
Abstract
Tissue kallikrein has protective function against various types of injury. In this study, we investigated whether exogenous pancreatic kininogenase (PK) conferred renoprotection in a rat model of unilateral ureteral obstruction (UUO) and H2O2-treated HK-2 cells in vitro. SD rats were subjected to UUO surgery, then PK (7.2 U/g per day, ip) was administered for 7 or 14 days. After the treatment, rats were euthanized; the obstructed kidneys were harvested for further examination. We found that PK administration significantly attenuated interstitial inflammation and fibrosis, and downregulated the expression of proinflammatory (MCP-1, TLR-2, and OPN) and profibrotic (TGF-β1 and CTGF) cytokines in obstructed kidney. UUO-induced oxidative stress, closely associated with excessive apoptotic cell death and autophagy via PI3K/AKT/FoxO1a signaling, which were abolished by PK administration. We further showed that PK administration increased the expression of bradykinin receptors 1 and 2 (B1R and B2R) mRNA and the production of NO and cAMP in kidney tissues. Coadministration with either B1R antagonist (des-Arg9-[Leu8]-bradykinin) or B2R antagonist (icatibant) abrogated the renoprotective effects of PK, and reduced the levels of NO and cAMP in obstructed kidney. In H2O2-treated HK-2 cells, addition of PK (6 pg/mL) significantly decreased ROS production, regulated the expression of oxidant and antioxidant enzymes, suppressed the expression of TGF-β1 and MCP-1, and inhibited cell apoptosis. Our data demonstrate that PK treatment protects against the progression of renal fibrosis in obstructed kidneys.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD) is the ninth leading cause of death in the United States and remains a major human health problem worldwide. The global prevalence of CKD is approximately of 13% for stages 1–5 and 10% for stages 3–5, and is increasing because of the increasing prevalence of obesity, diabetes mellitus, hypertension, and increased longevity [1]. Renal tubulointerstitial fibrosis (TIF), which is the final common end and histological amalgamation of most types of progressive CKD, ultimately results in end-stage renal disease. Unilateral ureteral obstruction (UUO) is a reproducible rodent experimental model of TIF that mimics the effects that are observed in human CKD. Histologically, UUO manifests as interstitial inflammation, substantial extracellular matrix deposition, tubular dilatation, atrophy, and fibrosis. This process involves a complex multistage procedure that is orchestrated by a network of chemokines, adhesion molecules, cytokines, growth factors, and signaling pathways [2]. Although the precise mechanism underlying UUO-induced renal TIF is unclear, oxidative stress-induced inflammation, transforming growth factor (TGF)-β1, and programmed cell death (apoptosis and autophagy) have been implicated [3, 4].
Tissue kallikrein, a serine proteinase, cleaves kininogen to produce bioactive kinin peptides. The binding of kinins to the B1 receptor and/or B2 receptor triggers multiple biological functions via an increase in intracellular Ca2+ mobilization, resulting in the formation of nitric oxide (NO), prostaglandin, 3′,5′-cyclic adenosine monophosphate (cAMP), and cyclic guanosine monophosphate [5]. Because of these activities, kallikrein confers a number of benefits for the regulation of hypertension, cardiovascular diseases, ischemic stroke, and diabetes mellitus [6]. An early study illustrated that delivery of the kallikrein gene alleviates serum glucose and improves lipid metabolism in streptozotocin (STZ)-induced diabetes [7]. In the kidney, a genetic deficiency in kallikrein exacerbates microalbuminuria in STZ-induced diabetic nephropathy (DN) and administration of exogenous kallikrein ameliorates not only hyperglycemia, but also albuminuria and renal fibrosis [8, 9]. Similar renoprotection by kallikrein or kallikrein gene transfer has also been observed in gentamicin-induced nephrotoxicity [10], renal ischemia-reperfusion injury [11], hypertensive nephropathy [12], and polycystic kidney disease [13].
It has not been determined whether kallikrein attenuates UUO-induced renal fibrosis. We used in vivo and in vitro studies to test: (1) whether exogenous treatment with pancreatic kininogenase (PK) conferred renoprotection in obstructed kidneys and (2) whether the effectiveness of PK was abolished by a bradykinin receptor (B1R or B2R) antagonist. The results of our study clearly demonstrated that PK is a potential treatment to manage renal fibrosis in UUO.
Materials and methods
Establishment of UUO and treatment schedule
The experimental protocol was approved by the Animal Care and Research Committee of Yanbian University of China (YBU 2018-10-8). Adult male Sprague-Dawley rats weighing 200–230 g were housed in individual cases with a 12-h artificial light–dark cycle environment and were permitted free access to standard chow and water. Two separate experiments were conducted as follows:
Protocol 1
This study was performed to evaluate the effect of exogenous PK on renal fibrosis caused by UUO. Induction of UUO was performed as described previously [14]. Briefly, weight-matched rats were anesthetized with pentobarbital (40 mg/kg) and a flank incision was made to expose the kidney and ureter. The left ureter was ligated with 4–0 silk, and then the incision was sutured. Sham surgery was performed as in the UUO procedure without ligation of the left ureter. After surgery, the rats were randomly assigned to five groups: sham operation (n = 6); UUO7 (n = 8); UUO + PK7 (n = 8); UUO14 (n = 8); and UUO + PK14 (n = 8). Administration of PK (7.2 U/g per day, intraperitoneal, Changzhou Qianhong Bio-pharma Co., Ltd. China,) was started 24 h after UUO and continued for 14 days. After 7 or 14 days of treatment, the rats were anesthetized and perfused, and the obstructed kidneys were rapidly harvested for further examination.
