inhibitor. Total protein concentrations were measured using a BCA protein assay kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Equal amounts of protein (30 μg) were separated on SDS-polyacrylamide gels (8, 10, 12%) and transferred to polyvinylidene difluoride membranes (Roche, Basel, Switzerland). The membranes were incubated with mouse anti-ANP (Cat# 515701) from Santa Cruz Biotechnology, rabbit anti-Hrd1 (Cat# ab170901) from Abcam (Cambridge, UK), rabbit anti—XBP-1s (Cat# 83418), rabbit anti-cleaved caspase-3 (Asp175) (Cat# 9664), rabbit anti-caspase-3 (Cat# 9662), rabbit anti-Bax (Cat# 2772), rabbit anti-Bcl-2 (Cat# 3498), and mouse anti-CHOP (Cat# 2895) from Cell Signaling Technology (Beverly, MA, USA), rabbit anti-GRP78 (Cat# AF0729), rabbit anti-ATF6 (Cat# DF6009), rabbit anti-PERK (Cat# AF5304), rabbit anti-phosphorylated PERK (Thr982) (Cat# DF7576), and rabbit anti-ATF4 (Cat# DF6008) from Affinity Biosciences (Cincinnati, OH, USA), and mouse anti-GAPDH from ZSGB-BIO (Beijing, China) (Cat# TA08) at a 1:1000 dilution at 4 °C overnight. Blots were incubated with HRP-labeled sheep anti-mouse or sheep anti-rabbit IgG (1:3000; Cell Signaling Technology Inc., Beverly, MA, USA) at room temperature for 1 h. The protein-antibody complexes were visualized by a Pierce ECL Western blotting substrate (Thermo Fisher Scientific Inc., Waltham, MA, USA). The specific molecular band intensity was determined by Quantity One software from Bio-Rad.
Statistical analysis
All data were analyzed using the statistical software GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). All data are expressed as the means ± SEM. One-way ANOVA was used to determine the significance between groups. Statistical significance was determined at P < 0.05.
Results
ZNS improved the body weight and blood glucose of T2DM mice
During the experiment, we monitored the body weight and fasting blood glucose of the mice every 2 weeks during the 3 weeks of HFD feeding. We found that T2DM mice suffered mild weight loss in response to STZ injection and HFD feeding for 4 weeks, and the body weight of the T2DM mice gradually increased after treatment with ZNS (Supplementary Fig. 1a). The fasting blood glucose levels of T2DM mice were significantly higher than those of the control mice but were slightly decreased by treatment with ZNS for 16 weeks (Supplementary Fig. 1b). To further evaluate the effect of ZNS on glucose metabolism, we measured glucose tolerance by the OGTT. The data showed that glucose concentrations were dramatically elevated in T2DM mice at all time points. The OGTT readings at 90 and 120 min in the ZNS treatment group were all lower than those in the T2DM group (Supplementary Fig. 1c). We also calculated the AUCs. Compared with the control group, the T2DM group had significantly higher AUC values, but ZNS treatment slightly reduced the AUC values (Supplementary Fig. 1d). These findings suggest that ZNS slightly ameliorated the blood glucose levels of T2DM mice.
ZNS ameliorated cardiac function in T2DM mice, as determined by echocardiography
Echocardiography was performed, and the results were compared with those of the control group (Fig. 1b–k). There was a significant reduction in the EF%, FS%, and peak E to peak A (E/A) ratio in T2DM mice, which was improved by ZNS treatment (Fig. 1c–e). In addition, diabetes decreased the LVIDs and LVIDd but increased the LVAWd and LVAWs and LVPWd and LVPWs, whereas ZNS administration for 16 weeks restored these abnormalities (Fig. 1f–k). These results suggest that ZNS protected against T2DM-induced cardiac remodeling and cardiac dysfunction.
