
Abstract
Excessive activation of NLRP3 inflammasome is associated with the pathogenesis of inflammatory diseases. Pristimerin (Pri) is a quinonoid triterpene derived from traditional Chinese medical herb Celastraceae and Hippocrateaceae. Pri has shown antifungal, antibacterial, antioxidant, and anticancer activities. In this study we investigated whether NLRP3 inflammasome was associated with the anti-inflammatory activity of Pri. We showed that Pri (0.1−0.4 μM) dose-dependently blocked caspase-1 activation and IL-1β maturation in LPS-primed mouse bone-marrow-derived macrophages (BMDMs). Pri specifically inhibited NLRP3 inflammasome activation, had no visible effects on NLRC4 and AIM2 inflammasome activation. Furthermore, we demonstrated that Pri blocked the assembly of the NLRP3 inflammasome via disturbing the interaction between NEK7 and NLRP3; the α, β-unsaturated carbonyl moiety of Pri was essential for NLRP3 inflammasome inactivation. In LPS-induced systemic inflammation mouse model and MSU-induced mouse peritonitis model, preinjection of Pri (500 μg/kg, ip) produced remarkable therapeutic effects via inhibition of NLRP3 inflammasome in vivo. In HFD-induced diabetic mouse model, administration of Pri (100 μg· kg−1 ·d−1, ip, for 6 weeks) reversed HFD-induced metabolic disorders via suppression of NLRP3 inflammasome activation. Taken together, our results demonstrate that Pri acts as a NLRP3 inhibitor, suggesting that Pri might be useful for the treatment of NLRP3-associated diseases.
Similar content being viewed by others


Introduction
The NOD-like receptor protein 3 (NLRP3) inflammasome is a cytosolic protein complex composed of NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and cysteinylaspartate-specific proteinase 1 (caspase-1) [1, 2]. After stimulation by diverse damage-associated molecular patterns and pathogen-associated molecular patterns, these proteins oligomerize followed by promotion of NLRP3 inflammasome assembly and activation, which leads to caspase-1 activation and the maturation of the pro-inflammatory cytokines IL-1β and IL-18 [3, 4]. Excessive activation and gain-of-function mutations of the NLRP3 inflammasome are essential for the pathogenesis of several inflammatory diseases, including endotoxin shock, ulcerative colitis, obesity, type-2 diabetes, Alzheimer’s disease, atherosclerosis, and gout [5,6,7,8,9,10,11]. Considerable evidence has demonstrated that pharmacological suppression of NLRP3 inflammasome activation is a promising therapeutic strategy for the treatment of inflammation-related diseases [12]. Therefore, activation of the NLRP3 inflammasome should be tightly controlled, and NLRP3 is an attractive target for therapeutic intervention in inflammatory diseases.
A few endogenous compounds have recently shown potential inhibition of NLRP3 inflammasome activation, including omega-3 fatty acids, dopamine, β-hydroxybutyrate, and bile acids [13,14,15,16]. Several small molecules directly or indirectly target the NLRP3 inflammasome and alleviate inflammasome-mediated disease, such as dimethyl sulfoxide [17], MCC950 [18], glyburide [19], CY-09 [20], and tranilast [21]. Many active components from traditional Chinese medicine exert preventive or therapeutic effects on NLRP3-related diseases. The natural diterpenoid andrographolide triggered mitophagy-mediated NLRP3 inflammasome inhibition in colitis and colitis-associated cancer models [22]. Cardamonin, a medicinal herb from Alpinia katsumadai (Caodoukou in Chinese), prevents septic shock via NLRP3 inflammasome inactivation [23]. The bioactive diterpenoid oridonin blocks the assembly and activation of the NLRP3 inflammasome by directly targeting the NLRP3 protein [24]. Therefore, the identification of NLRP3 inflammasome inhibitors from traditional Chinese medicine is important for clinical application.
Pristimerin (Pri) is a quinonoid triterpene isolated from Celastraceae and Hippocrateaceae [25]. Pri exhibits antifungal, antibacterial, antioxidant, and anticancer activities [26,27,28]. Pri also exerts anti-inflammatory activity via suppression of the NF-κB and MAPK signaling pathways [29,30,31,32]. Pri has exhibited protective effects against colitis, sepsis, and neuroinflammation [30, 33, 34]. Although the use of Pri for the treatment of inflammatory disorders is promising, the underlying mechanism is not well understood.
The present study showed that Pri selectively inhibited NLRP3 inflammasome activation in vitro and in vivo. Further studies demonstrated that Pri blocked the assembly of the NLRP3 inflammasome by disturbing the interaction of (never in mitosis gene a)-related kinase7 (NEK7) and NLRP3. Notably, Pri prevented acute inflammation and metabolic disorder by inhibiting NLRP3 inflammasome activation. Our findings suggest that the NLRP3 inflammasome is an important target of Pri to exert its anti-inflammatory activity.