Protocol 2
This study was designed to evaluate the effect of B1R or B2R antagonists on the renoprotective effects of PK in obstructed kidneys. The rats were randomly assigned to five groups: sham operation (n = 6); UUO14 (n = 8); UUO + PK14 (n = 8); UUO + PK14 + B1R (n = 8); and UUO + PK14 + B2R (n = 8). After 14 days of treatment, the rats were euthanized. The dose and route of administration of PK, B1R antagonist (250 μg subcutaneous injection twice daily, des-Arg9-[Leu8]-bradykinin, Sigma-Aldrich B6929, UK), or the B2R antagonist icatibant (200 μg subcutaneous injection twice daily, HOE 140, Sigma-Aldrich H157, St. Louis, Missouri, USA) were chosen based on previous studies [13, 15].
Antibodies
The antibodies used in this study for immunohistochemistry and immunoblotting were as follows: ED-1 (Serotec Inc., UK), osteopontin (OPN, #ab8448, Abcam, Cambridge, UK), TGF-β1 (R&D Systems, Minneapolis, MN, USA), connective tissue growth factor (CTGF, #ab125943, Abcam, Cambridge, UK), matrix metalloproteinase-2 (MMP-2 #ab38929, Abcam, Cambridge, UK), monocyte chemoattractant protein-1 (MCP-1, Santa Cruz Biotechnoogy, Dallas, TX, USA), Toll-like receptor-2 (TLR-2, Santa Cruz Biotechnoogy, Dallas, TX, USA), heme oxygenase-1 (#ab13248, Abcam, Cambridge, UK), superoxide dismutase 1 (SOD1, #ab16831, Abcam, Cambridge, UK), superoxide dismutase 2/manganese superoxide dismutase (SOD2/MnSOD, #ab13534, Abcam, Cambridge, UK), NADPH oxidase-2 (NOX-2, #ab31092, Abcam, Cambridge, UK), NADPH oxidase-4 (NOX-4, #ab109225, Abcam, Cambridge, UK), B-cell lymphoma-2 (Bcl-2, #ab59348, Abcam, Cambridge, UK), Bcl-2-associated X (Bax, #ab32503, Abcam, Cambridge, UK), cleaved caspase-3 (Millipore, Billerica, Mass), light chain 3B (LC3B, #ab192890, Abcam, Cambridge, UK), Beclin-1 (#ab62557, Abcam, Cambridge, UK), p62 (#ab56416, Abcam, Cambridge, UK), phospho-PI3K (#ab138364, Abcam, Cambridge, UK), PI3K (#ab180967, Abcam, Cambridge, UK), phospho-AKT (#ab38449, Abcam, Cambridge, UK), AKT (#ab8805, Abcam, Cambridge, UK), phospho-FoxO1a (#ab131339, Abcam, Cambridge, UK), and β-actin (#ab8226, Abcam, Cambridge, UK).
Preservation of renal specimens
Kidney specimens were preserved by in vivo perfusion. Rats were perfused using 0.01 mol/L phosphate-buffered saline (PBS) to flush the blood via the abdominal aorta, and the dissected kidneys were rapidly immersed in periodate-lysine-2% paraformaldehyde solution, and embedded in paraffin.
Histopathology
After dewaxing the embedded blocks, 4-μm sections were cut and stained with Masson’s trichrome and hematoxylin–eosin. Quantitative analysis of TIF was performed using a color image autoanalyzer (TDI Scope Eye Version 3.6 for Windows; Seoul, Korea) as previously described [16].
Immunohistochemistry
Dewaxed sections were incubated with 0.5% Triton X-100/PBS for 30 min and washed with PBS three times. Nonspecific binding sites were blocked with normal horse serum diluted 1:10 in 0.3% bovine serum albumin for 30–60 min, and then incubated for 2 h at 4 °C in primary antibodies diluted 1:1000 in a humid environment. After rinsing in Tris-buffered saline (TBS), the sections were incubated in peroxidase-conjugated goat anti-rabbit IgG (Amersham Pharmacia Biotech, Piscataway, NJ, USA) for 30 min. For coloration, the sections were incubated with a mixture of 0.05% 3,3′-diaminobenzidine containing 0.01% H2O2 at room temperature until a brown color was visible, washed with TBS, counterstained with hematoxylin, and examined under light microscopy. Quantitative analysis was conducted by calculating the positive area or percentage using histogram equalization (TDI Scope Eye Version 3.6 for Windows; Seoul, Korea).
Transmission electron microscopy
Kidney tissues were postfixed with 1% OsO4 and embedded in Epon 812, following fixation in 2.5% glutaraldehyde in 0.1 M phosphate buffer. Ultrathin sections were cut and stained with uranyl acetate/lead citrate, and photographed with a JEM-1200EX transmission electron microscope (JEOL Ltd., Tokyo, Japan).
Immunoblotting analysis
Immunoblotting was performed as described previously [17]. Briefly, renal tissue lysates were incubated with specific antibodies, and immunoblotting image analyses were performed using an image analyzer.
In situ TdT-mediated dUTP-biotin nick end labeling assay
Apoptosis in tissue sections was determined using the ApopTag in situ apoptosis detection kit (Sigma-Aldrich, Merck millipore). The number of TdT-mediated dUTP-biotin nick end labeling (TUNEL)-positive cells was counted in 20 randomly selected nonoverlapping fields in each section at ×200 magnification.