ZNS suppressed cardiac hypertrophy in T2DM hearts
At 16 weeks after diabetes induction, the hearts of T2DM mice showed severe cardiac hypertrophy. Histological analysis showed increased cross-sectional areas in cardiomyocytes from diabetic mice, which is consistent with a significant increase in the heart weight to tibia length ratio (HW/TL), while ZNS treatment decreased the HW/TL ratios and reversed cardiac hypertrophy (Fig. 2a–d). In addition, the increased cardiomyocyte diameter in diabetic mice was attenuated by ZNS (Fig. 2e). These changes were further confirmed by molecular marker analysis. Western blotting results showed that diabetes induced increased levels of ANP, which was reversed by ZNS administration (Fig. 2f, g). Taken together, these findings indicate that ZNS inhibits cardiac hypertrophy in the T2DM model.

a Representative heart size in the different groups. b Representative histological cross-sectional image illustrating the anatomy at the papillary muscle level (scale bar: 500 μm, n = 3). c Representative hematoxylin and eosin (HE) staining of LV transverse sections (scale bar: 50 μm, n = 3). d Quantitative analysis of heart weight to tibia length ratio (HW/TL). e Quantitative analysis of cardiomyocyte cell diameter. f, g Relative protein levels of ANP expressed as the fold change relative to the control. The data are presented as the means ± SEM (n = 7). *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. T2DM.
ZNS suppressed apoptosis in the hearts of T2DM mice
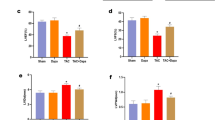
Cardiomyocyte apoptosis has been observed in the hearts of diabetic animal models in previous studies [4,5,6, 8]. In our study, we also detected apoptotic changes in the heart tissue. Using the TUNEL assay, we found that treatment with ZNS effectively counteracted the high ratio of apoptotic cells in the T2DM group (Fig. 3a, b). Western blotting results showed increased Bax and caspase-3 activity with corresponding downregulation of Bcl-2 activity in the hearts of the T2DM mice compared with the control groups, and ZNS effectively abolished these alterations (Fig. 3c–f). Thus, ZNS can decrease diabetes-induced myocardial apoptosis.

a, b TUNEL assay results and quantification of cell apoptosis in the Con, T2DM, and T2DM + ZNS groups (green, TUNEL staining; blue, DAPI staining). c–f Western blotting results and quantitative analysis of Bcl-2, Bax, caspase-3, and cleaved caspase-3 expression. All values are the fold changes normalized to their control group. The data are presented as the means ± SEM (n = 4-5). *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. T2DM.
ZNS alleviated HG-induced cardiac hypertrophy and apoptosis in cardiomyocytes
We also investigated the effect of ZNS on HG-incubated cardiomyocyte hypertrophy. The cardiomyocyte surface area was significantly increased in HG-induced NRCMs (Fig. 4a, b). Consistent with this finding, HG increased ANF mRNA expression and ANP levels (Fig. 4c, f, g), and HG resulted in the upregulation of β-MHC, with a corresponding downregulation of α-MHC mRNA expression (Fig. 4d, e). These effects were abolished after ZNS treatment (Fig. 4c–g).

a Representative micrographs showing immunofluorescence of phalloidin–tetramethylrhodamine isothiocyanate (phalloidin, red) and DAPI (blue) in cardiomyocytes. Scale bar = 50 μm. b Quantitative analysis of cell surface area by ImageJ software. c–e The mRNA levels of ANF, α-MHC, and β-MHC were determined by qPCR. f, g Relative ANP levels and their fold change relative to the control. The data are presented as the means ± SEM (n = 6). *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. HG. Con Control group; HG High glucose; HG + ZNS, High glucose-treated mice that were administered ZNS.
In addition, using the TUNEL assay, we also found that treatment with ZNS effectively counteracted the high ratio of apoptotic cells in HG-cultured cardiomyocytes (Fig. 5a, b). Moreover, Bax and cleaved Caspase 3 were increased after HG treatment. Consistent with this, the level of Bcl-2 was reduced. All of these changes were ameliorated by treatment with ZNS (Fig. 5c–e).

