
Abstract
Uveal melanoma (UM) is a rare ocular tumor. The loss of BRCA1-associated protein 1 (BAP1) and the aberrant activation of G protein subunit alpha q (GNAQ)/G protein subunit alpha 11 (GNA11) contribute to the frequent metastasis of UM. Thus far, limited molecular-targeted therapies have been developed for the clinical treatment of UM. However, an increasing number of studies have revealed the close relationship between the ubiquitin proteasome system (UPS) and the malignancy of UM. UPS consists of a three-enzyme cascade, i.e. ubiquitin-activating enzymes (E1s); ubiquitin-conjugating enzymes (E2s); and ubiquitin-protein ligases (E3s), as well as 26S proteasome and deubiquitinases (DUBs), which work coordinately to dictate the fate of intracellular proteins through regulating ubiquitination, thus influencing cell viability. Due to the critical role of UPS in tumors, we here provide an overview of the crosstalk between UPS and the malignancy of UM, discuss the current UPS-targeted therapies in UM and highlight its potential in developing novel regimens for UM.
Similar content being viewed by others

Introduction
Uveal melanoma (UM) is a rare malignancy of the eye [1, 2]. As the most common primary ocular cancer in adults, it is usually characterized by metastasis to other tissues, particularly the liver [3]. The survival of UM patients diagnosed with metastasis is less than one year and has not improved in decades [3]. This extremely low survival rate is attributed to the malignant biology of UM and the lack of adequate clinical treatments.
Genetic alterations and several oncogenetic pathways are involved in the malignancy of UM. The loss of chromosome 3 (the presence of monosomy 3) is a well-established characteristic of UM [4]. BRCA1-associated protein 1 (BAP1) is a ubiquitin carboxy-terminal hydrolase (UCH) that is located on chromosome 3 [4]. Thus, the loss of chromosome 3 can lead to the mutation and downregulation of BAP1 protein [4]. Notably, BAP1 functions as a tumor suppressor, removing ubiquitin (a 76-amino-acid residue) from histone H2A [5] and therefore inducing ferroptosis [6], growth suppression [7], and DNA repair by homologous recombination [8]. However, a recent study indicated that BAP1 mutation occurs in 81% of UM cases with monosomy 3 [4]. Therefore, the aberration of BAP1 is a major cause of the increasing occurrence of UM metastasis. In addition to such genetic alterations, there have been three major oncogenetic pathways involved in the malignancy of UM. First, G protein-coupled receptors (GPCRs) can sense extracellular signals and activate GNAQ and GNA11. GNAQ and GNA11 further interact with guanine nucleotide exchange factor (GEF), which activates ADP ribosylation factor 6 (ARF6) [9]. ARF6 subsequently induces the trio Rho guanine nucleotide exchange factor (TRIO) and the ras homolog family member A (RHOA)/Rac family small GTPase 1 (RAC1)-mediated nuclear translocation of yes-associated transcriptional regulator (YAP) and the YAP-dependent transcription of oncogenes [10, 11]. Activated ARF6 also triggers phospholipase C beta 4 (PLCB4) and then initiates phosphatidylinositol 4,5-bisphosphate (PIP2)-mediated production of the second messenger diacylglycerol (DAG). DAG phosphorylates and activates RAS guanyl releasing protein 3 (RASGRP3), stimulating the mitogen-activated protein kinase (MAPK) pathway and the phosphatidylinositol 3-kinase (PI3K)–AKT axis through the activation of RAS [12,13,14]. Consequently, all three pathways promote the growth and proliferation of UM cells.
In the past few years, UM treatment has generally been confined to excision, radiotherapy, and chemotherapy [15, 16]. However, genetic alterations and modulation of oncogenetic pathways lead to the distant metastasis and resistance of UM cells to chemotherapeutic agents. An increasing number of recent studies have shown that new therapies targeting specific molecular pathways might be a promising way to combat malignant UM [17,18,19].
The ubiquitin–proteasome system (UPS) is a potential therapeutic target of a variety of tumors, such as multiple myeloma [20], hepatocellular carcinoma [21], and acute myeloid leukemia [22]. Mechanistically, the ubiquitin-activating enzyme (E1) activates the C-terminal glycine residue of ubiquitin with the help of ATP. Then, the activated ubiquitin is transferred to the active cysteine residue of a ubiquitin-conjugating enzyme (E2), followed by the C-terminus of ubiquitin linked to lysine residues of a substrate catalyzed by a specific ubiquitin-protein ligase (E3). Consequently, the 26S proteasome hydrolyzes substrates ligated with polyubiquitin chains [23]. Since many deubiquitinases (DUBs) can remove polyubiquitin chains and stabilize these substrates, DUBs are also considered to function in the UPS [24]. It is known that a variety of tumor-related proteins are regulated by UPS, which results in the degradation of tumor suppressors (tumor protein p53, TP53 [25]; tumor protein p63, TP63 [26]; phosphatase and tensin homolog, PTEN [27]) and the accumulation of oncoproteins (Ras [28], c-MYC [29], and YAP [30]).
An increasing number of studies have suggested that the UPS is involved in the regulation of the three major UM-related signaling pathways, particularly the Hippo-YAP [31], MAPK [32], and PI3K–AKT axes [33]. This regulatory role of the UPS and the DUB activity of BAP1 indicate that the UPS might be a promising therapeutic target against UM. In this review, we highlight the function of the UPS in UM and reveal the potential of the UPS as a therapeutic target of UM.
The ubiquitin–proteasome system in UM
As described above, the UPS consists of a three-enzyme cascade (E1–E3), the 26S proteasome and DUBs. The frequent deletion of BAP1 is a characteristic of metastatic UM [34, 35] and can drive immune exclusion, leading to the resistance of immunotherapy in UM [36]. Notably, BAP1 generally serves as a DUB to suppress tumorigenesis and tumor development [6,7,8], which suggests that the UPS plays a key role in the malignancy of UM. Therefore, it is reasonable to explore the potential correlation between UM development and other components of the UPS, including the enzymatic cascade (E1–E3) and proteasomes. Thus, the development of novel UPS-related therapeutic targets against UM is promising.
Deubiquitinases
The deubiquitinase BAP1 belongs to the UCH family [37] and is involved in the formation of multiprotein complexes. BAP1 plays key roles in cell death, the cell cycle, cellular differentiation, and gluconeogenesis [38, 39]. The cellular localization of BAP1 is controlled by the ubiquitin-conjugating enzyme E2 O (UBE2O). UBE2O is involved in the multi-monoubiquitination of the nuclear localization signal (NLS) of BAP1, which sequesters BAP1 in the cytoplasm. Notably, BAP1 autoregulates its nuclear translocation through the deubiquitination of the same residues in the NLS [8], which indicates that BAP1 is functional in both the nucleus and cytoplasm.
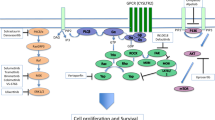
The loss of BAP1 expression in the nucleus frequently occurs in metastatic UM [34]. An inflammatory tumor microenvironment is observed in UM, which then potentiates metastasis. Thus, inflammation is considered a hallmark of UM [40]. Notably, the loss of BAP1 expression is associated with the inflammatory phenotype of UM [41, 42]. A recent study showed that the canonical nuclear factor kappa B (NF-κB) pathway is involved in the malignant inflammation of UM [43]. The analysis of 64 UM samples showed that the loss of BAP1 expression first activates the NF-κB pathway and then increases the expression of human leukocyte antigen (HLA) class I. HLA class I then promotes the secretion of cytokines and chemokines, which triggers immune cells to further induce the expression of HLA Class I. Furthermore, patients with elevated HLA Class I and activated NF-κB pathway have shortened overall survival (Fig. 1b) [43]. The molecular mechanism of BAP1-related regulation of the NF-κB pathway remains unclear. Inhibitors of NF-κB (IκBs) are considered lead molecules, since they can bind to NF-κB as well as inhibit the nuclear translocation and DNA binding of NF-κB [44]. However, the UPS mediates the degradation of IκBs, which is triggered by the signaling of two conserved serine residues phosphorylated at the N-terminus (Fig. 1c) [44]. This well-established function of IκBs indicates that BAP1 might block the NF-κB pathway by deubiquitinating and stabilizing IκBs. However, the repression of the NF-κB pathway might diminish in UM due to the loss of BAP1.