Enzyme-linked immunosorbent assay
The levels of 8-hydroxy-2′-deoxyguanosine (8-OHdG) were evaluated using a competitive enzyme-linked immunosorbent assay (ELISA) to determine the levels of the DNA adduct 8-OHdG in serum and urine (Japan Institute for the Control of Aging, Shizuoka, Japan).
To determine the concentrations of bradykinin, NO, and cAMP, kidney tissues were homogenized and analyzed using commercial ELISA kits (Shanghai Enzyme-lined Biotechnology Co., Ltd, Shanghai, China and Ray Biotech, Inc., Norcross, GA, USA). The samples and standards were added to microtiter plate wells and coated with the relevant purified rat antibodies. After labeling with a horseradish peroxidase conjugate, the wells were washed and developed using 3,3′,5,5′-tetramethylbenzidine substrate solution for 15 min in the dark. Reactions were terminated by adding sulfuric acid solution, and the absorbance was measured at a wavelength of 450 nm using a Synergy HTX Multi-Mode Microplate Reader (BioTek, USA). The concentrations were calculated using a standard curve of known standard concentrations.
Reverse transcription-quantitative polymerase chain reaction
Total RNA was extracted from rat kidney tissue using RNAiso Plus (TaKaRa, Shiga, Japan), and a cDNA was synthesized from 2 μg RNA with a cDNA synthesis kit (Zomanbio, Beijing, China). Quantitative polymerase chain reaction (qPCR) analysis was performed using SYBR master mix (Zomanbio, Beijing, China) and an ABI 7300 real-time PCR system according to the protocols provided by the manufacturer. The relative mRNA levels were calculated according to the 2∆CT method, in which ∆CT represents the difference in threshold cycle values between the target mRNA and the glyceraldehyde 3-phosphate dehydrogenase internal control. The primer sequences used were as follows:
GAPDH-F 5′-CCCCAATGTATCCGTTGTGGA-3′;
GAPDH-R 5′-GCCTGCTTCACCACCTTCTT-3′;
B1R-F 5′-GACGGCAAGCCCAAGCTA-3′;
B1R-R 5′-CGGGGAGGAGGAAACCCAAAA-3′;
B2R-F 5′-TGAGGAACAACGAGATGAAGAAG-3′; and
B2R-R 5′-GGAAACCAACACAGCACAAAGAG-3′.
Cell culture and treatment
Human kidney proximal tubular epithelial cells (HK-2 cells) were obtained from the American Type Culture Collection (Manassas, VA, USA). HK-2 cells were grown in Dulbecco’s modified Eagle’s medium/Nutrient F12 (HyClone; GE Healthcare Life Science, Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The cells were cultured in a humidified incubator with 5% CO2 and 37 °C. Cells were pretreated with PK at a concentration of 6 pg/mL for 1 h, and then treated with H2O2 (500 μM) with or without PK for 24 h.
Cell viability assay
The viability of HK-2 cells was evaluated using Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) according to the manufacturer’s protocol. Approximately 1.0 × 104 HK-2 cells/well were seeded in a 96-well plate. After the cells were treated with H2O2 (500 μM) with or without PK (6 pg/mL) for 24 h, 10 μL of CCK-8 solution was added to each well and incubated at 37 °C for 3 h. The absorbance was measured by determining the optical density at 450 nm (VersaMax Microplate Reader, Molecular Devices, LLC, Sunnyvale, CA, USA).
Measurement of reactive oxygen species production
The levels of intracellular reactive oxygen species (ROS) production were measured using 2′, 7′-dichlorodihydrofluorescein diacetate (H2-DCFDA, Invitrogen) according to the manufacturer’s instructions. HK-2 cells were seeded at a density of 2.0 × 105 cells/well in a six-well plate. After the treatment with H2O2 with or without PK for 24 h, the cells were washed three times in PBS and incubated with H2-DCFDA for 30 min. The cells were washed and collected in PBS, and fluorescence was measured using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
Apoptosis assay
Annexin V-positive HK-2 cells were detected using an Annexin V-FITC apoptosis detection kit (Biosharp, Hefei, China) according to the manufacturer’s protocol. Briefly, HK-2 cells were seeded and incubated with 500 μM H2O2 with or without PK (6 pg/mL) for 24 h. The cells were harvested, washed three times with PBS, and incubated with 1× binding buffer at a concerntration of 1 × 106 cells/mL. Then, the cells were incubated with 5 μL of Annexin V-FITC and 5 μL of propidium iodide (PI) at room temperature for 15 min in the dark. The samples were analyzed within 1 h using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA). Apoptotic cells were determind as a percentage of the total cell count. The percentage of apoptotic cells was calculated as the number of PI-positive and Annexin V-positive cells divided by the total number of cells. Three independent experiments were performed.
Statistical analysis
The data are presented as the mean ± SEM. Multiple comparisons among groups were performed by one-way analysis of variance with a Bonferroni post hoc test (SPSS software version 21.0, IBM, Armonk, NY, USA). Statistical significance was determined as P < 0.05.
