
Abstract
Camrelizumab, a programmed cell death 1 (PD-1) inhibitor, has been approved for the treatment of patients with relapsed or refractory classical Hodgkin lymphoma, nasopharyngeal cancer and non-small cell lung cancer. The aim of this study was to perform a population pharmacokinetic (PK) analysis of camrelizumab to quantify the impact of patient characteristics and to investigate the appropriateness of a flat dose in the dosing regimen. A total of 3092 camrelizumab concentrations from 133 patients in four clinical trials with advanced melanoma, relapsed or refractory classical Hodgkin lymphoma and other solid tumor types were analyzed using nonlinear mixed effects modeling. The PKs of camrelizumab were properly described using a two-compartment model with parallel linear and nonlinear clearance. Then, covariate model building was conducted using stepwise forward addition and backward elimination. The results showed that baseline albumin had significant effects on linear clearance, while actual body weight affected intercompartmental clearance. However, their impacts were limited, and no dose adjustments were required. The final model was further evaluated by goodness-of-fit plots, bootstrap procedures, and visual predictive checks and showed satisfactory model performance. Moreover, dosing regimens of 200 mg every 2 weeks and 3 mg/kg every 2 weeks provided similar exposure distributions by model-based Monte Carlo simulation. The population analyses demonstrated that patient characteristics have no clinically meaningful impact on the PKs of camrelizumab and present evidence for no advantage of either the flat dose or weight-based dose regimen for most patients with advanced solid tumors.
Similar content being viewed by others


Introduction
The programmed cell death 1 (PD-1) pathway is important for maintaining an immunosuppressive tumor microenvironment. The blockade of the PD-1 pathway has become the critical component of cancer immunotherapy [1]. Camrelizumab (SHR-1210, AiRuiKa™) is a humanized high-affinity IgG4-kappa monoclonal antibody (mAb) to PD-1 [2]. Since May 2019, it was approved by the National Medical Products Administration to treat patients with relapsed or refractory classical Hodgkin lymphoma, nasopharyngeal cancer, and non-small cell lung cancer [3,4,5]. Camrelizumab is now also being investigated as a treatment for gastric/gastroesophageal junction cancer and hepatocellular carcinoma [6, 7].
The pharmacokinetic (PK) characteristics of camrelizumab are consistent with those of other typical IgG4 antibodies [8]. Non-compartmental analysis indicated that the half-life of camrelizumab is 3–11 days after a single dose of 1 mg/kg-10 mg/kg. When the dose was increased from 1 to 10 mg/kg, the area under the concentration–time curve (AUC) increased supralinearly over the same dose range [8]. In a phase I clinical study of 60–400 mg infusions of camrelizumab, the coefficient of variation of the AUC was more than 30% [9]. Therefore, it is necessary to analyze the factors that affect the PK properties of camrelizumab and to investigate the effect of these factors on the dosing regimen.
Early clinical trials of camrelizumab employed weight-based dosing regimens of 1–10 mg/kg every 2 weeks (Q2W) and compared the 3 mg/kg Q2W regimen with a flat-dose regimen of 200 mg Q2W [5]. Although the flat dose was selected for the subsequent expansion phase based on the PK and receptor occupancy data, the relevance of body weight to the exposure of camrelizumab was not established. A dose adjustment of camrelizumab may be required when there is large variation in the weight of patients [9]. Population PK analyses of exposure data from multiple trials can be used to investigate this dose adjustment regimen [10].
The aim of this study was to (1) develop a population PK model of camrelizumab using pooled data from four clinical trials, (2) evaluate the covariate effects on PK parameters and (3) evaluate the appropriateness of a flat-dose regimen.
Materials and methods
Population pharmacokinetic data
A total of 133 patients with melanoma, advanced solid tumors, or relapsed/refractory classical Hodgkin lymphoma were pooled to conduct this population PK analysis (Table 1). The data of these patients came from three phase 1 trials (SHR-1210-101, SHR-1210-102 and SHR-1210-103) and one phase 2 trial (SHR-1210-II-204), which were registered at clinicaltrials.gov (NCT02721589, NCT02738489, NCT02742935 and NCT03155425, respectively). Informed consent was obtained from each patient before enrollment. The institutional review board and independent ethics committees of all trial centers approved the protocol and all amendments. All studies were conducted according to the principles defined in the Declaration of Helsinki (October 2013) [11].