Materials and methods
Mice
All mice were of a C57BL/6J background and housed under specific pathogen-free conditions. NLRP3 knockout (Nlrp3-/-) mice with a C57BL/6J background were kindly provided by Yu Li (Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai, China). All animal experiments were approved by the Animal Care Committee of the Hubei University of Medicine in China (approval number: 2016-005).
Reagents and antibodies
Adenosine triphosphate (ATP), monosodium urate (MSU) crystals, nigericin, lipopolysaccharide (LPS), dexamethasone (Dex), and D-glucose were purchased from Sigma (St. Louis, MO, USA). Poly (dA: dT) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Pri (B20098) was purchased from Yuanye Bio-Technology (Shanghai, China). Protein G agarose was supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-Flag (F2555) and anti-VSV (V4888) were purchased from Sigma. The anti-mouse caspase-1 (p20) (AG-20B-0042) antibody was purchased from AdipoGen (San Diego, CA, USA). Anti-mouse IL-1β (p17) (AF-401-NA) was purchased from R&D Systems (Minneapolis, MN, USA). Anti-NEK7 (SC-50756) and anti-ASC (sc-22514-R) antibodies were obtained from Santa Cruz Biotechnology. The anti-GAPDH antibody was obtained from Servicebio (Wuhan, China). A standard high-fat diet (HFD) (D12492, 60% kcal fat) was obtained from Research Diet Company (New Brunswick, NJ, USA).
Cell isolation and culture
Bone-marrow cells were collected from C57BL/6J mouse femurs and tibias and lysed in ACK lysis buffer. Bone-marrow cells were cultured and differentiated for 7 days in RPMI-1640 medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS, Biological Industries, Kibbutz Beit Haemek, Israel), 2-mM L-glutamine, 1-mM sodium pyruvate, and 50-ng/mL murine macrophage colony-stimulating factor (Antgene, Wuhan, China) to obtain bone-marrow-derived macrophages (BMDMs). Bone-marrow dendritic cells (BMDCs) were differentiated from bone-marrow cells in RPMI-1640 medium complemented with 10% FBS, 2-mM L-glutamine, 1-mM sodium pyruvate, and 50-ng/mL murine granulocyte-macrophage colony-stimulating factor (Antgene, Wuhan, China). To obtain peritoneal macrophages, C57BL/6J mice were intraperitoneally injected with 4% thioglycollate (MP Biologicals, Irvine, CA, USA). After 3 days, the cells were isolated from the peritoneal cavity and plated in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin for 6 h. Floating cells were removed, and the remaining adherent cells were used as peritoneal macrophages [35].
NLRP3 inflammasome activation
For activation of the NLRP3 inflammasome, 1 × 106 cells were preincubated with LPS (200 ng/mL) for 3 h. The cells were treated with the indicated concentrations of Pri for another 30 min and then stimulated with nigericin (10 μM) and ATP (1 mM) for 30 min or MSU (200 μg/mL) for 6 h. For NLRC4 inflammasomes, cells were infected with Salmonella typhimurium (Salmonella) for 3 h. For AIM2 inflammasome activation, poly (dA: dT) (1 μg/mL) was transfected for 4 h using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s protocol.
Enzyme-linked immunosorbent assay (ELISA)
Supernatants from cell culture, tissue culture, or serum for mouse IL-1β, IL-18, and TNF-α were detected as described previously [35].
Immunoprecipitation
For endogenous interaction, BMDMs were stimulated to activate the NLRP3 inflammasome and lysed in NP-40 buffer with a cocktail of protease inhibitors. The cell lysates were incubated with primary antibodies overnight at 4 °C. Protein G-agarose beads were added followed by incubation for another 4 h. Protein G beads pulled down the target protein for detection using Western blotting. For exogenous interaction, HEK-293T cells were transfected with plasmids using Lipofectamine 2000 according to the manufacturer’s instructions. After 24 h, cells were harvested and lysed in NP-40 buffer with a cocktail of protease inhibitors. Cell lysates were immunoprecipitated with the Flag-bead antibody and detected using Western blotting.