Results
PK attenuates UUO-induced TIF
Digital images of the gross findings clearly illustrated that by day 7, UUO resulted in an enlargement of the kidney with pyelectasis, destruction of the pelvis, and an extremely thin cortex, and these features were more pronounced by day 14 (Fig. 1a). This finding indicates that the UUO model was successfully established in these rats. Histological staining and electron microscopy showed that the major changes were localized to the tubulointerstitium at day 7, and were characterized by severe necrosis, vacuolization, mild atrophy and fibrosis, tubular basement membrane thickening, swelling of the tubular epithelium and interstitium, and inflammatory cell infiltration (Fig. 1b–d). After 14 days of UUO, extensive tubular vacuolization and atrophy, massive inflammatory cell infiltration, and severe TIF were observed. Of note, the sites of severe TIF were surrounded by areas of tubular vacuolization and atrophy (Fig. 1b, c). Electron microscopy clearly illustrated atrophied and vacuolated tubules (Fig. 1 d2) and collagenous fiber deposition (Fig. 1 d3). Our quantitative analysis system showed that the increase in TIF score seen in the UUO7 and UUO14 groups was significantly attenuated at both time points by PK treatment (Fig. 1e). Consistent with this result, immunoblotting analysis revealed that PK treatment inhibited the expression of TGF-β1, CTGF, and MMP-2 at days 7 and 14 compared with those of UUO alone (Fig. 3).

Representative photographs of gross findings a, photomicrographs of Masson trichrome b and hematoxylin–eosin c staining, transmission electron microscopy d, and quantitative analysis of tubulointerstitial fibrosis e. d1: normal kidney structure in the sham-operated group; d2: tubular vacuolization and atrophy in the obstructed kidney; d3: collagenous fiber deposition within the tubulointerstitium of the obstructed kidney (scatter); d4: tubular basement membrane thickening (arrows) and scattered interstitial collagen deposition (star); and d5: swelling of the tubulointerstitium (circle) and inflammatory cell infiltration (arrowheads); **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO14
PK attenuates UUO-induced interstitial inflammation
Inflammation is the predominant feature of UUO that precedes fibrosis [18]. Injured renal tubular cells overexpress chemoattractants that recruit monocytes and/or macrophages, while infiltrated inflammatory cells express chemotactic factors, leading to a vicious circle. To explore the effect of PK on inflammation, we studied the expression of inflammatory mediators using immunohistochemistry and immunoblotting. As shown in Figs. 2 and 3, UUO caused significant upregulation of MCP-1, TLR-2, and OPN expression, accompanied by an increase in the number of ED-1-positive cells at day 7, and this increased further by day 14. However, these alterations were all reversed following administration of PK.

Representative photomicrographs of immunohistochemical analysis of ED-1 a or OPN b, and transmission electron microscopy c with quantitative analyses d and e. c1: inflammatory cell infiltration within the tubulointerstitium (arrows); and c2: inflammasome (arrowhead). **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO14

Effect of PK on the expression of proinflammatory and profibrotic cytokines. Representative photomicrographs of immunoblotting analysis. Protein expression is represented using the sham-operated group as 100% reference and was normalized to β-actin. **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO14
PK attenuates UUO-induced oxidative stress
UUO has been closely linked to oxidative stress, as manifested by dysregulation of a variety of oxidant and antioxidant enzymes. Immunoblotting revealed that PK balanced oxidative stress by augmenting the expression of SOD1 and SOD2, and suppressing NOX-2 and NOX-4 expression (Fig. 4). In addition, the levels of 8-OHdG, a well-known marker of the oxidative stress, were significantly increased in a time-dependent manner in the serum and urine of the UUO group compared with those of the sham group (Fig. 4), whereas PK treatment decreased their levels. Therefore, PK seems to be a scavenger of oxidative stress-related molecules in this model of UUO.

Representative photomicrographs of immunoblotting analysis of a series of oxidative stress-related proteins a, and 8-OHdG concentrations in serum b and urine c. **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO14
PK attenuates UUO-induced programmed cell death
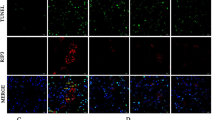
Both apoptosis (type I programmed cell death) and autophagy (type II programmed cell death) are involved in the progression of renal fibrosis in UUO. Using a TUNEL assay (Fig. 5a), we found that UUO led to a marked increase in the number of TUNEL-positive cells at both time points. This morphological change was paralleled by suppression of Bcl-2/Bax and upregulation of cleaved caspase-3 expression. PK treatment reduced the number of TUNEL-positive cells, and reversed the changes in Bcl-2/Bax and cleaved caspase-3 expression in favor of cell survival (Fig. 5b). In addition, electron microscopy showed that the induction of UUO was associated with the formation of autophagic components, such as lipid droplets, myelin bodies, autolysosomes, and autophagosomes (Fig. 6a). At a molecular level, the upregulated expression of an array of autophagy-related genes was downregulated by PK treatment (Fig. 6b).

Representative photomicrographs of the TUNEL assay a, transmission electron microscopy (a1) and immunoblotting analysis of apoptosis-related genes b. a1: apoptotic body (arrow). **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO14

Representative photomicrographs of transmission electron microscopy a and immunoblotting analysis of autophagy-related genes b. a1: scattered lipid droplets (lipophagy, stars); a2: myelin body (white arrows); a3: autolysosome formation (black arrows); and a4: autophagosome formation (ring). **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO14
PK attenuates UUO-induced activation of phosphoinositol 3-kinase/protein kinase B/forkhead box O1a signaling
To explore the effect of PK on signaling pathways that are related to renal fibrosis after UUO, we used immunoblotting to evaluate the expression of components of the phosphoinositol 3-kinase/protein kinase B/forkhead box O1a (PI3K/AKT/FoxO1a) pathway. Fig. 7 shows that UUO upregulated PI3K/AKT/FoxO1a expression in a time-dependent manner, which was suppressed by PK. This finding suggests that PK treatment attenuates renal fibrosis caused by UUO through PI3K/AKT/FoxO1a-dependent signaling.