Serum samples of camrelizumab were measured at prespecified time points during each clinical trial. In the first cycle of the three phase 1 trials, an intensive sampling strategy was employed, while a sparse sampling strategy was employed in the subsequent cycles of the phase 1 trials and in all cycles of the phase 2 trial. At each sampling time, 3 mL of whole blood was drawn and centrifuged at 2700 × g for 10 min at 4 °C. Then, the plasma of each sample was separated and stored at −60 °C until analysis. The details of the study design of each trial are listed in Table 1.
Camrelizumab concentrations were measured by enzyme-linked immunosorbent assays using a calibration range of 157–10,000 ng/mL for the three phase 1 trials and 180–10,000 ng/mL for the phase 2 trial [9].
Evaluable patients were defined as having ≥1 adequate dose and ≥1 corresponding concentration sample. Referring to the guidance for population modeling [12, 13], missing covariates were imputed using the median for continuous covariate variables or the mode for categorical covariate variables if <10% of the values were missing.
Population pharmacokinetic analyses
Population PK models were developed by nonlinear mixed effect modeling software (NONMEM, version 7.4.2, ICON Development Solutions, MD, USA) using first-order conditional estimation with interaction. The evaluation of the NONMEM outputs and graphical and statistical analyses were performed with Perl-speaks-NONMEM (PSN, version 4.7.0, Department of Pharmaceutical Biosciences, Uppsala University, Sweden), R (version 3.4.1, R Foundation for Statistical Computing, Vienna, Austria), and the R packages Xpose (version 4.5.3, Department of Pharmaceutical Biosciences, Uppsala University, Sweden) and Pirana (version 2.9.7, Certara, Inc., NJ, USA).
Base model
In the development of the structural PK model, the appropriateness of the models was assessed by fitting the concentration-time data with one and two-compartment models with linear or nonlinear clearance or parallel linear and nonlinear clearance. Nonlinear elimination pathways were explored by Michaelis–Menten kinetics [14] (Eq. 1):
where CLnonlinear is the nonlinear elimination rate, Vm is the maximum elimination rate, Km is the concentration reaching 50% of the maximum elimination rate, and C is the camrelizumab concentration.
Between-subject variability (BSV) was evaluated as a log-normal distribution [15] (Eq. 2):
where Pi is the post hoc or individual value of the parameter, Ppop is the mean of the population parameter, and ηi is the empirical Bayes estimates of BSV for the ith individual. It was considered that ηi in the population shows a normal distribution with mean zero and variance ω2.
The residual variabilities were assessed by a proportional or additive error or as a combination of both (Eq. 3):
where Y is the observed concentration, IPRED is the individual predicted concentration, εproportional is the proportional error component, and εadditive is the additive error component. Both εproportional and εadditive are normally distributed with mean zero and variance σ2.
According to Akaike’s information criterion (AIC) [16], precise parameter estimates, condition numbers and plots of goodness-of-fit, the base model selection was conducted.
Covariate model
Referring to the guidance for population modeling [13], the covariate analysis used a three-step approach. In the first step, the influence of covariates on PK parameters was evaluated by plotting potential covariates versus PK parameters. Linear regression analysis was used for continuous covariates, while analysis of variance testing was used for categorical covariates. Only those covariates that significantly influenced (R2 > 0.4, P < 0.01) the PK parameters were used in the next step [17, 18].
In the second step, a stepwise forward inclusion selection was implemented. Covariates identified as potentially influencing PK parameters were added cumulatively to the base model. The addition of one covariate resulted in a decrease in the objective function value by >3.84 (P < 0.05) until there was no further significant reduction.
In the third step, a stepwise backward elimination process was initiated. Each covariate was removed temporarily one at a time from the full model to observe the increase in the objective function value. The one with the least increase was removed, and the process was repeated. However, when there was an increase in objective function value by >6.63 (P < 0.01), the covariate was not removed during the stepwise backward elimination stage.