a BAP1/HCFC1-transactivated E2F1 target genes, together with the deubiquitination of histones H2A, induce the transcription of CCND1 and CCNE1 resulting in tumor growth and cell invasion. b The NF-κB pathway triggers HLA class I-mediated inflammation in UM resulting in poor survivals, which could be blocked by BAP1. c BAP1-mediated deubiquitination of ITPR3 induces the calcium release from the ER into the cytoplasm and mitochondria, directing cells to apoptosis.
In the cytoplasm, BAP1 localizes at the endoplasmic reticulum (ER), where apoptosis-related protein inositol 1,4,5-trisphosphate receptor type 3 (ITPR3) can be degraded through the UPS [45, 46]. ITPR3 is a receptor for inositol 1,4,5-trisphosphate, a second messenger mediating the release of intracellular calcium from the ER, which activates downstream apoptosis pathways [47]. A recent study reported that the protein expression of ITPR3 in fibroblasts with monosomy 3 is lower than that in BAP1 wild-type fibroblasts [46]. Interestingly, wild-type BAP1 exerts its DUB activity and removes ubiquitin from ITPR3 by directly interacting with ITPR3. The accumulation of ITPR3 releases calcium from the ER into the cytoplasm and mitochondria, which triggers apoptosis (Fig. 1c). However, the inactivating mutation of monosomy 3 frequently leads to malignant deletion of BAP1 in UM patients. The loss of BAP1 results in the degradation of ITPR3 with the assistance of UPS. Eventually, UM cells evade apoptosis and exhibit prolonged survival [46].
MicroRNAs (miRNAs) play important roles in the malignancy of UM [48]. A recent study revealed that 13 miRNAs are aberrantly overexpressed in UM [48]. These miRNAs regulate the transcription of genes involved in tumor-related signaling, such as epidermal growth factor and eukaryotic initiation factor 2 signaling [48]. In addition to the deletion of BAP1, UM has a large spectrum of BAP1 mutations, which might affect the miRNA regulatory network to promote UM malignancy [49]. BAP1 consists of five major domains: the UCH domain, BRCA1-associated RING domain 1 (BARD1) at the N-terminus, host cell factor C1 (HCFC1)-binding motif (HBM), BRCA1 interaction region, and NLS region at the C-terminus [50]. Although mutations are mostly observed in the UCH domain, the C-terminal mutations might primarily regulate the miRNA network, as the miRNA clusters are embedded in the 3′ UTR region of the BAP1 gene [49].
BAP1 also serves as a coregulator of the E2F transcription factor 1 (E2F1)-responsive promoter in UM [51]. BAP1, HCFC1, and E2F1 form a complex, where HCFC1 functions as a scaffold protein. This complex induces the transcription of E2F1 target genes, such as RB transcriptional corepressor like 1 (RBL1), cyclin A2 (CCNA2), and cell division cycle 25A (CDC25A), thus contributing to the expression of S-phase proteins, including cyclin D1 (CCND1) and cyclin E1 (CCNE1). Consequently, BAP1-mediated expression of these genes promotes the growth and invasion of UM cells (Fig. 1a). Notably, BAP1 also functions as a DUB and exerts its catalytic ability by deubiquitinating monoubiquitinated histone H2A (Lys119) on the E2F1-responsive promoter [5, 51]. Moreover, it is probable that BAP1 deubiquitinates histone H2A on the promoters of S-phase genes. Based on the epigenetic-silencing function of histone H2A ubiquitination, the deubiquitination of histone H2A induces the expression of S-phase genes in UM (Fig. 1a) [51, 52]. Interestingly, these studies revealed the oncogenetic functions of BAP1, which is quite different from the general tumor suppressor character of BAP1. It is plausible that BAP1 acts as a “double-edged sword” in tumor development. A recent study also showed that BAP1 plays a tumor-promoting role in myeloid neoplasms through the removal of H2A (Lys119) ubiquitination and subsequent posterior transcription of homeobox A cluster (HOXA) genes and interferon regulatory factor 8 (IRF8) [53]. Therefore, the tumor-promoting or tumor-suppressive function of BAP1 probably depends on the specific tumor type and different tumor characteristics. Moreover, BAP1 plays dual roles in UM, which might be attributable to the different substrates and cellular localization. In the nucleus, histone H2A is the major substrate of BAP1. Deubiquitinated histone H2A induces the transcription of some oncogenes. However, BAP1 mainly deubiquitinates and stabilizes specific tumor suppressors in the cytoplasm. Such DUB activity-dependent dual mechanisms may not be correlated with the loss or mutation of BAP1.
Other DUBs have not been shown to be involved in the malignancy of UM, with the exception of ubiquitin-specific peptidase 19 (USP19). The expression of USP19 is positively correlated with BAP1 in UM [54], which indicates that USP19 together with BAP1 might play a key role in the malignancy of UM.
Ubiquitin-protein ligases
To date, more than 600 human E3 ligases have been discovered [55]. The E3 ligases serving as both catalysts and molecular matchmakers facilitate the transfer of ubiquitin from E2 to a specific substrate [56]. Due to the great diversity and specificity of substrates, it is important to explore the correlation between E3 ligases and the malignancy of UM to discover novel therapeutic targets.
Murine double minute 2 (MDM2)
MDM2 is a well-known oncogene with RING domain-dependent E3 ligase activity [57, 58]. It interacts with canonical tumor suppressors such as TP53 and RB transcriptional corepressor 1 (RB1) to inhibit apoptosis [59], promote cell cycle progression [60, 61], and repair DNA damage [62]. It correlates with the RB1 and TP53 pathways. The phosphorylation of RB1 activates the E2F–ARF axis, which inhibits MDM2-mediated degradation of TP53, ultimately resulting in apoptotic cell death [63]. Mutations and dysregulation of the RB1 pathway lead to the inactivation of TP53 and enable cells to escape apoptosis, thus promoting UM development [63, 64]. Notably, a recent study indicated that p53 apoptosis effector related to PMP22 (PERP), a target gene of TP53, prevents TP53 from MDM2-mediated degradation through the UPS in a positive feedback loop [65]. Mechanistically, PERP elevates the protein expression of TP53 and recruits TP53 to the nucleus, where it transactivates tumor-suppressor genes (death receptor 4, DR4 and p53-induced death domain protein 1, PIDD1) to trigger apoptosis (Table 1, Fig. 2a) [65]. Furthermore, the phosphorylation of TP53 (Ser46) contributes to apoptotic progression [65,66,67]. TP53-driven apoptosis is unlikely to be involved in modulating canonical apoptosis regulators homeodomain-interacting protein kinase 2 (HIPK2) [68] and p38 [69], which indicates that other signaling pathways may participate in this process. In addition to the tumor suppressors mentioned above, MDM2 can also interact with miRNA [70, 71]. As reported in a recent study, MDM2 is a target gene of miR-17-3p. MiR-17-3p binds to MDM2 mRNA and suppresses the expression of MDM2, which inhibits MDM2-mediated degradation of TP53 (Fig. 2a). The stabilization of TP53 induces the transactivation of apoptotic genes to trigger apoptosis. However, MDM2 is overexpressed along with a higher level of the long noncoding RNA (lncRNA) PVT1 in UM cells compared to adjacent tissues. Mechanistically, lncRNA PVT1 binds to miR-17-3p, which interrupts the interaction between miR-17-3p and MDM2 mRNA (Fig. 2a). Overexpressed MDM2 inhibits the tumor suppressor function of TP53 and then induces the proliferation, migration, and invasion of UM cells [70].