ASC oligomerization assay
BMDMs were primed with 200-ng/mL LPS for 3 h and then stimulated with nigericin for 30 min. The supernatants were removed, and the cells were rinsed in ice-cold PBS and lysed in 200 μL of buffer containing 20-mM HEPES-KOH (pH 7.5), 150-mM KCl, 1% NP-40, 0.1-mM PMSF, and a protease inhibitor mixture followed by shearing ten times using a 21-gauge needle. Lysates were centrifuged at 330 × g for 10 min at 4 °C, and the pellets were washed twice with ice-cold PBS and resuspended in 500 μL of PBS. Disuccinimidyl suberate (2 mM) was added followed by incubation for 30 min with rotation at room temperature. Samples were centrifuged at 330 × g for 10 min at 4 °C. The cross-linked pellets were collected via centrifugation and boiled in 30 μL of SDS sample buffer [36].
Semidenaturing detergent agarose gel electrophoresis (SDD-AGE)
NLRP3 oligomerization was performed according to a published protocol [21]. BMDMs were primed with 200-ng/mL LPS for 3 h and then stimulated with nigericin for 30 min. Then, the cells were lysed with Triton X-100 lysis buffer (0.5% Triton X-100, 50-mM Tris–HCl, 150-mM NaCl, 10% glycerol, 1-mM PMSF, and protease inhibitor cocktail), resuspended in 1× sample buffer (0.5× TBE, 10% glycerol, 2% SDS, and 0.0025% bromophenol blue), and loaded onto a vertical 1.5% agarose gel followed by electrophoresis in running buffer (1× TBE and 0.1% SDS) for 1 h at 4 °C. Proteins were transferred to Immobilon membranes (Millipore) in 1× TBE buffer containing 89-mM Tris (pH 8.3), 89-mM boric acid, and 2-mM EDTA for Western blotting.
NLRP3 ATPase activity
For ATPase activity assays, human NLRP3 proteins were incubated with the indicated concentrations of Pri for 15 min at 37 °C. ATP (25 μM) was added followed by incubation for another 40 min. The ATPase activity was measured using an ADP-Glo kinase assay kit (Promega, Madison, MI, USA) according to the manufacturer’s protocol.
LPS-induced systemic inflammation
C57BL/6J mice were intraperitoneally injected with Pri (500 μg/kg), Dex (1 mg/kg), or vehicle 2 h before intraperitoneal injection of LPS (20 mg/kg) (n = 6 mice per group). After 4 h, the mice were euthanized, and blood was collected. The levels of serum IL-1β, IL-18, and TNF-α were measured using ELISA.
MSU-induced peritonitis
C57BL/6J mice were intraperitoneally injected with Pri (500 μg/kg), Dex (1 mg/kg), or vehicle 2 h before intraperitoneal injection of MSU (1 mg of MSU per mouse) (n = 6 mice per group). After 4 h, the mice were euthanized, the peritoneal cavities underwent lavage with PBS, blood was collected, and the number of neutrophils was determined. The levels of IL-1β in serum and peritoneal lavage fluid were measured by ELISA.
HFD model
C57BL/6J mice (6-week-old males) were randomized into various groups. For the HFD-induced diabetic mouse model, Nlrp3+/+ and Nlrp3-/- mice were fed a 60-kcal% HFD (Medicience Ltd, China) for 12 weeks and used in subsequent experiments. Pri (100-μg/kg body weight) was intraperitoneally injected daily for another 6 weeks. The mice were maintained on the HFD during Pri treatment.
Blood glucose assay
Glucose levels in blood collected from the tail vein were detected using a blood glucose test system kit.
Insulin tolerance test (ITT) and glucose tolerance test (GTT)
The ITT was performed via intraperitoneal injection of human recombinant insulin at a dose of 1 IU/kg for C57BL/6J mice after 4 h of fasting. The GTT was performed via intraperitoneal injection of glucose at 1.5 g/kg into C57BL/6J mice after 14 h of fasting. Blood glucose levels were collected from the tail veil at 15, 30, 60, 90, and 120 min after glucose or insulin injection.
Statistical analyses
The results are shown as the mean ± standard error of the mean. Statistical analyses were performed using GraphPad Prism software. Significant differences between the groups were assessed using Student’s unpaired data test or one-way ANOVA. P values < 0.05 were considered significant.
Results
Pri suppresses caspase-1 activation and IL-1β secretion
To determine the effects of Pri (Fig. 1a) on NLRP3 inflammasome activation, we examined whether Pri inhibited caspase-1 cleavage and IL-1β secretion. The results indicated that Pri dose-dependently blocked caspase-1 activation and IL-1β maturation in BMDMs (Fig. 1b, c). Pri also suppressed nigericin-induced IL-18 secretion (Fig. 1d). However, Pri did not affect the secretion of TNF-α, which suggests that Pri has no effect on LPS-induced priming under these conditions (Fig. 1e). In addition, we found that Pri did not affect cell growth (Fig. 1f), which suggested that the effects of Pri on IL-1β and IL-18 release are not due to its toxicity. Similar results were observed in mouse peritoneal macrophages (Fig. 1g, h) and BMDCs (Fig. 1i, j). These data indicate that Pri inhibits caspase-1 activation and IL-1β secretion.