a, b Representative images and the quantitative analysis of TUNEL staining showing the effects of HG and ZNS on apoptosis (red, TUNEL stain; blue, DAPI stain). c–e Representative Western blotting results showing Bax, Bcl-2, cleaved caspase-3, and full-length caspase-3 expression. All values are the fold changes normalized to their control group. The data are presented as the means ± SEM (n = 6). *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. HG.
ZNS alleviated HG-induced cardiac hypertrophy and apoptosis via suppression of activated ER stress
Many studies have reported that apoptosis initiated by ER stress is involved in the pathogenesis of DCM. Therefore, to further clarify the underlying mechanisms by which ZNS alleviates HG-induced cardiac hypertrophy, we measured the expression of ER stress-related proteins in HG-cultured primary neonatal rat cardiomyocytes. We found that HG caused a significant increase in expression of GRP78, XBP-1s, ATF6, p-PERK/ATF4/CHOP, and Hrd1, which was significantly inhibited by ZNS treatment, in addition to the upregulation of Hrd1 levels (Fig. 6a, b). These results suggest that ER stress was fully activated by HG stimulation and can be mostly suppressed by ZNS treatment. To further determine whether ZNS improved myocardial hypertrophy by directly inhibiting ER stress-mediated cardiomyocyte apoptosis, we utilized 4-PBA, a widely used ER stress inhibitor, to treat cultured NRCMs. 4-PBA alleviated the HG-induced increase in cardiomyocyte hypertrophy (Fig. 6c, d) and abolished the increase in expression of GRP78, ATF6, P-PERK, ATF4, and CHOP (Fig. 6g, h). As shown in Fig. 6e, f, HG-stimulated cardiomyocyte apoptosis was effectively abolished by 4-PBA. Moreover, Tm, a specific ER stress inducer, significantly blocked the inhibitory effects of ZNS on HG-induced cardiac hypertrophy, apoptosis (Fig. 6c–f) and ER stress (Fig. 6g, h), which demonstrated that ZNS-induced inhibition of myocardial hypertrophy is mediated by suppressing ER stress-induced apoptosis in cardiomyocytes.

NRCMs were pretreated with 5 mM 4-PBA (an inhibitor of ERS) or 10 ng/mL tunicamycin (Tm, an ERS inducer) for 2 h and then exposed to glucose (33 mM) in the presence or absence of ZNS (3 μM) for 24 h. a–b Representative and quantitative images showing the protein expression of ERS markers, including GRP78, XBP-1s, ATF6, p-PERK, PERK, ATF4, CHOP, and Hrd1. c Immunofluorescence staining of cardiomyocytes with phalloidin (red) and cell nuclei with DAPI (blue), Scale bar = 50 μm. d Quantitative analysis of cell surface area by ImageJ software. e–f Representative Western blotting and analysis of Bax and Bcl-2 expression. g–h Representative and quantitative images of GRP78, ATF6, p-PERK, PERK, ATF4, and CHOP expression. All values are the fold changes normalized to their control group. The results are presented as the means ± SEM (n = 6). *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. HG; $P < 0.05, $$P < 0.01 vs. HG + ZNS.
ZNS suppressed ER stress in the hearts of T2DM mice
To confirm the changes in ER stress in vitro, we further detected the effects of ER stress in vivo. Consistent with the in vitro findings in NRCMs, we found that the expression of GRP78, XBP-1s, ATF6, p-PERK, ATF4, CHOP, and Hrd1 was upregulated in T2DM mice and was inhibited by ZNS treatment, in addition to upregulation of Hrd1 levels (Fig. 7a, b).

a–b Representative and quantitative images showing the protein expression of ER stress markers, including GRP78, XBP-1s, ATF6, p-PERK, PERK, ATF4, CHOP, and Hrd1. All values are the fold changes normalized to their control group. The data are presented as the means ± SEM (n = 4–7). *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. T2DM.