Immunoblotting analysis of the PI3K/AKT/FoxO1a signaling pathway. **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO14
Effect of PK on H2O2-treated HK-2 cells
To confirm the renoprotective effects of PK that was observed in the rat model of UUO, we performed an in vitro study in which HK-2 cells were subjected to H2O2 treatment in the presence or absence of PK. Consistent with the results of the in vivo study, the addition of PK inhibited intracellular ROS production, and regulated SOD2 and NOX-4 expression (Figs. 8 and 9). Inhibition of the oxidative stress by PK was accompanied by suppression of TGF-β1 and MCP-1 expression, a reduction in HK-2 cell death and regulation of apoptosis- and autophagy-related genes (Figs. 8 and 9).

Effect of PK on intracellular ROS production a, apoptosis assay b, and CCK-8 cell viability of H2O2-treated HK-2 cells. ***P < 0.001 vs. control, ##P < 0.01 vs. H2O2

Representative photomicrographs of immunoblotting analysis of HK-2 cells that were treated with H2O2 and/or PK. ***P < 0.001 vs. control, ##P < 0.01 vs. H2O2
Effect of PK on the intrarenal kallikrein–kinin system
We evaluated the levels of intrarenal kallikrein–kinin system (KKS) components in rat kidneys with or without the PK treatment. UUO did not influence the concentrations of NO and cAMP, or the expression of B1R and B2R mRNA compared with those of the sham group, but these parameters were significantly increased by the addition of PK over the 14 day experimental period (Fig. 10). In contrast, there were no significant differences between groups in the concentration of bradykinin.

Levels of components of the KKS in kidney tissues in each of the experimental groups. a, b: qPCR analysis of B1R and B2R; c bradykinin; d nitric oxide; and e cAMP. **P < 0.01 vs. sham or UUO7; #P < 0.05 vs. sham or UUO14
Effect of B1R and B2R antagonists on the renoprotective effect of PK
Given that PK attenuates renal fibrosis by activating B1R and B2R, and subsequently producing NO and cAMP, we next evaluated the effect of B1R and B2R antagonists on the renoprotective effects of PK in a rat model of UUO. We found that either the B1R or B2R antagonist abolished the renoprotective effect of PK as shown by the TIF score (Fig. 11a–c), and immunoblotting for TGF-β1 and MCP-1 (Fig. 11d). Notably, the increase in NO and cAMP concentrations was abrogated in response to both the B1R and B2R antagonists (Fig. 12).

Representative photomicrographs of Masson trichrome a and hematoxylin–eosin b staining, and quantitative analysis of tubulointerstitial fibrosis, c and immunoblotting analysis of potent proinflammatory MCP-1 and profibrotic TGF-β1 expression d in the UUO + PK14 group that was administered either the B1R or B2R antagonist. d1: sham; d2: UUO14; d3: UUO + PK14; d4: UUO + PK14 + B1R; and d5: UUO + PK14 + B2R. **P < 0.01 vs. sham; #P < 0.05 vs. UUO7; $P < 0.05 vs. UUO + PK14

Levels of components of the KKS in kidney tissues of each of the experimental groups. a, b qPCR for B1R and B2R; c bradykinin; d cAMP; and e nitric oxide. **P < 0.01 vs. sham; #P < 0.05 vs. UUO + PK14
Discussion
Reducing the incidence of CKD has important clinical implications because of the consequent improvement in the quality of life of patients and decrease in the economic burden on society. The results of this study demonstrate that PK attenuates renal fibrosis in obstructed kidneys by interfering with oxidative stress and programmed cell death via activation of the intrarenal KKS. These findings suggest that PK has a protective effect against renal fibrosis caused by UUO.
Tissue kallikrein inhibits the production of inflammatory mediators, and abrogates TGF-β1 expression and collagen accumulation in myocardial infarction, choroidal neovascularization, and a variety of renal injuries [10, 15, 19, 20]. Thus, kallikrein may have both anti-inflammatory and antifibrotic potential. In the current study, we found that PK suppressed the expression of a series of proinflammatory (MCP-1, TLR-2, and OPN) and profibrotic (TGF-β1 and CTGF) cytokines, and decreased the number of ED-1-positive cells at both 7 and 14 days after UUO. These effects were accompanied by a significant decrease in MMP-2 expression and the severity of TIF. Moreover, in HK-2 cells treated with H2O2 to mimic the rat model of UUO, PK inhibited H2O2-induced upregulation of TGF-β1 and MCP-1 expression. Similar findings were reported by Bledsoe et al. [10] for gentamicin nephrotoxicity and Liu et al. [8] for STZ-induced DN.
