
Abstract
Antiepileptic drug zonisamide has been shown to be curative for Parkinson’s disease (PD) through increasing HMG-CoA reductase degradation protein 1 (Hrd1) level and mitigating endoplasmic reticulum (ER) stress. Hrd1 is an ER-transmembrane E3 ubiquitin ligase, which is involved in cardiac dysfunction and cardiac hypertrophy in a mouse model of pressure overload. In this study, we investigated whether zonisamide alleviated cardiac hypertrophy in rats by increasing Hrd1 expression and inhibiting ER stress. The beneficial effects of zonisamide were assessed in two experimental models of cardiac hypertrophy: in rats subjected to abdominal aorta constriction (AAC) and treated with zonisamide (14, 28, 56 mg · kg−1 · d−1, i.g.) for 6 weeks as well as in neonatal rat cardiomyocytes (NRCMs) co-treated with Ang II (10 μM) and zonisamide (0.3 μM). Echocardiography analysis revealed that zonsiamide treatment significantly improved cardiac function in AAC rats. We found that zonsiamide treatment significantly attenuated cardiac hypertrophy and fibrosis, and suppressed apoptosis and ER stress in the hearts of AAC rats and in Ang II-treated NRCMs. Importantly, zonisamide markedly increased the expression of Hrd1 in the hearts of AAC rats and in Ang II-treated NRCMs. Furthermore, we demonstrated that zonisamide accelerated ER-associated protein degradation (ERAD) in Ang II-treated NRCMs; knockdown of Hrd1 abrogated the inhibitory effects of zonisamide on ER stress and cardiac hypertrophy. Taken together, our results demonstrate that zonisamide is effective in preserving heart structure and function in the experimental models of pathological cardiac hypertrophy. Zonisamide increases Hrd1 expression, thus preventing cardiac hypertrophy and improving the cardiac function of AAC rats.
Similar content being viewed by others
Introduction
In mammals, physiological hypertrophy occurs in the heart under demanding conditions during normal daily living, such as repetitive exercise, while pathological hypertrophy is generally accepted to contribute to adverse ventricular events [1]. Pathological cardiac hypertrophy is typically characterized by systolic/diastolic dysfunction and interstitial fibrosis, resulting in irreversible cardiac remodeling and heart failure due to sustained stimulation of the heart associated with disease conditions, such as hypertension, valvular diseases, myocardial infarction, or excessive neurohormonal activation [2, 3]. Proteostasis is a cellular network that ensures proteome integrity [4]. The endoplasmic reticulum (ER) is an important multifunctional intracellular organelle that plays an important role in the synthesis, modification, folding, and translocation of proteins through reactions catalyzed by ER-resident factors at steady state in eukaryotic cells. These proteins include calcium-handling proteins, transmembrane receptors, growth factors, and hormones. When misfolded or unfolded proteins accumulate in the ER, an intracellular signaling network termed the unfolded protein response (UPR) is stimulated. The UPR involves several signaling pathways that ameliorate the accumulation of unfolded proteins by inhibiting protein translation, accumulating ER-resident chaperones and promoting the degradation of unfolded or misfolded proteins by ER-associated protein degradation (ERAD). During ERAD, unfolded or misfolded ER-transmembrane and luminal proteins are transported to the cytosol and then ubiquitylated by ER-transmembrane E3 ubiquitin ligases for degradation by the 26S proteasome [5,6,7]. Challenges related to ER protein-folding capacity, which impair cardiac development and function, have been documented in many diseases. If the adaptive response fails, cardiomyocytes undergo apoptosis [8,9,10,11,12].
One of the genes induced by activating transcription factor 6 (ATF6) in the heart is the ER-transmembrane E3 ubiquitin ligase Hrd1 [13]. Hrd1 was discovered in yeast and named for its ability to ubiquitylate the ER-transmembrane protein hydroxymethyl glutaryl-coenzyme A reductase [14]. Hrd1 overexpression improved cardiac function and attenuated cardiac hypertrophy in a mouse model of pressure overload by accelerating the degradation of aberrant proteins in the ER [13]. Zonisamide (1,2-benzisoxazole-3-methanesulfonamide), an antiepileptic agent, has beneficial effects on Parkinson’s disease at a dose that is 1/10 lower than that used to treat epilepsy in Japan [15]. Zonisamide prevented ER stress-induced cell death and activation of Caspase3 (an apoptosis-related cysteine peptidase) by increasing the Hrd1 level in SH-SY5Y cells [16], and our previous study also found that zonisamide attenuates diabetic cardiomyopathy in a mouse model of type 2 diabetes mellitus [17]. However, the effect of zonisamide on cardiac hypertrophy in rats remains unclear.
Therefore, we hypothesized that zonisamide attenuates cardiac hypertrophy by inhibiting ER stress and increasing Hrd1 expression in the hearts of rats subjected to pressure overload.
Materials and methods
Reagents and antibodies
Zonisamide was purchased from Target Molecule Corp. (Boston, MA, USA). Angiotensin II was purchased from Sigma Chemical Co. (St. Louis, MO, USA). The primary antibodies used in this study are listed in Table 1. Horseradish peroxidase (HRP)-conjugated goat anti-rat IgG secondary antibodies, horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibodies, Alexa 488-conjugated goat anti-rabbit IgG and Alexa 647-conjugated goat anti-mouse IgG were purchased from Abcam (Cambridge, MA, USA). The enhanced chemiluminescence (ECL) kit was obtained from Pierce (Rockford, IL, USA).