Overall, these results suggest that ZNS improved diabetes-induced cardiac hypertrophy and dysfunction by regulating ER stress.
Discussion
DCM is a critical complication and a leading cause of diabetes-associated death that is characterized by structural changes in the heart and diastolic/systolic dysfunction [31]. Consequently, effective drugs to prevent DCM are important. In the present study, we established a mouse model of type 2 diabetes by HFD feeding and low-dose STZ injection in vivo. Cardiac remodeling is the most important and representative change in DCM and an important predisposing factor for the development of heart failure in diabetic patients. Cardiac hypertrophy is the main characteristic of cardiac remodeling in diabetic hearts. The present study demonstrates for the first time that ZNS improves cardiac function and cardiac hypertrophy in T2DM by inhibiting ER stress.
In this study, we observed that ZNS can improve heart systolic and diastolic function in T2DM mice, as evidenced by increased EF%, FS%, LVIDs, LVIDd, and E/A ratios, as well as decreased LVAWd, LVAWs, LVPWd, and LVPWs, as determined by echocardiography. In addition, histological observations by HE staining showed decreased cardiomyocyte width, which was further confirmed by sharply reduced levels of the hypertrophic marker ANP in the ZNS group. In vitro, we found that HG (33 mM) was capable of provoking cardiac hypertrophy in NRCMs. Consistent with the in vivo results, the cardiomyocyte surface area obviously increased, the mRNA levels of the hypertrophic markers ANF and β-MHC were markedly elevated and α-MHC mRNA was reduced in HG-stimulated cardiomyocytes. These effects were abolished by administration of ZNS. These findings demonstrated that ZNS might represent a new strategy to block cardiac hypertrophy.
Hyperglycemia, which is a common feature of diabetes mellitus, is the major cause of DCM and a risk factor for heart injury [32]. In this study, ZNS treatment for 16 weeks weakly improved the fasting blood glucose level and OGTT results compared with those of the HFD group, suggesting that ZNS improved DCM in a glucose-independent manner. Previous studies showed that increased cardiomyocyte apoptosis was observed in T2DM [4,5,6,7,8]. In addition, the continuous loss of cardiomyocytes triggers myocardial hypertrophy, myocardial fibrosis and impaired systolic and diastolic function [14]. Consistent with previous studies, we found that the levels of Bax and cleaved Caspase 3 were increased and the levels of Bcl-2 were reduced in both T2DM mice and HG-stimulated NRCMs. The TUNEL assay results showed a high ratio of apoptotic cells in both T2DM mice and HG-stimulated NRCMs. ZNS treatment reversed the above changes, suggesting that ZNS alleviates myocardial damage in T2DM by decreasing cardiomyocyte apoptosis.
Overwhelming evidence supports that the ER stress-induced UPR is an essential step in the pathogenesis of DCM [10, 11]. A number of studies observed the upregulation of UPR pathway proteins, such as GRP78, IRE1α/XBP-1s, ATF6, and PERK/e IF2α/ATF4, in the hearts of diabetic models [4, 33,34,35,36,37]. ERAD is also activated by the UPR to clear irreparably misfolded proteins. Hrd1, an E3 ubiquitin ligase, plays a very broad role in the ubiquitination process of abnormal proteins in the ER. Hrd1 levels have been reported to be upregulated in response to induced ER stress in cultured cardiomyocytes, and Hrd1 contributes to preserving heart structure and function in a mouse model of pathological cardiac hypertrophy [38]. Moderate ER stress can enhance the ability of the ER to respond to stress. However, prolonged disruption of ER functions by ER stress or if ER functions could not be restored within a certain time, the apoptotic pathway would be activated [10, 38, 39]. Increasing evidence suggests that ER stress participates in apoptosis in DCM [40, 41]. ER-induced apoptosis occurs via three primary pathways, the IRE1/ASK1/JNK pathway, the caspase-12 kinase pathway, and