The mechanism by which the PK treatment abolishes renal fibrosis in this model of UUO may be multifactorial, but at least two possibilities should be mentioned. UUO induces oxidative stress, which subsequently leads to fibrosis [21, 22], and inhibiting oxidative stress by either antioxidants (e.g., vitamin E) or knocking out epoxide hydrolase in mice attenuates renal fibrosis. PK has been shown to alleviate myofibrosis and interstitial collagen accumulation in diabetic cardiomyopathy [23], and to decrease the thickness of the glomerular basement membrane and renal fibrosis in DN [8] through antioxidant mechanisms. In addition, apoptosis plays a critical role in the pathogenesis of UUO-associated fibrosis [24, 25]. UUO directly induces renal epithelial cell apoptosis [26] or indirectly induces apoptosis via oxidative stress [27] or TGF-β1 expression [28, 29]. Therefore, oxidative stress, TGF-β1, and apoptotic cell death are interrelated. Here, we found that PK decreased serum and urine concentrations of 8-OHdG, inhibited H2O2-induced intracellular ROS production, and balanced oxidant and antioxidant protein expression. These changes were followed by improved HK-2 cell viability, a reduced proportion of apoptotic cells (in vivo and in vitro), and modulation of the expression of apoptosis-related genes. Taken together, these findings indicate that the abrogation of renal fibrosis by PK that was observed in this study may be related to its actions on oxidative stress and apoptosis.
Autophagy plays an essential role in the progression of UUO because it is an intracellular defense process of degradation of damaged cytoplasmic components that is triggered by either physiological or pathological stimuli, particularly hypoxia and cellular stress. In the UUO model, genetic ablation of Atg7 in distal tubules or pharmacological blockade of autophagy exacerbates the progression of TIF [30, 31], indicating a protective role of autophagy [32, 33]. However, this self-cannibalistic potential may be deleterious if enough renal cells are lost. Livingston et al. reported that persistent activation of autophagy exacerbated renal fibrosis caused by UUO in mice with proximal-tubule knockout of Atg7 [34]. Moreover, excessive autophagy is associated with overproduction of profibrotic cytokines and extracellular matrix, myofibroblast differentiation, apoptosis, and mitochondrial damage, resulting in fibrosis [35, 36]. The reasons for this discrepancy in the role of autophagy in UUO are unknown, but may be related to the different cell types that are involved, complex cellular crosstalk, or the strain of rodent used. Therefore, autophagy may also play dual roles even within a single context. By electron microscopy, we observed an abundance of autophagic derivatives in obstructed kidneys, including lipid droplets, myelin bodies, autolysosomes, and autophagosomes. This was paralleled by overexpression of the microtubule-associated proteins 1A/1B LC3B, Beclin-1, and P62, which was reversed by administering PK. Thus, we propose that the attenuation of renal fibrosis by PK that was seen in this study may be attributed in part to the modulation of autophagy.
FoxO transcription factors are key mediators of oxidative stress and are regulated by posttranslational modifications, such as phosphorylation, acetylation, and ubiquitination through the PI3K/AKT pathway [37]. FoxO1 (FKHR) is a pleiotropic transcription factor that plays a pivotal role in a variety of physiological and pathological processes, including glycolipid metabolism, transcriptional translation, oxidative stress, inflammation, and apoptosis [38, 39]. Recently, evidence has emerged that the PI3K/AKT/FoxO1 signaling pathway is involved in various types of renal injury [40,41,42]. In this study, UUO strongly increased the expression of PI3K, phosphorylated AKT, and phosphorylated FoxO1, and this increase was inhibited by administration of PK. Furthermore, upregulation of SOD1 and SOD2 was observed in the obstructed kidneys of PK-treated rats and HK-2 cells. These findings demonstrate that PK contributes to resistance to UUO-induced oxidative stress by modulating PI3K/AKT/FoxO1 signaling.
Exploring the use of exogenous PK supplementation to achieve renoprotection in this model of UUO is of paramount interest, because tissue kallikrein directly activates kinin receptors, avoiding the requirement for kinin formation [43], and activation of the KKS has benefits in renal injury [44,45,46]. Based on the hypothesis that the KKS could be stimulated with exogenous PK, we measured the components of the KKS by reverse transcription-qPCR (RT-qPCR) and ELISA, and studied the impact of B1R and B2R antagonists on the effectiveness of PK. The results of this study demonstrated that PK upregulated B1R and B2R mRNA expression, and augmented NO and cAMP levels. However, the renoprotective effects of PK were abolished by either the B1R or B2R antagonist, accompanied by a decrease in NO and cAMP levels. Neither UUO nor PK influenced bradykinin concentrations. Collectively, our findings suggest that the renoprotective effect of PK on UUO-induced renal injury is mediated via activation of B1R and B2R, as was previously reported in mice with DN and diabetic retinopathy [8, 47].
In summary, this study demonstrated that PK has antifibrotic effects against UUO-induced renal injury. Stimulation of the intrarenal KKS followed by suppression of oxidative stress and programmed cell death may be one of the mechanisms underlying these renoprotective properties of PK. Our findings provide a potential rationale for the clinical use of PK in CKD patients.
References
Romagnani P, Remuzzi G, Glassock R, Levin A, Jager KJ, Tonelli M, et al. Chronic kidney disease. Nat Rev Dis Prim. 2017;3:17088.
Martinez-Klimova E, Aparicio-Trejo OE, Tapia E, Pedraza-Chaverri J. Unilateral ureteral obstruction as a model to investigate fibrosis-attenuating treatments. Biomolecules. 2019;9:141.
Xiong C, Guan Y, Zhou X, Liu L, Zhuang MA, Zhang W, et al. Selective inhibition of class IIa histone deacetylases alleviates renal fibrosis. FASEB J. 2019;33:8249–62.