Animals
Neonatal (1–3 days old) and adult male Sprague-Dawley rats (SD rats) (100–120 g) were purchased from the Medical Laboratory Animal Center of Guangzhou University of Chinese Medicine (Guangzhou, China). The animal use protocol was approved by the Committee on the Ethics of Animal Experiments of Guangzhou Medical University under approval reference number SYXK 2016-0168.
Rats were housed under controlled temperature (25 ± 2 °C) with a 12/12 h light/dark cycle and had free access to food and water. The rats were randomly divided into five groups (n = 7 per group): sham-operated group (sham), abdominal aortic constriction (AAC) group, AAC+zonisamide group (14 mg · kg−1 · d−1, A+Z 14), AAC+zonisamide group (28 mg · kg−1 · d−1, A+Z 28), and AAC+zonisamide group (56 mg · kg−1 · d−1, A+Z 56). Zonisamide was dissolved in 1% dimethyl sulfoxide (DMSO) (Sigma-Aldrich, MO, USA). Rats in the sham and AAC groups were administered 1% DMSO intragastrically 2 days after AAC surgery, and the other three groups were treated with different doses of zonisamide.
Induction of AAC
Rats (100–120 g) were anesthetized by intraperitoneal injection of sodium pentobarbital (50 mg · kg−1). The rats were subjected to AAC as described previously [18, 19]. In the sham group, the same surgery was performed without constriction of the aorta. All animals were housed in facilities with a constant temperature and had free access to water as well as standard rodent chow.
Neonatal rat cardiomyocytes (NRCMs)
NRCMs and cardiac fibroblasts were isolated as previously described [20]. NRCMs and cardiac fibroblasts were randomized into five groups: (i) control (Con), (ii) angiotensin II 10 μM (Ang II), (iii) angiotensin II 10 μM + zonisamide 0.1 μM (Ang II+Z 0.1), (iv) angiotensin II 10 μM + zonisamide 0.3 μM (Ang II+Z 0.3), and (v) angiotensin II 10 μM + zonisamide 1 μM (Ang II+Z 1). After 24 h of treatment, cells were harvested for analysis. Ang II and zonisamide were dissolved in ddH2O and 0.1% DMSO, respectively.
Echocardiography
Transthoracic echocardiography was performed noninvasively with Visual Sonics (Vevo 2100; Visual Sonics Inc., ON, Canada) equipped with a 25 MHz transducer as previously described [21, 22]. To minimize the influence of differences in heart rate (HR) among the rats on left ventricular (LV) function, HR was maintained at 350–400 beats per minute by adequately adjusting the flow of inhalational isoflurane (kept under 2% at an inflow rate of 0.5–1.5 mL · min−1) to anaesthetize the rats. LV function was evaluated noninvasively via parasternal long-axis and short-axis views at the platform of the papillary muscles with M-mode scanning reflecting LV systolic and diastolic function, LV internal diameters (LVID) at end-diastole and end-systole (LVID,D and LVID,S, respectively), and wall thicknesses. LV ejection fraction (EF) and fractional shorting (FS), early and late transmitral peak diastolic flow velocity (E; A waves), and the ratio of E wave velocity to A wave velocity were measured.
Real-time RT-PCR
Total RNA was extracted using TRIzol reagent (Life Technologies, Grand Island, NY, USA) according to the product specifications. cDNA was synthesized from 1 μg of RNA with a Prime Script RT reagent kit with gDNA Eraser (Takara Bio Inc., Kusatsu, Shiga, Japan). Real-time PCR amplification reactions were performed in triplicate using a SYBR Premix Ex Taq kit with ROX (Takara Bio Inc., Kusatsu, Shiga, Japan) and the ABI Step One Plus Real-Time PCR system. Gene expression was obtained via the 2−ΔΔCt calculation and quantified by normalization to the GAPDH mRNA level. The data are presented as the fold change in the expression of the gene of interest relative to that of the control groups. The primer sequences (Life Technologies) are shown in Table 2.
Histological examination and immunohistochemical staining
Hearts were quickly removed and perfused with 10% KCl to arrest the hearts in diastole. After being photographed, the hearts (left ventricle) (n = 3) were cut into three transverse blocks parallel to the atrioventricular groove. Transverse sections were fixed with 10% formalin, embedded in paraffin, cut into 5-μm sections, and then stained with hematoxylin and eosin (H&E) or Masson’s trichrome for histological examination. For immunohistochemical staining, paraffin-embedded sections were incubated with a primary antibody against Hrd1 (1:200 dilution) and then with a biotinylated secondary antibody. The sections were then visualized with diamino benzidine. The sections were examined by light microscopy (Leica, Germany). The percentages of fibrosis and positive expression of Hrd1 were calculated using ImageJ software (NIH, Bethesda, USA).
Cell surface area measurement
The cell surface area was measured using phalloidin-tetramethylrhodamine isothiocyanate to stain F-actin as described previously [23]. Briefly, after Ang II and zonisamide exposure for 24 h, NRCMs were fixed with 4% paraformaldehyde for 10 min at room temperature. Then, the cells were incubated with 1% Triton X-100 in PBS for 20 min. After being blocked with 10% normal goat serum for 1 h, the slides were incubated with 50 μg/mL phalloidin-TRITC (Sigma-Aldrich, MO, USA) to stain F-actin for 40 min at room temperature. Cell nuclei were counterstained with DAPI (Sigma-Aldrich, MO, USA). After the cells were washed with PBS three times, images were captured by laser scanning confocal microscopy (A1 si, Nikon, Japan), and the cell surface area was analyzed with ImageJ software (NIH, Bethesda, USA).