A linear function and a power function were used to evaluate continuous covariates (Eqs. 4 and 5) and categorical covariates as follows (Eq. 6):
where Pi is the parameter for the ith individual; Covcon is the continuous covariate value of the ith individual; Covmedian is the median value of the continuous covariate; Covcat is the categorical covariate value of the ith individual, which could be equal to 1 or 0; θ1 is the mean of the PK parameter in an individual with the covariate median value; and θ2 is the estimated typical value of the covariate effect.
The shrinkage derived from the final model in the third step was evaluated for each BSV term and residual variability.
Model evaluation
The model was evaluated using goodness-of-fit plots that included the observed concentration (DV) vs. the population predicted concentration (PRED) or individual predicted concentrations (IPRED) and conditional weighted residuals (CWRES) vs. PRED or time.
The model was also evaluated internally using bootstrap analysis [19]. During the bootstrap process, new datasets were established with patient data randomly sampled from the original dataset and contained the same number of samples as the original dataset. The median values and 2.5%–97.5% values of the population PK parameters from 1000 newly established bootstrap datasets were compared with those from the final model.
To evaluate the predictive performance of the final model, a visual predictive check was performed to compare the observed concentrations and model predictions. This process was repeated 1000 times.
Model-based simulation
Monte Carlo simulations based on the established model were used to assess the clinical relevance of significant covariates following the 200 mg Q2W regimen. The PK exposures calculated based on typical reference patients (population median) were compared to those of patients with various covariate levels.
Monte Carlo simulations were also used to compare a flat dose of 200 mg Q2W and a weight-based dose of 3 mg/kg Q2W since both regimens were shown to have similar efficacy and safety profiles in phase I clinical trials. Virtual patient datasets were created to predict the PKs of camrelizumab in each dosing regimen group with the replacement of covariate values that were randomly sampled from the pooled modeling data.
BSV was sampled in the model from the established distributions. The variabilities, PK parameters and covariate relationships for each virtual patient were used in the simulations to determine the steady-state trough concentration (Cmin,ss), steady-state peak concentration (Cmax,ss), and steady-state average concentration (Caverage,ss). Caverage,ss was calculated as (Eq. 7):
where AUCss is the steady-state AUC in one dosing interval. Summary statistics (median, 5%–95%) were determined using R software.
Results
Demographics
In total, 133 patients with 3298 plasma concentrations were collected, of which 206 samples were excluded: 203 (6.16%) samples were below the limit of quantification and 3 (0.09%) samples had missing values. Overall, in the population PK analysis, 3092 observations (93.75%) were used. Two-thirds of the patients were males. The tumor types included in the study were hematologic cancer (36%) and solid tumors (64%). The majority (97%) of the investigated population was taking opioids as concomitant medication. For covariates, no data were missing. A summary of the patient demographics used for the analysis dataset is presented in Table 2.
Population pharmacokinetic model
Base model
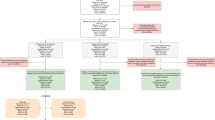
There was a decrease of >300 points in AIC for the PK of camrelizumab by the two-compartment model, which was better described than the one-compartment model. Compared with the linear model, the inclusion of first-order and nonlinear elimination resulted in a further decrease in AIC of 60 points. The final two-compartment base model with parallel linear and nonlinear clearance was parameterized by the clearance of linear elimination (CLlinear), intercompartmental clearance (Q), distribution volume of the central compartment (V1), distribution volume of the peripheral compartment (V2), Vm, and Km. The model structure is shown in Fig. 1.

k0, infusion rate; k12, elimination rate from central compartment to peripheral compartment; k21, elimination rate from peripheral compartment to central compartment; klinear, linear elimination rate; CLlinear, clearance of linear elimination; Q, intercompartmental clearance; V1, apparent distribution volume of central compartment; V2, apparent distribution volume of peripheral compartment; CLnonlinear, clearance of nonlinear elimination; C1, concentration of central compartment; Vm, maximum elimination rate; Km, Michaelis–Menten constant.
BSV was estimated for CLlinear, V1, and Vm with an acceptable precision. The residual error was best described by a combined proportional and additive error model.