a MiR-17-3p inhibits MDM2-mediated degradation of TP53 through the UPS. However, LncRNA PVT1 in UM could disturb miR-17-3p-mediated inhibition of MDM2. b SKP2-mediated degradation of CDKN1B through the UPS induces UM growth. c Hypoxia induces transactivation of HIF1A that could be degraded through VHL-mediated UPS in normoxia. d RNF2-mediated ubiquitination of histone H2A executes completely different regulation of downstream genes in UM versus other tissues. e MYCBP2 elevates the sensitivity of UM cells to Trail-induced apoptosis by conjugating to C-MYC. However, miR-92a-3p overexpressed in UM reduces the protein level of MYCBP2.
S-phase kinase associated protein 2 (SKP2)
SKP2 is a component of the SKP1–CUL1–ROC1–F-box (SCF) E3 ligases [72]. Serving as a substrate-specific adaptor, SKP2 is normally considered an oncogene in tumors [72, 73]. SKP2 plays a key role in promoting tumorigenesis by disturbing normal cell processes such as TP53-dependent apoptosis and cyclin-dependent kinase inhibitor 1B (CDKN1B)-mediated regulation of the cell cycle [73]. A recent study uncovered the aberrant overexpression of SKP2 in UM cells [74]. Intriguingly, the knockdown of SKP2 inhibits the proliferation of UM cells and suppresses the growth of UM tumors. Mechanistically, the inhibition of SKP2 reduces the ubiquitination of its substrate CDKN1B, which acts as a cell cycle regulator. The accumulation of CDKN1B then leads to G1-phase arrest (Table 1) [74]. CDKN1B can inhibit the enzymatic ability of cyclin-dependent kinases (CDKs), well-known cell division drivers [75], and therefore suppress aberrant cell proliferation (Fig. 2b) [76]. Altogether, SKP2 might inhibit the CDKN1B–CDK axis in UM.
Von Hippel–Lindau tumor suppressor (VHL)
The hypoxic microenvironment is a common characteristic of solid tumors [77, 78]. In normoxia, the E3 ligase VHL mediates the degradation of hypoxia inducible factor 1 subunit alpha (HIF1A), the hallmark of hypoxia, through the UPS. However, hypoxia decreases the expression of VHL but promotes the nuclear translocation of HIF1A, which transactivates the target oncogenes (Fig. 2c) [77]. Similarly, hypoxia also plays a key role in UM. A recent study illustrated that a higher level of VHL contributes to a longer survival of UM patients, while HIF1A performs oppositely. Such worsened survival is ascribed to HIF1A-induced transactivation of vascular growth-related genes, such as platelet and endothelial cell adhesion molecule 1 (PECAM1) and Von Willebrand factor (VWF) (Table 1, Fig. 2c) [79]. Furthermore, BAP1-negative UM tumors have a higher abundance of HIF1A and a lower level of VHL than BAP1-positive tumors, which indicates that the loss of BAP1 promotes the malignant progression of UM [79].
Ring finger protein 2 (RNF2)
The posttranslational modification of histones plays a key role in chromatin-dependent nuclear processes [80]. As a component of chromatin, histone H2A is frequently ubiquitinated and controls gene silencing [81]. RNF2 is an E3 ligase subunit of polycomb repressive complex 1 (PRC1) and a primary modifier of histone H2A [82]. As mentioned above, the loss of BAP1 is a biomarker of malignant UM [83]. In contrast, many other cells and tissues, including embryonic stem cells, fibroblasts, liver, and pancreatic tissues, progress to apoptosis in the same BAP1-inactivated background [84]. Mechanistically, the E3 ligase RNF2 monoubiquitinates histone H2A (Lys119), which suppresses the expression of prosurvival genes, such as B-cell leukemia/lymphoma-2 (BCL2) and myeloid cell leukemia 1 (MCL1). Consequently, these cells are intrigued to apoptosis incidence (Fig. 2d). In contrast, UM cells lacking BAP1 survive via elevated expression of the melanoma oncogene melanocyte-inducing transcription factor (MITF), which promotes malignancy (Table 1 and Fig. 2d) [84]. The mechanism behind this tissue difference remains unclear. It is plausible that the localization of RNF2 in the genome is distinct due to tissue-specific regulators [84], which suggests that discovering the function and genome localization of RNF2 in UM might contribute to developing UPS-targeted therapy.
MYC-binding protein 2 (MYCBP2)
CASP8 and FADD-like apoptosis regulator (CFLAR) is an apoptosis regulator that contributes to the resistance of UM cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis [85]. The threonine/serine-specific E3 ligase MYCBP2 binds to and activates c-MYC, which inhibits the transcription of CFLAR (Table 1) [86, 87]. Therefore, MYCBP2 is required for the c-MYC-mediated inhibition of CFLAR. However, the level of miR-92a-3p is higher in UM than in normal eyes. MiR-92a-3p binds to the 3′-UTR of MYCBP2 mRNA and suppresses the translation of MYCBP2, which consequently rescues UM cells from TRAIL-induced apoptosis (Fig. 2e) [86]. However, it is unclear whether the E3 ligase activity of MYCBP2 is involved in miR-92a-3p-mediated malignancy of UM.
Ubiquitin-activating enzymes, ubiquitin-conjugating enzymes and the proteasome
The proteasome is a key downstream effector in the UPS [88]. The G-protein-coupled receptor 5-hydroxytryptamine receptor 2B (HTR2B) is an oncogene in UM [89]. A recent study indicated that the activity of the proteasome is lower in metastatic UM than in nonmetastatic UM [90]. Thus, the degradation of HTR2B by the proteasome is less rapid in metastatic UM than in the primary tumor. Therefore, HTR2B accumulates in metastatic UM, which is considered a key predictive marker of UM at risk of liver metastasis and lesions [90]. However, the function of HTR2B in UM is still unclear and requires further exploration.
Similar to ubiquitination, neddylation also plays a vital role in the process of tumorigenesis [91, 92]. Neural precursor cell expressed developmentally downregulated 8 (NEDD8) is a ubiquitin-like protein activated by the E1 enzyme NEDD8-activating enzyme (NAE). After NEDD8 is transferred to the E2 enzyme, the cullin-RING ligases (CRLs) subsequently direct NEDD8 to substrates [93]. Ubiquitin-conjugating enzyme E2 M (UBE2M) is a DNA repair-related E2 enzyme that generally helps to maintain the integrity of genomes [94]. However, the level of UBE2M has been found to aberrantly decrease in a variety of tumor types, which contributes to cytogenetic stabilization [95]. Notably, a recent study investigated the difference in E2 enzymes between metastatic and nonmetastatic UM [90]. A reduction in UBE2M occurs in metastatic UM; however, the mechanism underlying UBE2M deregulation and UM metastasis remains unclear. It is postulated that UBE2M might be involved in the regulation of cytogenetic stabilization in UM.
Ubiquitin–proteasome system-targeted therapy of UM
UPS contributes to the malignancy of UM via the well-established mechanisms mentioned above, which form the basis of UPS-targeted therapy against UM. There are usually two E1s, a few E2s (~40), a number of E3s (>600), and DUBs (~100) involved in the UPS [23, 24, 55, 96,97,98]. A specific E3 ligase can selectively catalyze the ubiquitination of substrates [23]. Therefore, E3s are candidate targets for the development of small molecule agents.