the CHOP/GADD153 pathway. CHOP has been identified to play a key role in ER stress-induced apoptosis, which is regulated by PERK, ATF6, and IRE1. The PERK/ATF-4/CHOP pathway is critical in activating apoptosis [42]. CHOP decreases the antiapoptotic factor/proapoptotic factor ratio (Bcl-2/Bax), which leads to cell apoptosis [20, 43]. Increasing evidence indicates that upregulation of Hrd1 prevents apoptosis to improve some diabetic complications, including diabetic nephropathy and diabetic retinopathy [44,45,46]. Notably, it has been demonstrated that ZNS inhibits neuronal cell death in patients with Parkinson’s disease by increasing Hrd1 protein levels [23]. Our data demonstrated that levels of ER stress markers (GRP78, XBP-1s, ATF6, P-PERK, ATF4, CHOP, and Hrd1) were significantly increased in diabetic mice and HG-cultured NRCMs, which was similar to the results of previous reports [14, 34, 35, 40, 47,48,49,50,51]. After treatment with ZNS, other proteins were downregulated, in addition to the upregulation of Hrd1 expression. Tm, a specific ER stress inducer, significantly eliminated the inhibitory effects of ZNS on HG-induced cardiac hypertrophy, apoptosis, and ER stress, suggesting that ZNS improved cardiac hypertrophy in T2DM by suppressing ER stress-induced apoptosis in cardiomyocytes.
In conclusion, these findings provide compelling evidence for ZNS-mediated protection during DCM by inhibiting ER stress-induced cell death. Therefore, ZNS may become a new agent for the treatment of cardiovascular complications of diabetes.
References
Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52:313–20.
Fang WJ, Wang CJ, He Y, Zhou YL, Peng XD, Liu SK. Resveratrol alleviates diabetic cardiomyopathy in rats by improving mitochondrial function through PGC-1alpha deacetylation. Acta Pharmacol Sin. 2018;39:59–73.
Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–38.
Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome c-mediated caspase-3 activation pathway. Diabetes. 2002;51:1938–48.
Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol. 2003;3:219–28.
Adeghate E. Molecular and cellular basis of the aetiology and management of diabetic cardiomyopathy: a short review. Mol Cell Biochem. 2004;261:187–91.
An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2006;291:H1489–506.
Pappachan JM, Varughese GI, Sriraman R, Arunagirinathan G. Diabetic cardiomyopathy: pathophysiology, diagnostic evaluation and management. World J Diabetes. 2013;4:177–89.
Groenendyk J, Sreenivasaiah PK, Kim DH, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–97.
Xu J, Zhou Q, Xu W, Cai L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp Diabetes Res. 2012;2012:827971.
Yang L, Zhao D, Ren J, Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:209–18.
Cong XQ, Piao MH, Li Y, Xie L, Liu Y. Bis(maltolato)oxovanadium(IV) (BMOV) attenuates apoptosis in high glucose-treated cardiac cells and diabetic rat hearts by regulating the unfolded protein responses (UPRs). Biol Trace Elem Res. 2016;173:390–8.
Lian J, Chen J, Yuan Y, Chen J, Daud MSM, Luo L, et al. Cortex Mori Radicis extract attenuates myocardial damages in diabetic rats by regulating ERS. Biomed Pharmacother. 2017;90:777–85.
Wang XM, Wang YC, Liu XJ, Wang Q, Zhang CM, Zhang LP, et al. BRD7 mediates hyperglycaemia-induced myocardial apoptosis via endoplasmic reticulum stress signalling pathway. J Cell Mol Med. 2017;21:1094–105.
He Y, Zhou L, Fan Z, Liu S, Fang W. Palmitic acid, but not high-glucose, induced myocardial apoptosis is alleviated by N-acetylcysteine due to attenuated mitochondrial-derived ROS accumulation-induced endoplasmic reticulum stress. Cell Death Dis. 2018;9:568.
Kaneko M, Nomura Y. ER signaling in unfolded protein response. Life Sci. 2003;74:199–205.
Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–63.
Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–64.
Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–10.
Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation. 2010;122:361–9.