Immunofluorescence
For immunofluorescence, NRCMs were fixed in 4% paraformaldehyde at room temperature and permeabilized with 0.1% Triton X-100. The cells were blocked with 1% BSA for 1 h at 37 °C. Hrd1 and KDEL (KDEL (Lys-Asp-Glu-Leu) ER protein retention receptor) were applied at 4 °C overnight, and Alexa 488-conjugated goat anti-rabbit IgG (1:200 dilution) and Alexa 647-conjugated goat anti-mouse IgG (1:150 dilution) were added to the cells and incubated for 1 h at 37 °C. The cells were then stained with DAPI for 1 h and washed with PBS. Images were obtained via laser scanning confocal microscopy (A1 si, Nikon, Japan) and analyzed by ImageJ software (NIH, Bethesda, USA).
TUNEL staining
A terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay was performed using commercial apoptosis detection kits (No. 11684817910; Roche, IN, USA). Briefly, NRCMs were fixed with 4% paraformaldehyde for 1 h, permeabilized with 0.1% Triton X-100 and stained. The samples were treated with 20 μg/mL proteinase K for 15 min and incubated with 3% H2O2 for 15 min after washing with PBS. After adding equilibration buffer, the samples were stained with TUNEL reaction mixture for 1 h in a humidified chamber in the dark at 37 °C. DAPI staining was used to label nuclei. Apoptotic cells were analyzed using a laser scanning confocal microscope (A1si, Nikon, Japan). TUNEL-positive cells were quantitatively assessed using ImageJ software (NIH, Bethesda, USA).
Western blotting
Frozen heart tissues (n = 7 per group) and NRCM lysates were prepared by homogenization in lysis buffer as described previously [23]. Proteins were separated on SDS–polyacrylamide gels (8%–12%) and transferred to polyvinylidene difluoride membranes. The membranes were then blocked with 5% non-fat milk for 1 h and incubated with primary antibodies overnight (Table 1). After washing, the membranes were incubated with appropriate secondary antibodies for 1 h at room temperature. The protein–antibody complex was detected by ECL.
Statistical analysis
All data were analyzed using GraphPad Prism 7.0 statistical software (GraphPad Software, Inc., La Jolla, CA). All data are expressed as the mean ± SEM. One-way ANOVA was performed to determine the significance of differences between groups. P < 0.05 was considered statistically significant.
Results
Zonisamide alleviated cardiac hypertrophy and improved cardiac function in rats subjected to AAC
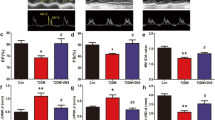
To investigate the effects of zonisamide on cardiac hypertrophy, we established a cardiac hypertrophy model by performing AAC in rats. Fig. 1a shows that vehicle or zonisamide was administered to the rats by intragastric administration for 6 weeks after sham surgery or AAC. Fig. 1b, c show representative images of the M-mode ECHO recordings of the LV and the mitral flow Doppler profile with E wave and A wave. The ventricular wall thickness (LVAW,S; LVAW,D; LVPW,S; LVPW,D) was increased, and the LVID were decreased after AAC, and these changes were significantly reversed by different doses of zonisamide (Fig. 1d–f). LV ejection fraction (EF) and fractional shortening (FS) were increased in the AAC group compared with the sham group, and zonisamide decreased EF and FS (Fig. 1g). In addition, the E/A ratio was decreased in the AAC group, and this change was reversed by zonisamide (Fig. 1h). There was no significant difference in HR in all groups (Fig. 1i). These results suggest that zonisamide treatment ameliorated cardiac hypertrophy and improved cardiac function in rats subjected to AAC.

a Experimental protocol for AAC and termination of the experiment. b, c Representative M-mode images (left panel) and the transmitral filling pattern (right panel) in the sham (vehicle), AAC (vehicle), A+Z 14 (AAC+Z 14 mg · kg−1 · d−1), A+Z 28 (AAC+Z 28 mg · kg−1 · d−1) and A+Z 56 (AAC+Z 56 mg · kg−1 · d−1) groups. d–i Quantitative analysis of LVAW,S; LVAW,D; LVPW,S; LVPW,D; LVID,S; LVID,D; EF%; FS%; MV E/A; and HR in five groups in the 6th week after treatment with zonisamide or vehicle (n = 7). Data were analyzed by one-way ANOVA as the mean ± SEM. **P < 0.01 vs. sham; #P < 0.05, ##P < 0.01 vs. AAC. AAC(A) abdominal aorta constriction, Z Zonisamide, A+Z 14 AAC+Z 14 mg · kg−1 · d−1, A+Z 28 AAC+Z 28 mg · kg−1 · d−1, A+Z 56 AAC+Z 56 mg · kg−1 · d−1, LVAW,S the left ventricular end-systolic anterior wall thickness, LVAW,D the left ventricular end-diastolic anterior wall thickness, LVPW,S the left ventricular end-systolic posterior wall thickness, LVPW,D the left ventricular end-diastolic posterior wall thickness, LVID,S the left ventricular end-systolic internal dimension, LVID,D the left ventricular end-diastolic internal dimension, EF ejection fraction, FS fractional shortening, MV E/A the ratio of E-wave velocity to A-wave velocity, HR heart rate.