Covariate model
The covariates investigated included sex, race, baseline age, weight, creatinine clearance, alanine aminotransferase, aspartate aminotransferase, total bilirubin, albumin, platelets, white blood cells (WBCs), tumor type, lean body weight (LWT), anti-drug antibody (ADA), and activated partial thromboplastin time (APTT). Initial graphical screening showed significant effects of albumin, platelets, and WBCs on CLlinear, effects of LWT, APTT on V1, effects of ADA on Vm, and effects of weight on Q. Then, a forward inclusion process was conducted to further test their effects. As a result, albumin, platelets, WBCs, LWT, APTT, ADA, and weight, which had significant effects (P < 0.05), were retained in the model. However, WBCs, LWT, APTT, ADA, and platelets were finally excluded after stepwise backward elimination (P < 0.01). In the end, albumin and weight were retained in the final model after the above covariate screening process. The main steps from the base model to the final model are summarized in Table S1.
The parameters of the final model are presented in Table 3 and listed below (Eq. 8):
There was a 49.3% decrease in CLlinear when albumin increased from 35 to 50 g/L. The BSV of CLlinear was reduced from 57.0% to 50.8% from the base model to the final model, which was attributed to albumin. Weight was found to affect Q in a power function.
The shrinkages of both BSV and residual variability were less than 30%, which indicated that the estimation of PK parameters was reliable (Table 3).
Model evaluation
There was good agreement between DV and PRED using the goodness-of-fit plots in the final model (Fig. 2). The random scatter around the identity line in the scatterplots of DV vs. PRED and DV vs. IPRED indicated the absence of systematic bias. In the scatterplots of DV vs. PRED and DV vs. IPRED, less than 1% of observations (n = 9) had a CWRES of ~6. These data were included in the model development and did not change the parameter estimates by >10%.

The upper left plot represents the observations versus the population predictions (a). The upper right plot represents the observations versus the individual predictions (b). The lower left plot represents the conditional weighted residuals versus the population predictions (c). The lower right plot represents the conditional weighted residuals versus the time after dosing (d). The red line represents the locally weighted scatterplot smoothing line
One thousand bootstrap analyses were performed for the final model with 85 runs (8.5%) terminated due to rounding errors. Overall, the estimated parameter values for the final model were close to the median and were within the 2.5%–97.5% interval of the bootstrap results. The 2.5%–97.5% interval of each parameter did not contain any null values. The bootstrap results are presented in Table 3.
According to the visual predictive check, the results of the median and 95% confidence intervals in the model-based predictions were in agreement with the observed data (Fig. 3).

Circles represent observed data. Lines represent the 5% (dashed), 50% (solid), and 95% (dashed) percentiles of the observed data. Shaded areas represent nonparametric 95% confidence intervals about the 5% (light blue), 50% (light red), and 95% (light blue) percentiles of the predicted concentrations
Model-based simulation
The simulated PK profiles of camrelizumab following the administration of 200 mg Q2W in patients with varying albumin levels and weights are shown in Fig. 4. As albumin levels increased from 35 to 50 g/L, Cmin,ss increased from 13.7 to 40.8 μg/mL. As weight increased from 40 to 100 kg, Cmin,ss increased from 26.4 to 31.0 μg/mL. Both of these levels were within the range of the 5%–95% percentiles of Cmin,ss(2.2–85.8 μg/mL).

a Cmin; b Cmax; c Caverage. Vertical reference lines represent the typical steady-state exposure value of a 62-kg patient with an albumin level of 44 g/L receiving 200 mg of camrelizumab every 2 weeks. The top bars in each plot represent the 5%–95% exposure values across the entire population. The labels at each of the lower bars indicate the range of the covariate values. The length of each bar describes the impact of that particular covariate on the observed PK parameter
The summary statistics for the simulated camrelizumab exposures with the 200 mg Q2W regimen and 3 mg/kg Q2W regimen are presented in Table 4. The results of 2.5%–97.5% of Caverage,ss for the 3 mg/kg Q2W regimen ranged from 12.81 to 113.87 μg/mL, which was similar to the results of the 200 mg Q2W regimen (15.28–112.08 μg/mL). The median Cmax,ss, Cmin,ss, and Caverage,ss for 200 mg Q2W were higher than those for 3 mg/kg Q2W.