Of E3 ligases, MDM2 is the most promising as a therapeutic target in UM. The MDM2–TP53 interaction contributes to the reduction of TP53, which accelerates malignancy of UM [65, 99]. The interplay of MDM2–TP53 is primarily mediated by the ~120 amino acid residues at the N-terminus of MDM2 and the 30 amino acid residues at the N-terminus of TP53 [100]. Based on the mechanisms mentioned above, blocking the interaction between MDM2 and TP53 is a promising strategy to treat UM. The MDM2–TP53 inhibitor NVP-CGM097 (Table 2) is considered a pivotal antitumor agent for UM treatment in clinical trials [100, 101]. Notably, the combination of NVP-CGM097 with the PKC inhibitor (AEB071) significantly inhibits tumor growth in UM patient-derived xenografts (PDXs) [102]. In addition, an increasing number of studies have revealed the function of peptide transduction in targeted therapies of tumors [103, 104], which suggests the potential clinical applications of MDM2-targeted transducible peptides. In accordance, Harbor and his colleagues [105] designed a transducible peptide named Tat-αMDM2 (YGRKKRRQRRRG-QETFSDLWKLLP) (Table 2). This peptide conjugates the Tat transduction sequence (YGRKKRRQRRRG) to a short peptide (QETFSDLWKLLP) derived from the binding domain of TP53 to MDM2. Interestingly, Tat-αMDM2 selectively binds to MDM2 and stabilizes TP53 by inhibiting MDM2-mediated degradation via UPS. The accumulation of TP53 induces the transcription of target genes, including cyclin-dependent kinase inhibitor 1A (CDKN1A), tumor protein p53 inducible protein 3 (TP53I3), and BCL2-associated X, apoptosis regulator (BAX), which promotes apoptosis of primary UM cells. However, Tat-αMDM2 exerts little effect on normal cells [105]. These studies collectively indicate that MDM2–TP53 inhibition is a promising therapeutic strategy for malignant UM.
SKP2 also acts as an E3 ligase. The inhibition of SKP2 is considered a novel antitumor strategy [73]. Small molecules can inhibit the proteolytic and/or nonproteolytic function of SKP2 by inhibiting SKP2 expression, SKP2–SCF complex formation, SKP2–cyclin-dependent kinase regulatory subunit 1 (CKS1, the accessory protein of SCF-SKP2) interaction and other components of the SCF complex [106]. As mentioned above, the downregulation of SKP2 inhibits the ubiquitination of CDKN1B and consequently leads to G1-phase arrest in UM [74]. This result suggested that SKP2 is a potential target against UM. The SKP2 inhibitor C1 (SKPin C1) (Table 2) is a small molecule that specifically blocks the interaction of SKP2 and CDKN1B through counteracting at the interface of SKP2–CKS1 [107]. Interestingly, SKPin C1 can target SKP2 and suppress the proliferation of UM cells by stabilizing CDKN1B in a concentration-dependent manner [74].
In addition to direct inhibition of E3 ligases such as MDM2 and SKP2, the upstream regulation of E3 ligases is also a potential therapeutic strategy for UM. Based on the well-established regulatory mechanism of the miR-92a-3p–MYCBP2–c-MYC axis mentioned above, the histone deacetylase inhibitor entinostat (Table 2) enhances the sensitivity of UM cells to TRAIL-induced apoptosis by downregulating miR-92a-3p [86]. However, it is still unclear whether this anti-UM function of histone deacetylase inhibitors depends on the E3 ligase activity of MYCBP2. It is plausible that histone deacetylases (HDACs) might be involved in the entinostat-mediated inhibition of miR-92a-3p due to the close relationship between HDACs and miRNA in tumors [108,109,110]. In addition to these preclinical studies of targeted therapies, a clinical study (NCT02697630) is ongoing to explore the effectiveness of coadministered entinostat and pembrolizumab in UM patients.
The 26S proteasome is a critical downstream effector of the UPS that degrades multiple ubiquitinated substrates. Although its selectivity is not as good as that of an E3 ligase, the antitumor efficacy of proteasome-targeted therapies is clinically preferred. Bortezomib (Table 2) is a well-known proteasome inhibitor [111] that binds to the catalytic site of the 26S proteasome to execute the slowly reversible inhibition of the proteasome [111]. Interestingly, a preclinical study showed that bortezomib augments the sensitivity of metastatic melanoma cells to the chemotherapeutic agent temozolomide [112]. Although no preclinical evidence regarding the anti-UM function of bortezomib has been achieved, the combination of bortezomib with carboplatin/paclitaxel is undergoing a phase II clinical trial (NCT00288041) for metastatic UM [113]. Multiple signaling pathways, including the NF-κB pathway, contribute to the limited efficacy of chemotherapies in UM [114, 115]. Due to the potency of proteasome inhibitors in blocking NF-κB signaling, it is anticipated that a combination of bortezomib with other chemotherapies might improve the clinical outcome in treating UM [116, 117]. However, future studies are needed to explore the mechanism underlying the anti-UM effect of bortezomib.
Similar to ubiquitination, neddylation also plays a vital role in the malignancy of UM [93]. NAE1 overexpression is observed in UM [118]. Pevonedistat (MLN4924) (Table 2), a first-in-class NAE1 inhibitor [119], stabilizes several CRL substrates, such as nuclear factor, erythroid 2 like 2 (NFE2L2) and phospho-IκBs, in a concentration-dependent manner in UM. The accumulation of these CRL substrates inhibits the expression of the prosurvival gene B-cell lymphoma-extra large (BCLXL) and induces the proapoptotic gene BCL-2-interacting mediator of cell death (BIM) to promote apoptosis in UM cells. Meanwhile, these CRL substrates activate the ataxia-telangiectasia-mutated (ATM)–checkpoint kinase 1 (CHK1)–cell division cycle 25C (CDC25C) axis to trigger the DNA damage response and G2/M-phase arrest of UM cells. Consequently, pevonedistat can significantly suppress the hepatic metastasis of UM [118].
Discussion
Currently, there are 152 UM-related clinical trials (https://clinicaltrials.gov/ct2/results?cond=uveal+melanoma&term=&cntry=&state=&city=&dist=). A wide range of agents have been tested in UM. Among them, canonical chemotherapeutic agents such as cisplatin and paclitaxel have been coadministered with other molecular-targeted agents to ameliorate the resistance of UM to chemotherapies (NCT00329641, NCT00288041). In addition to chemotherapeutic agents, bortezomib, sorafenib, and sunitinib deserve more attention due to their extensive involvement in the UPS-targeted therapies [111, 120]. Bortezomib, the first-in-class proteasome inhibitor, is shown to be a potent UPS-targeted therapy in treating multiple tumors, such as UM [112], lung cancer [121], liver cancer [21], and osteosarcoma [122]. However, most studies emphasize the ability of bortezomib to sensitize cells to other antitumor agents [112, 122]. Therefore, more attention should be paid to the antitumor function of bortezomib as a single agent, especially in treating UM. In addition, there are several preclinical studies and clinical trials of sorafenib/sunitinib in patients with metastatic UM [123, 124]. However, few studies have explored the application of sorafenib/sunitinib-mediated UPS-targeted therapy in primary UM. Indeed, several tumors have developed resistance to sorafenib/sunitinib by regulating the ubiquitination of key proteins [125, 126]. Whether such resistance is pertinent to UM remains unknown. Alternatively, combined treatment of sorafenib/sunitinib with other UPS inhibitors that regulate ubiquitination of the key proteins contributing to drug resistance might be a lead in treating UM.
Proteolysis-targeting chimeras (PROTACs) are novel UPS-dependent therapeutic strategies in drug discovery [127]. PROTAC molecules can bind proteins of interest and specific E3 ligases simultaneously, which promotes ubiquitination and degradation of some oncoproteins [128]. Interestingly, the E3 ligases VHL and MDM2 are prioritized in PROTACs [129, 130]. VHL/MDM2-recruiting PROTAC molecules facilitate the ligation of oncoproteins and VHL/MDM2, which indicates that VHL and MDM2 are potential targets in tumor treatment. However, the abundance of VHL might be too low for the application of VHL-mediated PROTACs because UM tissues sometimes display a hypoxic microenvironment, which is characteristic of solid tumors [79]. Moreover, MDM2 is involved in the degradation of the tumor suppressor TP53, as mentioned above [65]. Therefore, MDM2 is currently defined as an oncoprotein in UM. It seems to be difficult to treat UM by utilizing the MDM2-recruiting PROTAC. However, a recent study showed that the MDM2-recruiting PROTAC inhibits the proliferation of many cancer cells through simultaneously degrading bromodomain-containing protein 4 (BRD4) and stabilizing TP53 [131]. This enables a customized PROTAC molecule to execute MDM2-mediated degradation of oncoproteins and simultaneously block the interaction of MDM2–TP53 in UM.