Zonisamide reduced myocardial fibrosis in rats subjected to AAC
Myocardial fibrosis often accompanies pathological cardiac hypertrophy, leading to loss of function of the myocardium due to abnormal diastolic stiffness [24]. Heart size was increased in the AAC group, and this change was reversed by different doses of zonisamide (Fig. 2a). The increased heart weight-to-body weight ratio (HW/BW) in the AAC group was attenuated by zonisamide (Fig. 2b). In addition, H&E staining of cross sections revealed a decreased cardiomyocyte surface area in the zonisamide group compared with the AAC group (Fig. 2c, d). Masson trichrome staining showed that zonisamide reduced the accompanying interstitial fibrosis in the hearts from the AAC group (Fig. 2e, f). These data indicate that zonisamide administration dramatically attenuated the increase in cardiomyocyte surface area and cardiac remodeling in rats with pressure overload.

Rats in the sham (vehicle), AAC (vehicle), A+Z 14, A+Z 28, and A+Z 56 groups were treated with vehicle or zonisamide for 6 weeks. a Original heart images from different groups. b Quantitative analysis of the HW/BW (n = 7). c Representative H&E staining from hearts in different groups (n = 3) (scale bar = 50 μm). d Quantitative analysis of cardiomyocyte cell surface area (n = 3). e Representative Masson trichrome staining from hearts in different groups (n = 3) (scale bar = 50 μm). f Statistical results for fibrotic area in different groups (n = 3). Data were analyzed by one-way ANOVA as the mean ± SEM. **P < 0.01 vs. sham; #P < 0.05, ##P < 0.01 vs. AAC.
Zonisamide alleviated cardiac hypertrophy and fibrosis in vitro
Cardiomyocyte hypertrophy and fibrosis were established in vitro by exposing NRCMs and cardiac fibroblasts to Ang II (10 μM) for 24 h, respectively. Compared to the Ang II treatment, zonisamide (0.1, 0.3, 1 μM for 24 h) decreased the expression of atrial natriuretic factor (ANF) and cardiomyosin heavy chain β (β-MHC) but increased the expression of cardiac myosin heavy chain α (α-MHC) in NRCMs. Zonisamide also decreased cardiac expression of the fibrosis-related gene Collagen 1A1 (Col1A1) in cardiac fibroblasts (Fig. 3a). The cardiomyocyte surface area was significantly increased in the Ang II group, and this change was reversed by zonisamide treatment (0.3 μM) (Fig. 3b, c). According to the above results, 0.3 μM zonisamide was chosen for further studies.

NRCMs and cardiac fibroblasts were incubated with zonisamide (0.1, 0.3, 1 μM dissolved in 0.1% DMSO) and Ang II for 24 h. a mRNA levels of ANF, β-MHC, and α-MHC in NRCMs and Collagen 1A1 in cardiac fibroblasts were assessed by RT-PCR (n = 5). b NRCMs were stained with phalloidin (phalloidin-tetramethylrhodamine isothiocyanate) and DAPI to evaluate cell surface area. c Quantitative analysis of cell surface area with ImageJ software (n = 4) (scale = 50 µm). Data were analyzed by one-way ANOVA. Values are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. Ang II.
Zonisamide attenuated apoptosis in rats subjected to AAC and hypertrophic primary cardiomyocytes
Once apoptosis is initiated, the transcription factors C/EBP homologous protein (CHOP) and B-cell CLL/lymphoma 2 (Bcl-2) family proteins are recruited, eliciting organellar cross-talk with the mitochondria [25]. We investigated the effects of zonisamide on apoptotic cell death in the heart. TUNEL analysis showed that apoptosis of NRCMs was initiated in Ang II group, which was reversed by zonisamide treatment (Fig. 4a, b). Among the three concentrations of zonisamide used in the in vivo assay, 14 mg · kg−1 · d−1 was the most effective and was chosen for further studies. CHOP, a key mediator of ER stress, was upregulated in the AAC group and reversed by zonisamide treatment. Bcl-2-associated X protein (Bax) and Caspase12 (an inducer of apoptosis and a marker of maladaptive ER stress) were significantly activated, but Bcl-2 was decreased in the AAC group, and this change was reversed by zonisamide treatment (Fig. 4c, d). These findings obtained in NRCMs were in agreement with those obtained in rats (Fig. 4e, f).

Rats in the sham (vehicle), AAC (vehicle), and A+Z 14 groups were treated with vehicle or zonisamide for 6 weeks. NRCMs were incubated with zonisamide and Ang II for 24 h, cell apoptosis was measured by TUNEL staining (green, TUNEL staining; blue, DAPI staining), and the protein levels of CHOP, Bax, Bcl-2, and Caspase12 were measured by Western blotting. a, b NRCMs were analyzed for apoptosis by TUNEL analysis and then quantified to determine the percentage of TUNEL-positive nuclei (n = 4) (scale = 50 µm). c, d Western blotting results and quantitative analysis of CHOP, Bax, Bcl-2, and Caspase12 expression in rats hearts (n = 7). e, f Western blot results and quantitative analysis of CHOP, Bax, Bcl-2, and Caspase12 expression in NRCMs (n = 7). Data were analyzed by one-way ANOVA. Values are presented as the mean ± SEM. For b, f, *P < 0.05, **P < 0.01 vs. Con; #P < 0.05, ##P < 0.01 vs. Ang II. For d, *P < 0.05, **P < 0.01 vs. sham; #P < 0.05, ##P < 0.01 vs. AAC.