Kassan M, Ait-Aissa K, Radwan E, Mali V, Haddox S, Gabani M, et al. Essential role of smooth muscle STIM1 in hypertension and cardiovascular dysfunction. Arterioscler Thromb Vasc Biol. 2016;36:1900–9.
Murata M, Horiuchi E, Kanazawa I. Zonisamide has beneficial effects on Parkinson’s disease patients. Neurosci Res. 2001;41:397–9.
Omura T, Asari M, Yamamoto J, Kamiyama N, Oka K, Hoshina C, et al. HRD1 levels increased by zonisamide prevented cell death and caspase-3 activation caused by endoplasmic reticulum stress in SH-SY5Y cells. J Mol Neurosci. 2012;46:527–35.
Tsujii S, Ishisaka M, Shimazawa M, Hashizume T, Hara H. Zonisamide suppresses endoplasmic reticulum stress-induced neuronal cell damage in vitro and in vivo. Eur J Pharmacol. 2015;746:301–7.
Luo J, Quan J, Tsai J, Hobensack CK, Sullivan C, Hector R, et al. Nongenetic mouse models of non-insulin-dependent diabetes mellitus. Metabolism. 1998;47:663–8.
Zhu WF, Liu XP, Cao Y, Kong HY, Liu AM, Shi G, et al. Optimization and evaluation of type 2 diabetes induced by high-fat diet and low dose of STZ in C57BL/6J mouse model. China Trop Med. 2010;10:529–613.
Yokoyama H, Yano R, Kuroiwa H, Tsukada T, Uchida H, Kato H, et al. Therapeutic effect of a novel anti-parkinsonian agent zonisamide against MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) neurotoxicity in mice. Metab Brain Dis. 2010;25:305–13.
Hou N, Cai B, Ou CW, Zhang ZH, Liu XW, Yuan M, et al. Puerarin-7-O-glucuronide, a water-soluble puerarin metabolite, prevents angiotensin II-induced cardiomyocyte hypertrophy by reducing oxidative stress. Naunyn Schmiedebergs Arch Pharmacol. 2017;390:535–45.
Schumacher-Bass SM, Vesely ED, Zhang L, Ryland KE, McEwen DP, Chan PJ, et al. Role for myosin-V motor proteins in the selective delivery of Kv channel isoforms to the membrane surface of cardiac myocytes. Circ Res. 2014;114:982–92.
Chen K, Rekep M, Wei W, Wu Q, Xue Q, Li S, et al. Quercetin prevents in vivo and in vitro myocardial hypertrophy through the proteasome-GSK-3 pathway. Cardiovasc Drugs Ther. 2018;32:5–21.
Friedberg MK, Margossian R, Lu M, Mercer-Rosa L, Henderson HT, Nutting A, et al. Systolic-diastolic functional coupling in healthy children and in those with dilated cardiomyopathy. J Appl Physiol. 2016;120:1301–18.
Fang ZY, Prins JB, Marwick TH. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev. 2004;25:543–67.
Li Z, Zhang T, Dai H, Liu G, Wang H, Sun Y, et al. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J Clin Biochem Nutr. 2007;41:58–67.
Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T, et al. The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of unfolded protein response (UPR) signaling proteins. Int J Biochem Cell Biol. 2013;45:438–47.
Liu ZW, Zhu HT, Chen KL, Dong X, Wei J, Qiu C, et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:158.
Ozturk N, Olgar Y, Ozdemir S. Trace elements in diabetic cardiomyopathy: an electrophysiological overview. World J Diabetes. 2013;4:92–100.
Varga ZV, Giricz Z, Liaudet L, Hasko G, Ferdinandy P, Pacher P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2015;1852:232–42.
Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–9.
Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827–30.
Liu X, Xu Q, Wang X, Zhao Z, Zhang L, Zhong L, et al. Irbesartan ameliorates diabetic cardiomyopathy by regulating protein kinase D and ER stress activation in a type 2 diabetes rat model. Pharmacol Res. 2015;93:43–51.