Discussion
This is the first study to report a population PK model of camrelizumab in subjects with advanced solid tumors, advanced melanoma, and relapsed or refractory classical Hodgkin lymphoma. In this study, the PKs of camrelizumab were comprehensively described using a two-compartment model with first-order and Michaelis–Menten clearance from the central compartment.
The final model in this study showed a result that agrees with the known characteristics of antibody PKs. The nonlinear characteristic of mAb clearance is related to saturable target-mediated mechanisms, while the linear characteristic is related to unsaturated clearance pathways, such as Fc-mediated elimination [20]. Target-mediated elimination contributes to a substantial fraction of the total elimination at camrelizumab concentrations lower than the Michaelis–Menten constant, which was 1.38 μg/mL in the model. In addition, as camrelizumab concentrations increase, the target-mediated elimination pathway becomes saturated, and its impact on total mAb clearance weakens. When reaching the median of the simulated concentration in the 200 mg Q2W regimen, the total clearance approached the first-order elimination, and no significant effects of the nonlinear elimination pathway were observed.
The impact of albumin on the PK of mAbs has been previously reported for atezolizumab [21], durvalumab [22], nivolumab [23], and pembrolizumab [24]. This study also showed that camrelizumab clearance will decrease as albumin levels increase because of the same Fc receptor salvaging pathway shared by albumin and IgG. Albumin and IgG are protected from intracellular catabolism by this Fc receptor, which plays a significant role in the homeostasis of albumin [25]. The number of Fc receptors will increase when albumin concentrations increase, which will result in a decreasing rate of camrelizumab elimination [26]. Although albumin had a statistically significant influence on CLlinear, the magnitude of its effect on camrelizumab exposure was limited according to the simulation analyses (Fig. 4). Therefore, a dose adjustment for albumin is not necessary.
Therapeutic mAb dosing is normally based on body weight [27]. However, this dosing strategy has recently been re-evaluated because of the stable therapeutic efficacy and tolerability of camrelizumab over a wide dosing range. Thus, a flat dose was considered and applied in clinical settings due to its convenience, safety, elimination of wastage and better compliance [9]. Our study showed that weight only has an impact on the Q of camrelizumab, and its effect on camrelizumab exposure is limited. Meanwhile, the mean exposure with a flat dose of the 200 mg Q2W regimen was essentially similar to the exposure with the 3 mg/kg Q2W regimen. Although patients with higher weight have lower simulated exposures with the 200 mg Q2W regimen than with the 3 mg/kg Q2W regimen, the obtained distribution of concentrations from patients was still within the range of that from prior clinical reports [28]. Therefore, both weight-based dosing and flat dosing are appropriate for camrelizumab, and neither regimen demonstrates a significant PK advantage over the other.
Our study showed that tumor type (i.e., solid tumors or hematologic malignancies) has no impact on the PKs of camrelizumab. However, several previous studies have shown that tumor type might affect the PKs of PD-1 inhibitors, such as durvalumab [29] and nivolumab [30]. During population PK analysis, the effects of tumor type on PKs could be confounded by the study design (single or multiple dosing regimens), disease status, PD-1 expression, and concomitant drugs. Therefore, this subject needs further investigation in a large sample size of patients.
Several limitations in this study are noteworthy. The study was based solely on dose-exposure analysis, and all patient data came from China. Whether the results can be applied to populations in North American or European countries remains to be elucidated. In addition, there was a lack of data on comprehensive safety and efficacy. Further research, including an exposure-response study, is needed to inform clinical dosing strategies.
Conclusions
In this study, a population PK model of camrelizumab was developed to quantify the impact of patient characteristics on PKs. Patient demographic and pathological characteristics did not have a clinically meaningful impact on the PKs of camrelizumab. Simulations based on the established population PK model indicated that that dosing regimens of 200 mg Q2W and 3 mg/kg Q2W provided similar concentration distributions, and neither regimen demonstrated a significant advantage over the other.