Zonisamide alleviated ER stress in rats subjected to AAC and hypertrophic primary cardiomyocytes
The protein levels of phosphorylation of eukaryotic translation initiation factor 2-alpha kinase 3 (p-PERK), activating transcription factor 4 (ATF4), ATF6, X-box binding protein 1 (XBP1), heat shock 70 kDa protein 5 (GRP78) and heat shock protein 90 kDa beta (GRP94) were dramatically increased in the AAC group, and these changes were abrogated by zonisamide treatment (Fig. 5a, b). In addition, consistent with the findings in rats, the protein levels of p-PERK, ATF4, ATF6, and XBP1, along with the levels of GRP78 and GRP94, were increased in the NRCMs in Ang II group and were reduced by zonisamide treatment (Fig. 5c, d). These data suggest that zonisamide alleviated ER stress in hearts subjected to AAC and hypertrophic primary cardiomyocytes.

Rats in the sham (vehicle), AAC (vehicle), and A+Z 14 groups were treated with vehicle or zonisamide for 6 weeks. NRCMs were incubated with zonisamide and Ang II for 24 h. a, b Protein levels of phosphorylation of PERK (p-PERK), PERK, ATF4, ATF6, XBP1, GRP78, and GRP94 in rats hearts were measured by Western blotting and densitometric quantification after normalization to the GAPDH level (n = 7). c, d Protein levels of phosphorylation of PERK (p-PERK), PERK, ATF4, ATF6, XBP1, GRP78, and GRP94 in NRCMs were measured by Western blotting and densitometric quantification after normalization to the GAPDH level (n = 7). Data were analyzed by one-way ANOVA. Values are presented as the mean ± SEM. For b, *P < 0.05, **P < 0.01 vs. sham; #P < 0.05 vs. AAC. For d, *P < 0.05 vs. Con; #P < 0.05, ##P < 0.01 vs. Ang II.
Zonisamide upregulated Hrd1 expression and accelerated ERAD in the hearts
Immunohistochemical analysis (Fig. 6a, b) and Western blotting (Fig. 6c–f) showed that zonisamide increased Hrd1 expression compared to that in the hearts from the AAC group and the NRCMs in the Ang II group. To further examine whether there was increased accumulation of Hrd1 in the ER of NRCMs, we examined the colocalization of Hrd1 (green) with KDEL (red, an ER stress marker). Double-labeling immunofluorescence analyses for Hrd1 and KDEL were performed in NRCMs. Zonisamide treatment increased Hrd1 but inhibited KDEL expression (Fig. 6g, h). T-cell antigen receptor alpha-chain (TCR-α), an ER transmembrane protein, misfolds and is degraded by the ERAD process [26]. ERAD was assessed by examining the rate of TCR-α degradation after inhibiting protein synthesis via cycloheximide (CHX) treatment. Since the role of Hrd1 in ERAD is well known and a previous study showed that knockdown of Hrd1 dramatically decreased ERAD in ER stress-stimulated NRCMs [13], we then examined the effects of zonisamide on ERAD in NRCMs by using TCR-α as a model misfolded ER protein. The TCR-α level was decreased from 1 to 0 h, and the rate of ERAD (ratio of TCR-α at 0 to 1 h) was increased after CHX treatment for 1 h in the Ang II group compared with the Con group, while zonisamide accelerated ERAD (Fig. 6i–k). These findings demonstrate that zonisamide increased Hrd1 and the rate of degradation of misfolded proteins.

a, b The expression of Hrd1 was measured by immunohistochemistry, and the relative protein expression of Hrd1 was analyzed with ImageJ software (n = 3) (scale bar = 50 μm). c, d The protein level of Hrd1 in rats hearts was measured by Western blotting and densitometric quantification after normalization to the GAPDH level (n = 7). e, f The protein level of Hrd1 in NRCMs was measured by Western blotting and densitometric quantification after normalization to the GAPDH level (n = 7). g, h Hrd1 and KDEL in NRCMs were assessed by double immunofluorescence staining and quantitative analysis with ImageJ software (n = 4) (scale bar = 20 μm). i, j NRCMs were incubated with zonisamide and Ang II for 24 h. The cells were then treated with CHX for 1 h. Western blotting of TCR-α and densitometric quantification were performed (n = 6). k ERAD is displayed here as the ratio of TCR-α at 1 to 0 h after CHX treatment (n = 6). Data were analyzed by one-way ANOVA. Values are presented as the mean ± SEM. For b, d, *P < 0.05, **P < 0.01 vs. sham; ##P < 0.01 vs. AAC. For f, h, j, k, *P < 0.05 vs. Con; #P < 0.05, ##P < 0.01 vs. Ang II.