The lack of intervention targets in the UPS remains a burning issue in UM treatment. Therefore, some prospective studies of UPS-based targets and inhibitors need to be mentioned to guide future research. A recent study reported that CDK7/9 are highly expressed in metastatic UM [132]. The CDK7/9 inhibitor SNS-032 represses cancer stem-like cell properties and inhibits UM cell motility by regulating different transcription factors, such as Kruppel-like factor 4 (KLF4) and c-MYC [132]. This indicates that the degradation of CDK7/9 through the UPS could be a potential therapeutic strategy for UM. Notably, a study reported that a series of PROTACs targeting CDK9 can recruit the E3 ligase cereblon to the natural product Wogonin, which provides a potential application of CDK7/9 degradation in treating UM [133]. Another study indicated that the histone-lysine N-methyltransferase enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) increases the percentage of cancer stem-like cells through miR-29c-disheveled segment polarity protein 2 (DVL2)-β-catenin signaling and accelerates the migration of UM cells through the Rho GDP-dissociation inhibitor gamma (RhoGDIγ)–RAC1 axis [134]. Intriguingly, the EZH2 inhibitor GSK126 can abrogate these malignant phenotypes [134]. Therefore, EZH2 is considered a druggable target in metastatic UM. Furthermore, an increasing number of DUBs, including USP21 [135], UPS44 [136], and DUB3 [137], have been shown to deubiquitinate and stabilize EZH2, resulting in malignancy, which suggests that inhibition of various DUBs and subsequent degradation of EZH2 might be promising targeted therapies in UM.
In addition to UPS-related proteins, other classical antitumor targets, including HDACs [138, 139], insulin-like growth factor 1 receptor (IGF1R) [140], MAPK [141], poly(ADP-ribose) polymerase 1 (PARP1) [142], and vascular endothelial growth factor (VEGF) [143], could also be potential drug targets of UM. Among them, HDAC is more favorable, as its inhibitor entinostat has entered two UM-related clinical trials (Table 2). Moreover, some preclinical studies have shown that HDAC inhibitors, such as entinostat [85], tenovin-6 [138], and JSL-1 [139] execute anti-UM activity through suppressing migration, inducing apoptosis, eliminating cancer stem cells and ameliorating chemoresistance. Compared to other inhibitors, UPS-targeted small molecules may have the following advantages. First, there are only 18 HDACs that control complex downstream signaling [144], while more than 600 E3 ligases regulate specific substrates [145]. Second, other targeted therapies generally modulate the function of protein targets, and mutation of these targets might lead to resistance. However, UPS-targeted therapies not only alter the activity of specific cancer-causing proteins but also lead to the degradation of oncogenic proteins, thus lowering the risk of mutation-associated resistance [146]. Third, UPS-targeted therapies can also target undruggable oncogenic driver proteins [146]. In summary, although the therapeutic potential of other molecular targets, such as HDACs, cannot be underestimated, an increasing amount of evidence has shown that UPS-targeted therapies are clinically significant in treating UM.
Conclusion
Although the evidence above demonstrated the correlation between the UPS and the malignancy of UM, there are still unsolved problems. For example, it is known that there are three major pathways, in particular the MAPK [32], PI3K–AKT [33], and Hippo-YAP pathways [31], that are involved in the UPS. The roles of these pathways in UPS-targeted therapies against UM have yet to be explored.
In summary, UM is a rare and highly metastatic tumor, and the dysregulation of UPS plays a key role in the malignancy of UM. Based on the existing knowledge regarding the UPS, UPS-targeted therapies have become promising therapeutic strategies against UM.
References
Kivela T. The epidemiological challenge of the most frequent eye cancer: Retinoblastoma, an issue of birth and death. Br J Ophthalmol. 2009;93:1129–31.
Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110:956–61.
Amaro A, Gangemi R, Piaggio F, Angelini G, Barisione G, Ferrini S, et al. The biology of uveal melanoma. Cancer Metastasis Rev. 2017;36:109–40.
Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of bap1 in metastasizing uveal melanomas. Science. 2010;330:1410–3.
Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone h2a deubiquitinase activity of the polycomb repressive complex pr-dub. Nature. 2010;465:243–7.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. Bap1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92.
Chen XX, Yin Y, Cheng JW, Huang A, Hu B, Zhang X, et al. Bap1 acts as a tumor suppressor in intrahepatic cholangiocarcinoma by modulating the erk1/2 and jnk/c-jun pathways. Cell Death Dis. 2018;9:1036.
Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, et al. Tumor suppressor and deubiquitinase bap1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA. 2014;111:285–90.
Yoo JH, Shi DS, Grossmann AH, Sorensen LK, Tong Z, Mleynek TM, et al. Arf6 is an actionable node that orchestrates oncogenic gnaq signaling in uveal melanoma. Cancer Cell. 2016;29:889–904.
Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, et al. Hippo-independent activation of yap by the gnaq uveal melanoma oncogene through a trio-regulated rho gtpase signaling circuitry. Cancer Cell. 2014;25:831–45.
Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, et al. Mutant gq/11 promote uveal melanoma tumorigenesis by activating yap. Cancer Cell. 2014;25:822–30.
Chen X, Wu Q, Depeille P, Chen P, Thornton S, Kalirai H, et al. Rasgrp3 mediates mapk pathway activation in gnaq mutant uveal melanoma. Cancer Cell. 2017;31:685–96.e6.
Moore AR, Ran L, Guan Y, Sher JJ, Hitchman TD, Zhang JQ, et al. Gna11 q209l mouse model reveals rasgrp3 as an essential signaling node in uveal melanoma. Cell Rep. 2018;22:2455–68.
Li Y, Sun D, Sun W, Yin D. Ras-pi3k-akt signaling promotes the occurrence and development of uveal melanoma by downregulating h3k56ac expression. J Cell Physiol. 2019;234:16032–42.
Dogrusoz M, Jager MJ, Damato B. Uveal melanoma treatment and prognostication. Asia Pac. J Ophthalmol (Phila). 2017;6:186–96.
Schinzari G, Rossi E, Cassano A, Dadduzio V, Quirino M, Pagliara M, et al. Cisplatin, dacarbazine and vinblastine as first line chemotherapy for liver metastatic uveal melanoma in the era of immunotherapy: a single institution phase ii study. Melanoma Res. 2017;27:591–5.
Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, et al. Combined pkc and mek inhibition in uveal melanoma with gnaq and gna11 mutations. Oncogene. 2014;33:4724–34.
Chua V, Aplin AE. Novel therapeutic strategies and targets in advanced uveal melanoma. Curr Opin Oncol. 2018;30:134–41.
Croce M, Ferrini S, Pfeffer U, Gangemi R. Targeted therapy of uveal melanoma: recent failures and new perspectives. Cancers. 2019;11:846.
Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017;36:561–84.
Chen YJ, Wu H, Shen XZ. The ubiquitin-proteasome system and its potential application in hepatocellular carcinoma therapy. Cancer Lett. 2016;379:245–52.
Weisberg EL, Schauer NJ, Yang J, Lamberto I, Doherty L, Bhatt S, et al. Inhibition of usp10 induces degradation of oncogenic flt3. Nat Chem Biol. 2017;13:1207–15.
Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79.
Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat Rev Drug Discov. 2018;17:57–78.
Niazi S, Purohit M, Niazi JH. Role of p53 circuitry in tumorigenesis: A brief review. Eur J Med Chem. 2018;158:7–24.
Armstrong SR, Wu H, Wang B, Abuetabh Y, Sergi C, Leng RP. The regulation of tumor suppressor p63 by the ubiquitin-proteasome system. Int J Mol Sci. 2016;17:2041.
Lee MS, Jeong MH, Lee HW, Han HJ, Ko A, Hewitt SM, et al. Pi3k/akt activation induces pten ubiquitination and destabilization accelerating tumourigenesis. Nat Commun. 2015;6:7769.
Baietti MF, Simicek M, Abbasi Asbagh L, Radaelli E, Lievens S, Crowther J, et al. Otub1 triggers lung cancer development by inhibiting ras monoubiquitination. EMBO Mol Med. 2016;8:288–303.