Younce CW, Burmeister MA, Ayala JE. Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a. Am J Physiol Cell Physiol. 2013;304:C508–18.
Toth A, Nickson P, Mandl A, Bannister ML, Toth K, Erhardt P. Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc Hematol Disord Drug Targets. 2007;7:205–18.
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90.
Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, et al. IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat Cell Biol. 2015;17:1546–55.
Huang Y, Sun Y, Cao Y, Sun H, Li M, You H, et al. HRD1 prevents apoptosis in renal tubular epithelial cells by mediating eIF2alpha ubiquitylation and degradation. Cell Death Dis. 2017;8:3202.
Yan S, Zheng C, Chen ZQ, Liu R, Li GG, Hu WK, et al. Expression of endoplasmic reticulum stress-related factors in the retinas of diabetic rats. Exp Diabetes Res. 2012;2012:743780.
Li Z, Zhang T, Dai H, Liu G, Wang H, Sun Y, et al. Endoplasmic reticulum stress is involved in myocardial apoptosis of streptozocin-induced diabetic rats. J Endocrinol. 2008;196:565–72.
Wu T, Dong Z, Geng J, Sun Y, Liu G, Kang W, et al. Valsartan protects against ER stress-induced myocardial apoptosis via CHOP/Puma signaling pathway in streptozotocin-induced diabetic rats. Eur J Pharm Sci. 2011;42:496–502.
Barr LA, Shimizu Y, Lambert JP, Nicholson CK, Calvert JW. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide. 2015;46:145–56.
Liu Z, Zhao N, Zhu H, Zhu S, Pan S, Xu J, et al. Circulating interleukin-1beta promotes endoplasmic reticulum stress-induced myocytes apoptosis in diabetic cardiomyopathy via interleukin-1 receptor-associated kinase-2. Cardiovasc Diabetol. 2015;14:125.
Pei SJ, Zhu HY, Guo JH, Zhang X, Deng ZJ. Knockout of CNR1 prevents metabolic stress-induced cardiac injury through improving insulin resistance (IR) injury and endoplasmic reticulum (ER) stress by promoting AMPK-alpha activation. Biochem Biophys Res Commun. 2018;503:744–51.
Acknowledgements
This work was supported by Natural Science Foundation of Guangdong Province (2014A030313485), Scientific and Technological Planning Program of Guangzhou (2017071010458), Municipal Education Bureau Program of Guangzhou (1201610286), Guangzhou Education Bureau (1201410365), and Natural Science Foundation of Guangdong Province (2018A030313719).
Author information
Authors and Affiliations
Contributions
JHT and QW designed the research; JHT carried out the study and wrote the paper; JHT, QW, YXH, QYS, MR, and GPZ performed the experiments; QX and JDL helped in discussing the data and writing the paper; YHL and QX conceived, designed, and supervised the study and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Tian, Jh., Wu, Q., He, Yx. et al. Zonisamide, an antiepileptic drug, alleviates diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress. Acta Pharmacol Sin 42, 393–403 (2021). https://doi.org/10.1038/s41401-020-0461-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0461-z
Keywords
This article is cited by
-
Zonisamide attenuates pressure overload-induced myocardial hypertrophy in mice through proteasome inhibition
Acta Pharmacologica Sinica (2024)
-
Casting Light on the Janus-Faced HMG-CoA Reductase Degradation Protein 1: A Comprehensive Review of Its Dualistic Impact on Apoptosis in Various Diseases
Molecular Neurobiology (2024)
-
DCGG: drug combination prediction using GNN and GAE
Progress in Artificial Intelligence (2024)
-
Diabetic cardiomyopathy: Early diagnostic biomarkers, pathogenetic mechanisms, and therapeutic interventions
Cell Death Discovery (2023)
-
Zonisamide alleviates cardiac hypertrophy in rats by increasing Hrd1 expression and inhibiting endoplasmic reticulum stress
Acta Pharmacologica Sinica (2021)