Zonisamide ameliorated ER stress and cardiac hypertrophy by increasing Hrd1 levels in hypertrophic primary cardiomyocytes
To determine whether zonisamide improves Ang II-induced cardiomyocyte hypertrophy by upregulating Hrd1, NRCMs were transiently transfected with siRNA (siCon or siHrd1). As a result, there was no difference in Hrd1 expression between the Con group and the siCon group (Fig. 7a, b). The protein level of Hrd1 was reduced significantly in the siHrd1 group (Fig. 7c, d). Zonisamide upregulated Hrd1 expression, which was inhibited by siHrd1 in NRCMs. In addition, zonisamide decreased GRP78 (an ER stress marker) in NRCMs incubated with Ang II, and this change was abrogated by Hrd1 knockdown (Fig. 7e, f). Moreover, Hrd1 knockdown led to increased KDEL (Fig. 7g, h) and cardiomyocyte surface area (Fig. 7i, j).

NRCMs were treated with siCon or siHrd1 for 24 h and then treated with zonisamide and Ang II for 24 h. a–d Protein level of Hrd1 were detected by Western blotting after siCon and siRNA interference (n = 4). e, f Protein levels of Hrd1 and GRP78 were detected by Western blotting after Hrd1 knockdown (n = 4). g, h Hrd1 and KDEL levels in NRCMs were assessed by double immunofluorescence staining and quantitative analysis with ImageJ software (n = 4) (scale bar = 20 μm). i, j NRCMs were stained with phalloidin and DAPI to measure cell surface area, and quantitative analysis of cell surface area was performed with ImageJ software (n = 4) (scale bar = 50 µm). Data were analyzed by one-way ANOVA. Values are presented as the mean ± SEM. *P < 0.05, **P < 0.01 vs. siCon; #P < 0.05, ##P < 0.01 vs. Ang II+siCon; $P < 0.05, $$P < 0.01 vs. Ang II+Z 0.3+siCon.
Discussion
Zonisamide which was originally synthesized in Japan, has been used since 1989 and was approved by the Food and Drug Administration (FDA) in 2000, was developed as an adjunctive treatment for partial seizures in the United States [27,28,29]. Zonisamide has neuroprotective effects on PD and was approved for PD adjunctive treatment in Japan. The underlying molecular mechanisms of zonisamide are related to the upregulation of Hrd1 and the inhibition of ER stress and cell death [16, 30, 31]. The present study aimed to determine whether zonisamide improves cardiac hypertrophy by inhibiting ER stress and increasing Hrd1 level.
We established a model of cardiac hypertrophy by using Ang II-stimulated cardiomyocytes and rats subjected to AAC. We found that treatment with zonisamide relieved cardiac hypertrophy and improved cardiac function in the AAC group. This anti-hypertrophic effect was indicated by reductions in LV wall thickness, EF, and FS and increases in LVID and E wave to A wave ratios (E/A) in the hearts of pressure-overloaded rats. These well-documented beneficial cardiovascular effects were accompanied by reductions in the cell surface area, interstitial collagen, and heart weight-to-body weight ratio in the zonisamide-treated groups. Pathological cardiac hypertrophy of the adult heart is correlated with downregulation of α-MHC and upregulation of β-MHC [32]. The process of cardiac hypertrophy is usually characterized by an increase in the re-expression of embryonic genes such as β-MHC and ANF [33]. After treatment with zonisamide, the mRNA expression of ANF, β-MHC, and Collagen 1A1 and the cell surface area were decreased, but the mRNA expression of α-MHC was increased in the in vitro assay.
It has been shown previously that cardiomyocyte apoptosis plays a crucial role during the transition of hypertrophy to heart failure in Ang II-treated cardiomyocytes and pressure-overloaded rats [34,35,36]. Proteins in the Bcl-2 family, which have been suggested to be involved in apoptosis and determine the commitment of cells to apoptosis, can be divided into two categories based on their biological effects [37, 38]. The anti-apoptotic proteins include Bcl-2, Bcl-XL, and Bcl-W. The proapoptotic proteins include Bax, Bak, and Bcl-XS. Previous studies have shown that ER stress serves as one of the factors that triggers cell apoptosis; in addition, CHOP is often associated with maladaptive ER stress and cell death and directly regulates death effectors such as Bcl-2 [39]. ER stress-mediated activation of Caspase12 is a marker of maladaptive ER stress and an inducer of apoptosis [13]. The present study demonstrated that the expression of the proapoptotic protein Bax was decreased, and that of the anti-apoptotic protein Bcl-2 was increased in zonisamide-treated rats hearts and NRCMs. Zonisamide alleviated ER stress-related cell apoptosis, as indicated by decreased protein levels of CHOP and Caspase12 in rats subjected to AAC and Ang II-treated NRCMs. This was also confirmed by TUNEL assay results in NRCMs, which showed a low ratio of positive cells in zonisamide-treated NRCMs compared with the Ang II group.
The UPR is mediated by three major ER-spanning transmembrane proteins, inositol requiring enzyme 1 (IRE1), PERK, and ATF6, which activate IRE1-mediated splicing of XBP1, the PERK–eIF2α (eukaryotic translation initiation factor 2 subunit 1 alpha)–ATF4–CHOP signaling pathway, and cleavage of pro-ATF6 to ATF6, respectively [40, 41]. Each sensor’s luminal domains bind to a chaperone, GRP78, which locks them in an inactive state under “stress-free” conditions while rendering them active by permitting their oligomerization and release to correct ER homeostasis. If the ER stress is prolonged or unresolved, insufficient clearance results in apoptosis [42, 43]. Pressure overload induces upregulation of the genes for ER chaperones and apoptosis of cardiomyocytes during the development of pathological cardiac hypertrophy [44]. ER chaperones are markedly induced in pressure-overloaded mice with hypertrophic hearts induced by TAC [44]. In the present study, we evaluated ER stress changes, and our data demonstrated the upregulation of GRP78 in the AAC group and Ang II group. Zonisamide treatment decreased the protein levels of p-PERK, ATF4, ATF6, XBP1, GRP78, and GRP94.