Sun XX, He X, Yin L, Komada M, Sears RC, Dai MS. The nucleolar ubiquitin-specific protease usp36 deubiquitinates and stabilizes c-myc. Proc Natl Acad Sci USA. 2015;112:3734–9.
Sun X, Ding Y, Zhan M, Li Y, Gao D, Wang G, et al. Usp7 regulates hippo pathway through deubiquitinating the transcriptional coactivator yorkie. Nat Commun. 2019;10:411.
Nguyen TH, Kugler JM. Ubiquitin-dependent regulation of the mammalian hippo pathway: therapeutic implications for cancer. Cancers. 2018;10:121.
Nguyen LK, Kolch W, Kholodenko BN. When ubiquitination meets phosphorylation: A systems biology perspective of egfr/mapk signalling. Cell Commun Signal. 2013;11:52.
He L, Liu X, Yang J, Li W, Liu S, Liu X, et al. Imbalance of the reciprocally inhibitory loop between the ubiquitin-specific protease usp43 and egfr/pi3k/akt drives breast carcinogenesis. Cell Res. 2018;28:934–51.
Carvajal RD, Schwartz GK, Tezel T, Marr B, Francis JH, Nathan PD. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br J Ophthalmol. 2017;101:38–44.
Smit KN, Jager MJ, de Klein A, and Kili E Uveal melanoma: Towards a molecular understanding. Prog Retin Eye Res. 2019 Sep. https://doi.org/10.1016/j.preteyeres.2019.100800.
Figueiredo CR, Kalirai H, Sacco JJ, Azevedo RA, Duckworth A, Slupsky JR, et al. Loss of bap1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J Pathol. 2020;250:420–39.
Fang Y, Shen X. Ubiquitin carboxyl-terminal hydrolases: Involvement in cancer progression and clinical implications. Cancer Metastasis Rev. 2017;36:669–82.
Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. Bap1 and cancer. Nat Rev Cancer. 2013;13:153–9.
Eletr ZM, Yin L, Wilkinson KD. Bap1 is phosphorylated at serine 592 in s-phase following DNA damage. FEBS Lett. 2013;587:3906–11.
de Waard-Siebinga I, Hilders CG, Hansen BE, van Delft JL, Jager MJ. Hla expression and tumor-infiltrating immune cells in uveal melanoma. Graefes Arch Clin Exp Ophthalmol. 1996;234:34–42.
Maat W, Ly LV, Jordanova ES, de Wolff-Rouendaal D, Schalij-Delfos NE, Jager MJ. Monosomy of chromosome 3 and an inflammatory phenotype occur together in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49:505–10.
Bronkhorst IH, Vu TH, Jordanova ES, Luyten GP, Burg SH, Jager MJ. Different subsets of tumor-infiltrating lymphocytes correlate with macrophage influx and monosomy 3 in uveal melanoma. Invest Ophthalmol Vis Sci. 2012;53:5370–8.
Souri Z, Wierenga APA, van Weeghel C, van der Velden PA, Kroes WGM, Luyten GPM, et al. Loss of bap1 is associated with upregulation of the nfkb pathway and increased hla class i expression in uveal melanoma. Cancers. 2019;11:1102.
Kanarek N, Ben-Neriah Y. Regulation of nf-kappab by ubiquitination and degradation of the ikappabs. Immunol Rev. 2012;246:77–94.
Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. Pml regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330:1247–51.
Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, et al. Bap1 regulates ip3r3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature. 2017;546:549–53.
Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, et al. Pten counteracts fbxl2 to promote ip3r3- and Ca2+-mediated apoptosis limiting tumour growth. Nature. 2017;546:554–8.
Smit KN, Chang J, Derks K, Vaarwater J, Brands T, Verdijk RM, et al. Aberrant microrna expression and its implications for uveal melanoma metastasis. Cancers. 2019;11:815.
Sharma A, Biswas A, Liu H, Sen S, Paruchuri A, Katsonis P, et al. Mutational landscape of the bap1 locus reveals an intrinsic control to regulate the mirna network and the binding of protein complexes in uveal melanoma. Cancers. 2019;11:1600.
Ventii KH, Devi NS, Friedrich KL, Chernova TA, Tighiouart M, Van Meir EG, et al. Brca1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008;68:6953–62.
Pan H, Jia R, Zhang L, Xu S, Wu Q, Song X, et al. Bap1 regulates cell cycle progression through e2f1 target genes and mediates transcriptional silencing via h2a monoubiquitination in uveal melanoma cells. Int J Biochem Cell Biol. 2015;60:176–84.
Scheuermann JC, Gutierrez L, Muller J. Histone h2a monoubiquitination and polycomb repression: The missing pieces of the puzzle. Fly (Austin). 2012;6:162–8.
Asada S, Goyama S, Inoue D, Shikata S, Takeda R, Fukushima T, et al. Mutant asxl1 cooperates with bap1 to promote myeloid leukaemogenesis. Nat Commun. 2018;9:2733.
Shahriyari L, Abdel-Rahman M, Cebulla C. Bap1 expression is prognostic in breast and uveal melanoma but not colon cancer and is highly positively correlated with rbm15b and usp19. PLoS One. 2019;14:e0211507.
Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, et al. Genome-wide and functional annotation of human e3 ubiquitin ligases identifies mulan, a mitochondrial e3 that regulates the organelle’s dynamics and signaling. PLoS One. 2008;3:e1487.
Berndsen CE, Wolberger C. New insights into ubiquitin e3 ligase mechanism. Nat Struct Mol Biol. 2014;21:301–7.
Cahilly-Snyder L, Yang-Feng T, Francke U, George DL. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3t3 cell line. Somat Cell Mol Genet. 1987;13:235–44.
Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9.
Wade M, Li YC, Wahl GM. Mdm2, mdmx and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96.
Lu X, Nguyen TA, Zhang X, Donehower LA. The wip1 phosphatase and mdm2: cracking the “wip” on p53 stability. Cell Cycle. 2008;7:164–8.
Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908.
Carson DA, Lois A. Cancer progression and p53. Lancet. 1995;346:1009–11.
Brantley MA Jr, Harbour JW. Deregulation of the rb and p53 pathways in uveal melanoma. Am J Pathol. 2000;157:1795–801.
Lai H, Ma F, Lai S. Identification of the novel role of prb in eye cancer. J Cell Biochem. 2003;88:121–7.
Davies L, Spiller D, White MR, Grierson I, Paraoan L. Perp expression stabilizes active p53 via modulation of p53-mdm2 interaction in uveal melanoma cells. Cell Death Dis. 2011;2:e136.
Mayo LD, Seo YR, Jackson MW, Smith ML, Rivera Guzman J, Korgaonkar CK, et al. Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J Biol Chem. 2005;280:25953–9.
Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, et al. P53aip1, a potential mediator of p53-dependent apoptosis, and its regulation by ser-46-phosphorylated p53. Cell. 2000;102:849–62.
D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at ser 46 and mediates apoptosis. Nat Cell Biol. 2002;4:11–9.
Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, et al. Phosphorylation of human p53 by p38 kinase coordinates n-terminal phosphorylation and apoptosis in response to uv radiation. EMBO J. 1999;18:6845–54.
Wu S, Chen H, Han N, Zhang C, Yan H. Long noncoding rna pvt1 silencing prevents the development of uveal melanoma by impairing microrna-17-3p-dependent mdm2 upregulation. Invest Ophthalmol Vis Sci. 2019;60:4904–14.
Al-Khalaf HH, Aboussekhra A. P16 controls p53 protein expression through mir-dependent destabilization of mdm2. Mol Cancer Res. 2018;16:1299–308.
Lough L, Sherman D, Ni E, Young LM, Hao B, Cardozo T. Chemical probes of skp2-mediated p27 ubiquitylation and degradation. Medchemcomm. 2018;9:1093–104.
Lee Y, Lim HS. Skp2 inhibitors: Novel anticancer strategies. Curr Med Chem. 2016;23:2363–79.
Zhao H, Pan H, Wang H, Chai P, Ge S, Jia R, et al. Skp2 targeted inhibition suppresses human uveal melanoma progression by blocking ubiquitylation of p27. Onco Targets Ther. 2019;12:4297–308.