Borgeson E, Docherty NG, Murphy M, Rodgers K, Ryan A, O’Sullivan TP, et al. Lipoxin A4 and benzo-lipoxin A4 attenuate experimental renal fibrosis. FASEB J. 2011;25:2967–79.
Chao J, Guo Y, Chao L. Protective role of endogenous kallistatin in vascular injury and senescence by inhibiting oxidative stress and inflammation. Oxid Med Cell Longev. 2018;2018:4138560.
Chao J, Bledsoe G, Yin H, Chao L. The tissue kallikrein-kinin system protects against cardiovascular and renal diseases and ischemic stroke independently of blood pressure reduction. Biol Chem. 2006;387:665–75.
Montanari D, Yin H, Dobrzynski E, Agata J, Yoshida H, Chao J, et al. Kallikrein gene delivery improves serum glucose and lipid profiles and cardiac function in streptozotocin-induced diabetic rats. Diabetes. 2005;54:1573–80.
Liu W, Yang Y, Liu Y, Lu X, Guo S, Wu M, et al. Exogenous kallikrein protects against diabetic nephropathy. Kidney Int. 2016;90:1023–36.
Bodin S, Chollet C, Goncalves-Mendes N, Gardes J, Pean F, Heudes D, et al. Kallikrein protects against microalbuminuria in experimental type I diabetes. Kidney Int. 2009;76:395–403.
Bledsoe G, Crickman S, Mao J, Xia CF, Murakami H, Chao L, et al. Kallikrein/kinin protects against gentamicin-induced nephrotoxicity by inhibition of inflammation and apoptosis. Nephrol Dial Transpl. 2006;21:624–33.
Khanmoradi M, Ali Mard S, Aboutaleb N, Nobakht M, Mahmoudian M. The protective activity of noscapine on renal ischemia-reperfusion injury in male Wistar rat. Iran J Basic Med Sci. 2014;17:244–9.
Figueroa CD, Bhoola KD, MacIver AG, Mackenzie JC. An ontogenic study of renal tissue kallikrein in Okamoto spontaneously hypertensive rats: comparisons with human hypertensive nephropathy. Nephrol Dial Transpl. 1992;7:516–25.
Braun C, Kleemann T, Hilgenfeldt U, Riester U, Rohmeiss P, van der Woude FJ. Activity and functional significance of the renal kallikrein-kinin-system in polycystic kidney disease of the rat. Kidney Int. 2002;61:2149–56.
Grados OB. The laboratory in programs for enteric infection control. Bol Oficina Sanit Panam. 1975;78:318–22.
Zhu D, Zhang L, Cheng L, Ren L, Tang J, Sun D. Pancreatic kininogenase ameliorates renal fibrosis in streptozotocin induced-diabetic nephropathy rat. Kidney Blood Press Res. 2016;41:9–17.
Zhang LY, Jin J, Luo K, Piao SG, Zheng HL, Jin JZ, et al. Shen-Kang protects against tacrolimus-induced renal injury. Korean J Intern Med. 2019;34:1078–90.
Lim SW, Shin YJ, Luo K, Quan Y, Ko EJ, Chung BH, et al. Effect of Klotho on autophagy clearance in tacrolimus-induced renal injury. FASEB J. 2019;33:2694–706.
Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta. 2013;1832:1049–60.
Yao YY, Yin H, Shen B, Chao L, Chao J. Tissue kallikrein infusion prevents cardiomyocyte apoptosis, inflammation and ventricular remodeling after myocardial infarction. Regul Pept. 2007;140:12–20.
Fukuhara J, Noda K, Murata M, Namba S, Kinoshita S, Dong Z, et al. Tissue kallikrein attenuates choroidal neovascularization via cleavage of vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 2013;54:274–9.
Wu M, Li R, Hou Y, Song S, Han W, Chen N, et al. Thioredoxin-interacting protein deficiency ameliorates kidney inflammation and fibrosis in mice with unilateral ureteral obstruction. Lab Invest. 2018;98:1211–24.
Ren Y, Du C, Shi Y, Wei J, Wu H, Cui H. The Sirt1 activator, SRT1720, attenuates renal fibrosis by inhibiting CTGF and oxidative stress. Int J Mol Med. 2017;39:1317–24.
Wu M, Yang Y, Wang M, Zeng F, Li Q, Liu W, et al. Exogenous pancreatic kallikrein improves diabetic cardiomyopathy in streptozotocin-induced diabetes. Front Pharmacol. 2018;9:855.
Yoon HE, Kim SJ, Kim SJ, Chung S, Shin SJ. Tempol attenuates renal fibrosis in mice with unilateral ureteral obstruction: the role of PI3K-Akt-FoxO3a signaling. J Korean Med Sci. 2014;29:230–7.
Chen Q, Yu S, Zhang K, Zhang Z, Li C, Gao B, et al. Exogenous H2S inhibits autophagy in unilateral ureteral obstruction mouse renal tubule cells by regulating the ROS-AMPK signaling pathway. Cell Physiol Biochem. 2018;49:2200–13.
Lu J, Li X, Zhang M, Chen Z, Wang Y, Zeng C, et al. Regulation of MUTYH, a DNA repair enzyme, in renal proximal tubular epithelial cells. Oxid Med Cell Longev. 2015;2015:682861.