It has been reported that adequate level of Hrd1, an ER-transmembrane E3 ubiquitin ligase that has been studied in pressure overload-induced cardiac hypertrophy, contribute to ER protein quality control via ERAD of misfolded proteins that induce ER stress [13]. Our findings showed that increased Hrd1 expression was observed in both zonisamide-treated rats and NRCMs, while zonisamide increased Hrd1 but decreased KDEL expression in NRCMs. KDEL represents the LysAsp-Glu-Leu (KDEL) ER retrieval sequence and suggests an accumulation of ER chaperones due to misfolded proteins [45]. KDEL is an important marker of ER stress that was found to be increased in NRCMs treated with Ang II and the hearts of mice subjected to TAC and recognizes both GRP78 and GRP94 [44]. Intracellularly accumulated Hrd1 colocalizes with KDEL, further suggesting intracellular accumulation of Hrd1 in the ER [13]. Here, we also observed that KDEL colocalized with Hrd1. Quantitative analysis revealed that compared with no treatment, Ang II increased the number of KDEL-positive cells, and compared with Ang II treatment, zonisamide decreased the number of KDEL-positive cells. The prevalence of Hrd1-positive cells in the zonisamide group was significantly higher than that in the Ang II group. ERAD was measured using a version of the model substrate, T-cell antigen receptor α-chain (TCR-α), essentially as described previously [26]. A high level of Hrd1 was accompanied by accelerated degradation of misfolded proteins, as evidenced by the increased rate of ERAD. All of these findings suggest that the cardiac effects of zonisamide may arise from inhibition of ER stress and upregulation of Hrd1.
We also confirmed that a decrease in ER stress was correlated with the upregulation of Hrd1 during Ang II-induced cardiac hypertrophy in NRCMs. To verify whether zonisamide inhibits ER stress by triggering Hrd1 expression, we performed small interfering RNA-mediated Hrd1 knockdown in NRCMs. After Hrd1 was knocked down, an augmented cell surface area and exacerbated cardiac hypertrophy were observed. In addition, ER stress (increased GRP78 and KDEL levels) dramatically increased. We also found that zonisamide decreased the number of GRP78 and KDEL-positive cells, and this effect was inhibited when Hrd1 was knocked down. These results suggest that the antihypertrophic and anti-ER stress effects of zonisamide were abrogated in the absence of Hrd1. These findings suggest that zonisamide may emerge as an effective therapeutic strategy to prevent ER stress and ER-related cell apoptosis in the hypertrophic cardiomyocytes by increasing Hrd1 level.
In summary, the present study provides a novel evidence that zonisamide improved cardiac hypertrophy by inhibiting ER stress and increasing Hrd1. Therefore, zonisamide may become a new agent to prevent ER stress associated with cardiac hypertrophy.
References
Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–9.
van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45.
Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–65.
Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–9.
Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3:246–55.
Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin–ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–73.
Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harbor Perspect Biol 2013;5:a013185. https://doi.org/10.1101/cshperspect.a013185.
Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–42.
Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–82.
Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol. 2013;55:85–91.
Millott R, Dudek E, Michalak M. The endoplasmic reticulum in cardiovascular health and disease. Can J Physiol Pharmacol. 2012;90:1209–17.
Glembotski CC. Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J Mol Cell Cardiol. 2014;71:11–5.
Doroudgar S, Volkers M, Thuerauf DJ, Khan M, Mohsin S, Respress JL, et al. Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ Res. 2015;117:536–46.
Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–44.
Murata M, Horiuchi E, Kanazawa I. Zonisamide has beneficial effects on Parkinson’s disease patients. Neurosci Res. 2001;41:397–9.
Omura T, Asari M, Yamamoto J, Kamiyama N, Oka K, Hoshina C, et al. HRD1 levels increased by zonisamide prevented cell death and caspase-3 activation caused by endoplasmic reticulum stress in SH-SY5Y cells. J Mol Neurosci. 2012;46:527–35.
Tian JH, Wu Q, He YX, Shen QY, Rekep M, Zhang GP, et al. Zonisamide, an antiepileptic drug, alleviates diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress. Acta Pharmacol Sin 2020. https://doi.org/10.1038/s41401-020-0461-z. [online ahead of print].
Barton CH, Ni Z, Vaziri ND. Enhanced nitric oxide inactivation in aortic coarctation-induced hypertension. Kidney Int. 2001;60:1083–7.
Kobayashi S, Yano M, Kohno M, Obayashi M, Hisamatsu Y, Ryoke T, et al. Influence of aortic impedance on the development of pressure-overload left ventricular hypertrophy in rats. Circulation. 1996;94:3362–8.