McGrath DA, Fifield BA, Marceau AH, Tripathi S, Porter LA, Rubin SM. Structural basis of divergent cyclin-dependent kinase activation by spy1/ringo proteins. EMBO J. 2017;36:2251–62.
Ray A, James MK, Larochelle S, Fisher RP, Blain SW. P27kip1 inhibits cyclin d-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol. 2009;29:986–99.
Semenza GL. Targeting hif-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32.
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408.
Brouwer NJ, Wierenga APA, Gezgin G, Marinkovic M, Luyten GPM, Kroes WGM, et al. Ischemia is related to tumour genetics in uveal melanoma. Cancers. 2019;11:1004.
Zhang Z, Jones AE, Wu W, Kim J, Kang Y, Bi X, et al. Role of remodeling and spacing factor 1 in histone h2a ubiquitination-mediated gene silencing. Proc Natl Acad Sci USA. 2017;114:E7949–E58.
Klusmann I, Wohlberedt K, Magerhans A, Teloni F, Korbel JO, Altmeyer M, et al. Chromatin modifiers mdm2 and rnf2 prevent rna:DNA hybrids that impair DNA replication. Proc Natl Acad Sci USA. 2018;115:E11311–E20.
Taherbhoy AM, Huang OW, Cochran AG. Bmi1-ring1b is an autoinhibited ring e3 ubiquitin ligase. Nat Commun. 2015;6:7621.
Field MG, Kuznetsov JN, Bussies PL, Cai LZ, Alawa KA, Decatur CL, et al. Bap1 loss is associated with DNA methylomic repatterning in highly aggressive class 2 uveal melanomas. Clin Cancer Res. 2019;25:5663–73.
He M, Chaurushiya MS, Webster JD, Kummerfeld S, Reja R, Chaudhuri S, et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase bap1. Science. 2019;364:283–5.
Venza I, Visalli M, Oteri R, Teti D, Venza M. Class i-specific histone deacetylase inhibitor ms-275 overrides trail-resistance in melanoma cells by downregulating c-flip. Int Immunopharmacol. 2014;21:439–46.
Venza M, Visalli M, Beninati C, Benfatto S, Teti D, Venza I. Mir-92a-3p and mycbp2 are involved in ms-275-induced and c-myc-mediated trail-sensitivity in melanoma cells. Int Immunopharmacol. 2016;40:235–43.
Pao KC, Wood NT, Knebel A, Rafie K, Stanley M, Mabbitt PD, et al. Activity-based e3 ligase profiling uncovers an e3 ligase with esterification activity. Nature. 2018;556:381–5.
Ao N, Chen Q, Liu G. The small molecules targeting ubiquitin-proteasome system for cancer therapy. Comb Chem High Throughput Screen. 2017;20:403–13.
Benhassine M, Guerin SL. Transcription of the human 5-hydroxytryptamine receptor 2b (htr2b) gene is under the regulatory influence of the transcription factors nfi and runx1 in human uveal melanoma. Int J Mol Sci. 2018;19:3272.
Le-Bel G, Benhassine M, Landreville S, Guerin SL. Analysis of the proteasome activity and the turnover of the serotonin receptor 2b (htr2b) in human uveal melanoma. Exp Eye Res. 2019;184:72–7.
Jiang Y, Liang Y, Li L, Zhou L, Cheng W, Yang X, et al. Targeting neddylation inhibits intravascular survival and extravasation of cancer cells to prevent lung-cancer metastasis. Cell Biol Toxicol. 2019;35:233–45.
Zhou Q, Li H, Li Y, Tan M, Fan S, Cao C, et al. Inhibiting neddylation modification alters mitochondrial morphology and reprograms energy metabolism in cancer cells. JCI insight. 2019;4:e121582.
Watson IR, Irwin MS, Ohh M. Nedd8 pathways in cancer, sine quibus non. Cancer Cell. 2011;19:168–76.
Cukras S, Morffy N, Ohn T, Kee Y. Inactivating ube2m impacts the DNA damage response and genome integrity involving multiple cullin ligases. PLoS One. 2014;9:e101844.
Shields CL, Say EAT, Hasanreisoglu M, Saktanasate J, Lawson BM, Landy JE, et al. Cytogenetic abnormalities in uveal melanoma based on tumor features and size in 1059 patients: The 2016 w. Richard green lecture. Ophthalmology. 2017;124:609–18.
Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS. E2 enzymes: more than just middle men. Cell Res. 2016;26:423–40.
Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403.
Hyer ML, Milhollen MA, Ciavarri J, Fleming P, Traore T, Sappal D, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med. 2018;24:186–93.
Coupland SE, Anastassiou G, Stang A, Schilling H, Anagnostopoulos I, Bornfeld N, et al. The prognostic value of cyclin d1, p53, and mdm2 protein expression in uveal melanoma. J. Pathol. 2000;191:120–6.
Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the mdm2-p53 protein-protein interaction (mdm2 inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58:1038–52.
Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, et al. Discovery of a dihydroisoquinolinone derivative (nvp-cgm097): A highly potent and selective mdm2 inhibitor undergoing phase 1 clinical trials in p53wt tumors. J Med Chem. 2015;58:6348–58.
Carita G, Frisch-Dit-Leitz E, Dahmani A, Raymondie C, Cassoux N, Piperno-Neumann S, et al. Dual inhibition of protein kinase C and p53-mdm2 or pkc and mtorc1 are novel efficient therapeutic approaches for uveal melanoma. Oncotarget. 2016;7:33542–56.
Snyder EL, Meade BR, Saenz CC, Dowdy SF. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004;2:E36.
Snyder EL, Saenz CC, Denicourt C, Meade BR, Cui XS, Kaplan IM, et al. Enhanced targeting and killing of tumor cells expressing the cxc chemokine receptor 4 by transducible anticancer peptides. Cancer Res. 2005;65:10646–50.
Harbour JW, Worley L, Ma D, Cohen M. Transducible peptide therapy for uveal melanoma and retinoblastoma. Arch Ophthalmol. 2002;120:1341–6.
Cai Z, Moten A, Peng D, Hsu CC, Pan BS, Manne R, et al. The skp2 pathway: A critical target for cancer therapy. Semin Cancer Biol. 2020 Feb. https://doi.org/10.1016/j.semcancer.2020.01.013.
Wu L, Grigoryan AV, Li Y, Hao B, Pagano M, Cardozo TJ. Specific small molecule inhibitors of skp2-mediated p27 degradation. Chem Biol. 2012;19:1515–24.
Bharathy N, Berlow NE, Wang E, Abraham J, Settelmeyer TP, Hooper JE, et al. The hdac3-smarca4-mir-27a axis promotes expression of the pax3:Foxo1 fusion oncogene in rhabdomyosarcoma. Sci Signal. 2018;11:eaau7632.
Lai TH, Ewald B, Zecevic A, Liu C, Sulda M, Papaioannou D, et al. Hdac inhibition induces microrna-182, which targets rad51 and impairs hr repair to sensitize cells to sapacitabine in acute myelogenous leukemia. Clin Cancer Res. 2016;22:3537–49.
Meyers-Needham M, Ponnusamy S, Gencer S, Jiang W, Thomas RJ, Senkal CE, et al. Concerted functions of hdac1 and microrna-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol Med. 2012;4:78–92.
Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14:417–33.
Amiri KI, Horton LW, LaFleur BJ, Sosman JA, Richmond A. Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: implication for bortezomib (velcade, ps-341) as a therapeutic agent for malignant melanoma. Cancer Res. 2004;64:4912–8.
Triozzi PL, Eng C, Singh AD. Targeted therapy for uveal melanoma. Cancer Treat Rev. 2008;34:247–58.
Ambrosini G, Do C, Tycko B, Realubit RB, Karan C, Musi E, et al. Inhibition of nf-kappab-dependent signaling enhances sensitivity and overcomes resistance to bet inhibition in uveal melanoma. Cancer Res. 2019;79:2415–25.