References
Huang J, Xu B, Mo H, Zhang W, Chen X, Wu D, et al. Safety, activity, and biomarkers of SHR-1210, an anti-PD-1 antibody, for patients with advanced esophageal carcinoma. Clin Cancer Res. 2018;24:1296–304.
Markham A, Keam SJ. Camrelizumab: first global approval. Drugs. 2019;79:1355–61.
AiRuiKa®. [package insert], Lianyungang, China: Jiangsu Hengrui Medicine Co. Ltd. 2019.
Song Y, Wu J, Chen X, Lin T, Cao J, Liu Y, et al. A single-arm, multicenter, phase II study of camrelizumab in relapsed or refractory classical hodgkin lymphoma. Clin Cancer Res. 2019;25:7363–9.
Fang W, Yang Y, Ma Y, Hong S, Lin L, He X, et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single-arm, phase 1 trials. Lancet Oncol. 2018;19:1338–50.
Huang J, Mo H, Zhang W, Chen X, Qu D, Wang X, et al. Promising efficacy of SHR-1210, a novel anti-programmed cell death 1 antibody, in patients with advanced gastric and gastroesophageal junction cancer in China. Cancer. 2019;125:742–9.
Zhang Z, Zhou Y, Hu K, Li Z, Wang Z, Huang Y. Complete response of early stage hepatocellular carcinoma in a patient treated with combination therapy of camrelizumab (SHR-1210) and apatinib. Dig Liver Dis. 2019;51:1488–90.
Lickliter JD, Gan HK, Voskoboynik M, Arulananda S, Gao B, Nagrial A, et al. A first-in-human dose finding study of camrelizumab in patients with advanced or metastatic cancer in Australia. Drug Des Devel Ther. 2020;14:1177–89.
Mo H, Huang J, Xu J, Chen X, Wu D, Qu D, et al. Safety, anti-tumour activity, and pharmacokinetics of fixed-dose SHR-1210, an anti-PD-1 antibody in advanced solid tumours: a dose-escalation, phase 1 study. Br J Cancer. 2018;119:538–45.
Wang Y, Booth B, Rahman A, Kim G, Huang SM, Zineh I. Toward greater insights on pharmacokinetics and exposure-response relationships for therapeutic biologics in oncology drug development. Clin Pharmacol Ther. 2017;101:582–4.
Czarkowski M. [Helsinki Declaration–next version]. Pol Merkur Lekarski. 2014;36:295–7.
Keizer RJ, Zandvliet AS, Beijnen JH, Schellens JH, Huitema AD. Performance of methods for handling missing categorical covariate data in population pharmacokinetic analyses. AAPS J. 2012;14:601–11.
Byon W, Smith MK, Chan P, Tortorici MA, Riley S, Dai H, et al. Establishing best practices and guidance in population modeling: an experience with an internal population pharmacokinetic analysis guidance. CPT Pharmacomet Syst Pharmacol. 2013;2:e51.
Fajszi C, Endrenyi L. New linear plots for the separate estimation of Michaelis-Menten parameters. FEBS Lett. 1974;44:240–6.
Ahamadi M, Largajolli A, Diderichsen PM, de Greef R, Kerbusch T, Witjes H, et al. Operating characteristics of stepwise covariate selection in pharmacometric modeling. J Pharmacokinet Pharmacodyn. 2019;46:273–85.
Donohue MC, Overholser R, Xu R, Vaida F. Conditional Akaike information under generalized linear and proportional hazards mixed models. Biometrika. 2011;98:685–700.
Bonate PL. The art of modeling. In: Bonate PL, editor. Pharmacokinetic-pharmacodynamic modeling and simulation.v 20, 2nd ed. Boston, MA: Springer; 2011. p. 17.
Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic–pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20:511–28.
Ette EI, Williams PJ, Kim YH, Lane JR, Liu MJ, Capparelli EV. Model appropriateness and population pharmacokinetic modeling. J Clin Pharmacol. 2003;43:610–23.
Bensalem A, Ternant D. Pharmacokinetic variability of therapeutic antibodies in humans: a comprehensive review of population pharmacokinetic modeling publications. Clin Pharmacokinet. 2020;59:857–74.