Hou N, Cai B, Ou CW, Zhang ZH, Liu XW, Yuan M, et al. Puerarin-7-O-glucuronide, a water-soluble puerarin metabolite, prevents angiotensin II-induced cardiomyocyte hypertrophy by reducing oxidative stress. Naunyn Schmiedebergs Arch Pharmacol. 2017;390:535–45.
Yang K, Zhang TP, Tian C, Jia LX, Du J, Li HH. Carboxyl terminus of heat shock protein 70-interacting protein inhibits angiotensin II-induced cardiac remodeling. Am J Hypertens. 2012;25:994–1001.
Samuel SM, Thirunavukkarasu M, Penumathsa SV, Koneru S, Zhan L, Maulik G, et al. Thioredoxin-1 gene therapy enhances angiogenic signaling and reduces ventricular remodeling in infarcted myocardium of diabetic rats. Circulation. 2010;121:1244–55.
Schumacher-Bass SM, Vesely ED, Zhang L, Ryland KE, McEwen DP, Chan PJ, et al. Role for myosin-V motor proteins in the selective delivery of Kv channel isoforms to the membrane surface of cardiac myocytes. Circ Res. 2014;114:982–92.
Assayag P, Carre F, Chevalier B, Delcayre C, Mansier P, Swynghedauw B. Compensated cardiac hypertrophy: arrhythmogenicity and the new myocardial phenotype. I. Fibrosis. Cardiovasc Res. 1997;34:439–44.
Walter L, Hajnoczky G. Mitochondria and endoplasmic reticulum: the lethal interorganelle cross-talk. J Bioenerg Biomembr. 2005;37:191–206.
Yu H, Kaung G, Kobayashi S, Kopito RR. Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J Biol Chem. 1997;272:20800–4.
Ojemann LM, Shastri RA, Wilensky AJ, Friel PN, Levy RH, McLean JR, et al. Comparative pharmacokinetics of zonisamide (CI-912) in epileptic patients on carbamazepine or phenytoin monotherapy. Ther Drug Monit. 1986;8:293–6.
Uno H, Kurokawa M, Masuda Y, Nishimura H. Studies on 3-substituted 1,2-benzisoxazole derivatives. 6. Syntheses of 3-(sulfamoylmethyl)-1,2-benzisoxazole derivatives and their anticonvulsant activities. J Med Chem. 1979;22:180–3.
Shinoda M, Akita M, Hasegawa M, Hasegawa T, Nabeshima T. The necessity of adjusting the dosage of zonisamide when coadministered with other anti-epileptic drugs. Biol Pharm Bull. 1996;19:1090–2.
Murata M. Novel therapeutic effects of the anti-convulsant, zonisamide, on Parkinson’s disease. Curr Pharm Des. 2004;10:687–93.
Murata M, Hasegawa K, Kanazawa I. Japan Zonisamide on PDSG. Zonisamide improves motor function in Parkinson disease: a randomized, double-blind study. Neurology. 2007;68:45–50.
Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, et al. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997;100:2315–24.
Chien KR, Knowlton KU, Zhu H, Chien S. Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. FASEB J. 1991;5:3037–46.
Chatterjee A, Mir SA, Dutta D, Mitra A, Pathak K, Sarkar S. Analysis of p53 and NF-kappaB signaling in modulating the cardiomyocyte fate during hypertrophy. J Cell Physiol. 2011;226:2543–54.
Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S. Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013;4:e582.
Wu QQ, Xu M, Yuan Y, Li FF, Yang Z, Liu Y, et al. Cathepsin B deficiency attenuates cardiac remodeling in response to pressure overload via TNF-alpha/ASK1/JNK pathway. Am J Physiol Heart Circ Physiol. 2015;308:H1143–54.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl-2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–59.
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–32.
Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111.
Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29.
Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–12.
Yamamoto K, Fujii R, Toyofuku Y, Saito T, Koseki H, Hsu VW, et al. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001;20:3082–91.
Acknowledgements
This work was supported by Natural Science Foundation of Guangdong Province (2014A030313485), Scientific and Technological Planning Program of Guangzhou (2017071010458), Municipal Education Bureau Program of Guangzhou (1201610286), Natural Science Foundation of Guangdong Province (2018A030313719). We are thankful to Dr. Xiao-yan Dai for her help in the figures’ rearrangement and language editing.
Author information
Authors and Affiliations
Contributions
QW and JHT designed the research; QW carried out the study and wrote the paper; QW, JHT, YXH, YYH, YQH, and GPZ performed the experiments; QX and JDL helped in discussing the data and writing the paper; YHL, XYY, QX conceived, designed, and supervised the study and wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Wu, Q., Tian, Jh., He, Yx. et al. Zonisamide alleviates cardiac hypertrophy in rats by increasing Hrd1 expression and inhibiting endoplasmic reticulum stress. Acta Pharmacol Sin 42, 1587–1597 (2021). https://doi.org/10.1038/s41401-020-00585-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00585-1
Keywords
This article is cited by
-
Zonisamide attenuates pressure overload-induced myocardial hypertrophy in mice through proteasome inhibition
Acta Pharmacologica Sinica (2024)
-
Casting Light on the Janus-Faced HMG-CoA Reductase Degradation Protein 1: A Comprehensive Review of Its Dualistic Impact on Apoptosis in Various Diseases
Molecular Neurobiology (2024)
-
HRD1 reduction promotes cholesterol-induced vascular smooth muscle cell phenotypic change via endoplasmic reticulum stress
Molecular and Cellular Biochemistry (2023)