Hu S, Luo Q, Cun B, Hu D, Ge S, Fan X, et al. The pharmacological NF-kappaB inhibitor Bay11-7082 induces cell apoptosis and inhibits the migration of human uveal melanoma cells. Int J Mol Sci. 2012;13:15653–67.
Pordanjani SM, Hosseinimehr SJ. The role of NF-kappaB inhibitors in cell response to radiation. Curr Med Chem. 2016;23:3951–63.
Yang J, LeBlanc FR, Dighe SA, Hamele CE, Olson TL, Feith DJ, et al. Trail mediates and sustains constitutive NF-kappaB activation in lgl leukemia. Blood. 2018;131:2803–15.
Jin Y, Zhang P, Wang Y, Jin B, Zhou J, Zhang J, et al. Neddylation blockade diminishes hepatic metastasis by dampening cancer stem-like cells and angiogenesis in uveal melanoma. Clin Cancer Res. 2018;24:3741–54.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of nedd8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–6.
Gossage L, Eisen T. Alterations in vhl as potential biomarkers in renal-cell carcinoma. Nat Rev Clin Oncol. 2010;7:277–88.
Qu YQ, Gordillo-Martinez F, Law BYK, Han Y, Wu A, Zeng W, et al. 2-aminoethoxydiphenylborane sensitizes anti-tumor effect of bortezomib via suppression of calcium-mediated autophagy. Cell Death Dis. 2018;9:361.
Punzo F, Tortora C, Di Pinto D, Pota E, Argenziano M, Di Paola A, et al. Bortezomib and endocannabinoid/endovanilloid system: a synergism in osteosarcoma. Pharmacol Res. 2018;137:25–33.
Mouriaux F, Servois V, Parienti JJ, Lesimple T, Thyss A, Dutriaux C, et al. Sorafenib in metastatic uveal melanoma: Efficacy, toxicity and health-related quality of life in a multicentre phase ii study. Br J Cancer. 2016;115:20–4.
Valsecchi ME, Orloff M, Sato R, Chervoneva I, Shields CL, Shields JA, et al. Adjuvant sunitinib in high-risk patients with uveal melanoma: comparison with institutional controls. Ophthalmology. 2018;125:210–7.
Azzariti A, Mancarella S, Porcelli L, Quatrale AE, Caligiuri A, Lupo L, et al. Hepatic stellate cells induce hepatocellular carcinoma cell resistance to sorafenib through the laminin-332/alpha3 integrin axis recovery of focal adhesion kinase ubiquitination. Hepatology. 2016;64:2103–17.
Huang H, Gao Y, Liu A, Yang X, Huang F, Xu L, et al. Eif3d promotes sunitinib resistance of renal cell carcinoma by interacting with grp78 and inhibiting its degradation. EBioMedicine. 2019;49:189–201.
Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by protacs. Pharmacol Ther. 2017;174:138–44.
Paiva SL, Crews CM. Targeted protein degradation: Elements of protac design. Curr Opin Chem Biol. 2019;50:111–9.
Han X, Zhao L, Xiang W, Qin C, Miao B, Xu T, et al. Discovery of highly potent and efficient protac degraders of androgen receptor (ar) by employing weak binding affinity vhl e3 ligase ligands. J Med Chem. 2019;62:11218–31.
Yang J, Li Y, Aguilar A, Liu Z, Yang CY, Wang S. Simple structural modifications converting a bona fide mdm2 protac degrader into a molecular glue molecule: A cautionary tale in the design of protac degraders. J Med Chem. 2019;62:9471–87.
Hines J, Lartigue S, Dong H, Qian Y, Crews CM. Mdm2-recruiting protac offers superior, synergistic antiproliferative activity via simultaneous degradation of brd4 and stabilization of p53. Cancer Res. 2019;79:251–62.
Zhang J, Liu S, Ye Q, Pan J. Transcriptional inhibition by cdk7/9 inhibitor sns-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol Cancer. 2019;18:140.
Bian J, Ren J, Li Y, Wang J, Xu X, Feng Y, et al. Discovery of wogonin-based protacs against cdk9 and capable of achieving antitumor activity. Bioorg Chem. 2018;81:373–81.
Jin B, Zhang P, Zou H, Ye H, Wang Y, Zhang J, et al. Verification of EZH2 as a druggable target in metastatic uveal melanoma. Mol Cancer. 2020;19:52.
Chen Y, Zhou B, Chen D. Usp21 promotes cell proliferation and metastasis through suppressing ezh2 ubiquitination in bladder carcinoma. Onco Targets Ther. 2017;10:681–9.
Park JM, Lee JE, Park CM, Kim JH. Usp44 promotes the tumorigenesis of prostate cancer cells through EZH2 protein stabilization. Mol Cell. 2019;42:17–27.
Luo F, Zhou Z, Cai J, Du W. Dub3 facilitates growth and inhibits apoptosis through enhancing expression of EZH2 in oral squamous cell carcinoma. Onco Targets Ther. 2020;13:1447–60.
Dai W, Zhou J, Jin B, Pan J. Class iii-specific hdac inhibitor tenovin-6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci Rep. 2016;6:22622.
Wang Y, Liu M, Jin Y, Jiang S, Pan J. In vitro and in vivo anti-uveal melanoma activity of jsl-1, a novel hdac inhibitor. Cancer Lett. 2017;400:47–60.
Economou MA, Andersson S, Vasilcanu D, All-Ericsson C, Menu E, Girnita A, et al. Oral picropodophyllin (ppp) is well tolerated in vivo and inhibits igf-1r expression and growth of uveal melanoma. Acta Ophthalmol. 2008;86(Thesis 4):35–41.
Chua V, Lapadula D, Randolph C, Benovic JL, Wedegaertner PB, Aplin AE. Dysregulated gpcr signaling and therapeutic options in uveal melanoma. Mol Cancer Res. 2017;15:501–6.
de Koning L, Decaudin D, El Botty R, Nicolas A, Carita G, Schuller M, et al. Parp inhibition increases the response to chemotherapy in uveal melanoma. Cancers (Basel). 2019 May. https://doi.org/10.3390/cancers11060751.
Dithmer M, Kirsch AM, Gräfenstein L, Wang F, Schmidt H, Coupland SE, et al. Uveal melanoma cell under oxidative stress - influence of vegf and vegf-inhibitors. Klin Monbl Augenheilkd. 2019;236:295–307.
McClure JJ, Li X, Chou CJ. Advances and challenges of hdac inhibitors in cancer therapeutics. Adv Cancer Res. 2018;138:183–211.
Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: Expanding the toolbox. Nat Rev Drug Discov. 2019;18:949–63.
Salami J, Crews CM. Waste disposal-an attractive strategy for cancer therapy. Science. 2017;355:1163–7.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81973349 to HZ), the Young Talent’s Subsidy Project in Science and Education of the Department of Public Health of Jiangsu Province (QNRC2016627 to KW), and the Six Talent Peaks Project in Jiangsu Province (WSW-047 to KW).
Author information
Authors and Affiliations
Contributions
CXZ was responsible for the conception and design of the review. CMZ and KW collected the literature. QJH, BY, HZ, and FFZ analyzed the literature and summarized the results. CXZ drafted the manuscript and generated the figures. HZ and FFZ revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Zhao, Cx., Zeng, Cm., Wang, K. et al. Ubiquitin–proteasome system-targeted therapy for uveal melanoma: what is the evidence?. Acta Pharmacol Sin 42, 179–188 (2021). https://doi.org/10.1038/s41401-020-0441-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0441-3
Keywords
This article is cited by
-
The multi-kinase inhibitor afatinib serves as a novel candidate for the treatment of human uveal melanoma
Cellular Oncology (2022)
-
MYH9-dependent polarization of ATG9B promotes colorectal cancer metastasis by accelerating focal adhesion assembly
Cell Death & Differentiation (2021)
-
UBQLN4 is activated by C/EBPβ and exerts oncogenic effects on colorectal cancer via the Wnt/β-catenin signaling pathway
Cell Death Discovery (2021)