Nilsson L, Madsen K, Krag S, Frokiaer J, Jensen BL, Norregaard R. Disruption of cyclooxygenase type 2 exacerbates apoptosis and renal damage during obstructive nephropathy. Am J Physiol Ren Physiol. 2015;309:F1035–48.
Fujiki K, Inamura H, Sugaya T, Matsuoka M. Blockade of ALK4/5 signaling suppresses cadmium- and erastin-induced cell death in renal proximal tubular epithelial cells via distinct signaling mechanisms. Cell Death Differ. 2019;26:2371–85.
Seo E, Kim WY, Hur J, Kim H, Nam SA, Choi A, et al. The Hippo-Salvador signaling pathway regulates renal tubulointerstitial fibrosis. Sci Rep. 2016;6:31931.
Nam SA, Kim WY, Kim JW, Park SH, Kim HL, Lee MS, et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-beta and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019;10:78.
Li H, Peng X, Wang Y, Cao S, Xiong L, Fan J, et al. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy. 2016;12:1472–86.
Kawaoka K, Doi S, Nakashima A, Yamada K, Ueno T, Doi T, et al. Valproic acid attenuates renal fibrosis through the induction of autophagy. Clin Exp Nephrol. 2017;21:771–80.
Du C, Ren Y, Yao F, Duan J, Zhao H, Du Y, et al. Sphingosine kinase 1 protects renal tubular epithelial cells from renal fibrosis via induction of autophagy. Int J Biochem Cell Biol. 2017;90:17–28.
Livingston MJ, Ding HF, Huang S, Hill JA, Yin XM, Dong Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy. 2016;12:976–98.
Bernard M, Dieude M, Yang B, Hamelin K, Underwood K, Hebert MJ. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy. 2014;10:2193–207.
Noh MR, Woo CH, Park MJ, In Kim J, Park KM. Ablation of C/EBP homologous protein attenuates renal fibrosis after ureteral obstruction by reducing autophagy and microtubule disruption. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1634–41.
Maiese K, Chong ZZ, Shang YC, Hou JA. “FOXO” in sight: targeting Foxo proteins from conception to cancer. Med Res Rev. 2009;29:395–418.
Kandula V, Kosuru R, Li H, Yan D, Zhu Q, Lian Q, et al. Forkhead box transcription factor 1: role in the pathogenesis of diabetic cardiomyopathy. Cardiovasc Diabetol. 2016;15:44.
Ponugoti B, Dong G, Graves DT. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp Diabetes Res. 2012;2012:939751.
Jin J, Jin L, Lim SW, Yang CW. Klotho deficiency aggravates tacrolimus-induced renal injury via the phosphatidylinositol 3-kinase-Akt-forkhead box protein O pathway. Am J Nephrol. 2016;43:357–65.
Bai X, Geng J, Li X, Wan J, Liu J, Zhou Z, et al. Long noncoding RNA LINC01619 regulates microRNA-27a/forkhead box protein O1 and endoplasmic reticulum stress-mediated podocyte injury in diabetic nephropathy. Antioxid Redox Signal. 2018;29:355–76.
Hong YA, Bae SY, Ahn SY, Kim J, Kwon YJ, Jung WY, et al. Resveratrol ameliorates contrast induced nephropathy through the activation of SIRT1-PGC-1alpha-Foxo1 signaling in mice. Kidney Blood Press Res. 2017;42:641–53.
Hecquet C, Tan F, Marcic BM, Erdos EG. Human bradykinin B(2) receptor is activated by kallikrein and other serine proteases. Mol Pharmacol. 2000;58:828–36.
Kakoki M, McGarrah RW, Kim HS, Smithies O. Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc Natl Acad Sci USA. 2007;104:7576–81.
Aburto A, Barria A, Cardenas A, Carpio D, Figueroa CD, Burgos ME, et al. Pre-stimulation of the kallikrein system in cisplatin-induced acute renal injury: an approach to renoprotection. Toxicol Appl Pharmacol. 2014;280:216–23.
Yiu WH, Wong DW, Wu HJ, Li RX, Yam I, Chan LY, et al. Kallistatin protects against diabetic nephropathy in db/db mice by suppressing AGE-RAGE-induced oxidative stress. Kidney Int. 2016;89:386–98.
Cheng Y, Yu X, Zhang J, Chang Y, Xue M, Li X, et al. Pancreatic kallikrein protects against diabetic retinopathy in KK Cg-A(y)/J and high-fat diet/streptozotocin-induced mouse models of type 2 diabetes. Diabetologia. 2019;62:1074–86.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 81560125; no. 81760293; no. 81760132; and no. 81760668) and the Korean Health Technology Research and Development Project, Ministry for Health and Welfare (grants HI14C3417 and HI16C1641).
Author information
Authors and Affiliations
Contributions
CL designed the research; LYZ, YJJ, HLZ, YSJ, JZJ, JJ, and SGP performed the research; HYL conducted the in vitro study; SC, KL, and YQ analyzed the data and performed molecular work; XHS, YLW, JCX, and CL wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Jin, Jz., Li, Hy., Jin, J. et al. Exogenous pancreatic kininogenase protects against renal fibrosis in rat model of unilateral ureteral obstruction. Acta Pharmacol Sin 41, 1597–1608 (2020). https://doi.org/10.1038/s41401-020-0393-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0393-7
Keywords
This article is cited by
-
DKK3 promotes oxidative stress injury and fibrosis in HK-2 cells by activating NOX4 via β-catenin/TCF4 signaling
Molecular and Cellular Biochemistry (2023)