Shemesh CS, Chanu P, Jamsen K, Wada R, Rossato G, Donaldson F, et al. Population pharmacokinetics, exposure-safety, and immunogenicity of atezolizumab in pediatric and young adult patients with cancer. J Immunother Cancer. 2019;7:314.
Baverel PG, Dubois VFS, Jin CY, Zheng Y, Song X, Jin X, et al. Population pharmacokinetics of durvalumab in cancer patients and association with longitudinal biomarkers of disease status. Clin Pharmacol Ther. 2018;103:631–42.
Osawa M, Hasegawa M, Bello A, Roy A, Hruska MW. Population pharmacokinetics analysis of nivolumab in Asian and non-Asian patients with gastric and gastro-esophageal junction cancers. Cancer Chemother Pharmacol. 2019;83:705–15.
Ahamadi M, Freshwater T, Prohn M, Li CH, de Alwis DP, de Greef R, et al. Model-based characterization of the pharmacokinetics of pembrolizumab: a humanized anti-PD-1 monoclonal antibody in advanced solid tumors. CPT Pharmacomet Syst Pharmacol. 2017;6:49–57.
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:633–59.
Cho J, Park J, Tae G, Jin MS, Kwon I. The minimal effect of linker length for fatty acid conjugation to a small protein on the serum half-life extension. Biomedicines. 2020;8:96.
Temrikar ZH, Suryawanshi S, Meibohm B. Pharmacokinetics and clinical pharmacology of monoclonal antibodies in pediatric patients. Paediatr Drugs. 2020;22:199–216.
Freshwater T, Kondic A, Ahamadi M, Li CH, de Greef R, de Alwis D, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5:43.
Ogasawara K, Newhall K, Maxwell SE, Dell’Aringa J, Komashko V, Kilavuz N, et al. Population pharmacokinetics of an anti-PD-L1 antibody, durvalumab in patients with hematologic malignancies. Clin Pharmacokinet. 2020;59:217–27.
Wang X, Ludwig EA, Passarell J, Bello A, Roy A, Hruska MW. Population pharmacokinetics and exposure-safety analyses of nivolumab in patients with relapsed or refractory classical Hodgkin lymphoma. J Clin Pharmacol. 2019;59:364–73.
Acknowledgements
ZJ is supported by Key Innovative Team of Shanghai Top-Level University Capacity Building in Clinical Pharmacy and Regulatory Science at Shanghai Medical college of Fudan University, HJW-R-2019-66-19, Shanghai Municipal Education Commission, China. We thank Wei-wei Wang, Qing Yang, and Xiao-jing Zhang from Jiangsu Hengrui Medicine Co. Ltd for critical review of the manuscript. We would like to thank Editage (www.editage.cn) for English language editing. This study was sponsored by Jiangsu Hengrui Medicine Co. Ltd.
Author information
Authors and Affiliations
Contributions
CYW: Methodology, software, formal analysis, validation, visualization, writing—original draft, writing—review & editing. CCS: Methodology, software, formal analysis, writing—review & editing. GLM: Methodology, data curation, supervision, writing—review & editing. DX: Data curation, writing—review & editing. XQL: Software, formal analysis. YYW, LZ, CLC, BHX, YQS, and JZ: Data curation. ZJ: Conceptualization, methodology, writing—original draft, writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
GLM, DX and YYW are employees of Jiangsu Hengrui Medicine Co. Ltd.
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, Cy., Sheng, Cc., Ma, Gl. et al. Population pharmacokinetics of the anti-PD-1 antibody camrelizumab in patients with multiple tumor types and model-informed dosing strategy. Acta Pharmacol Sin 42, 1368–1375 (2021). https://doi.org/10.1038/s41401-020-00550-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00550-y
Keywords
This article is cited by
-
Correlation analysis between camrelizumab trough concentration levels and efficacy or safety in East Asian patients with advanced lung cancer
Cancer Chemotherapy and Pharmacology (2024)
-
Therapeutic drug monitoring of immune checkpoint inhibitors: based on their pharmacokinetic properties and biomarkers
Cancer Chemotherapy and Pharmacology (2023)
-
Safety, activity, and pharmacokinetics of camrelizumab in advanced Asian melanoma patients: a phase I study
BMC Cancer (2022)