a The chemical structure of Pri. b LPS-primed BMDMs were treated with various concentrations of Pri for 30 min and then stimulated with nigericin for 30 min. Western blotting analysis of cleaved IL-1β and caspase-1 (p20) in culture supernatants (SN) and pro-IL-1β, pro-caspase-1 and NLRP3 in lysates (input). Protein levels were quantified, and GAPDH was used as the loading control. ELISA of IL-1β (c), IL-18 (d), and TNF-α (e) in the culture SN from LPS-primed BMDMs stimulated with nigericin for 30 min with or without Pri. f LPS-primed BMDMs were treated with the indicated concentrations of Pri for 12 h. After that, cell growth was analyzed. g, h LPS-primed mouse peritoneal macrophages were treated with various concentrations of Pri for 30 min, followed by treatment with nigericin for 30 min. SN and input were analyzed using Western blotting (g). Protein levels were quantified, and GAPDH was used as the loading control. Supernatants were analyzed using ELISA for IL-1β (h). i, j LPS-primed BMDCs were treated with the indicated concentrations of Pri for 30 min followed by treatment with nigericin for 30 min. SN and input were analyzed using Western blotting (i). Protein levels were quantified, and GAPDH was used as the loading control. Supernatants were analyzed using ELISA for IL-1β (j). Data are expressed as the means ± SEM (n = 6) from three independent experiments (c–f, h, j) or are representative of three independent experiments (b, g, i). *P < 0.05, **P < 0.01, ***P < 0.001.
Pri specifically inhibits NLRP3 inflammasome activation
Nigericin, ATP, and MSU activate the NLRP3 inflammasome [1]. To assess whether Pri only inhibited nigericin-triggered NLRP3 inflammasome activation, we examined other NLRP3 agonists. The results demonstrated that Pri also inhibited ATP- and MSU-induced caspase-1 cleavage and IL-1β secretion (Fig. 2a, b), which suggests that Pri is a broad inhibitor of NLRP3 inflammasome activation. To further examine whether the inhibitory effects of Pri were specific to the NLRP3 inflammasome, we measured the activation of the NLRC4 and AIM2 inflammasomes, which were triggered by Salmonella typhimurium infection and poly (dA: dT) transfection, respectively. The results indicated that Pri did not exhibit any visible inhibitory effects on NLRC4 (Fig. 2c, d) or AIM2 inflammasome activation (Fig. 2e, f). These results suggest that Pri specifically inhibits NLRP3 inflammasome activation.

a, b LPS-primed BMDMs were treated with various concentrations of Pri for 30 min and then stimulated with MSU, nigericin, or ATP. Western blotting analysis of cleaved IL-1β and caspase-1 (p20) in the SN and pro-IL-1β, pro-caspase-1, and NLRP3 in the input (a). Protein levels were quantified, and GAPDH was used as the loading control. Supernatants were analyzed using ELISA for IL-1β (b). c, d LPS-primed BMDMs were treated with various concentrations of Pri for 30 min and then stimulated with nigericin or infected with Salmonella typhimurium (Salmonella). SN and input were analyzed using Western blotting (c). Protein levels were quantified, and GAPDH was used as the loading control. Supernatants were analyzed using ELISA for IL-1β (d). e, f LPS-primed BMDMs were treated with various concentrations of Pri for 30 min and then stimulated with nigericin or transfected with poly(dA:dT). SN and input were analyzed using Western blotting (e). Protein levels were quantified, and GAPDH was used as the loading control. Supernatants were analyzed using ELISA for IL-1β (f). Data are expressed as the means ± SEM (n = 6) from three independent experiments (b, d, f) or are representative of three independent experiments (a, c, e). ***P < 0.001.
Pri inhibits NLRP3 inflammasome assembly
We examined the mechanism of Pri inhibition of NLRP3 inflammasome activation. ASC oligomerization is a key step in caspase-1 activation during NLRP3 inflammasome activation [37, 38]. The results showed that Pri markedly attenuated nigericin-induced ASC oligomerization (Fig. 3a). The interaction between NLRP3 and NEK7 plays a critical role in NLRP3 oligomerization and ASC recruitment [39, 40]. Therefore, we then investigated whether Pri affected NLRP3 inflammasome complex formation and found that Pri suppressed the endogenous interaction between NEK7 and NLRP3 (Fig. 3b). Pri also blocked the endogenous NLRP3-ASC interaction (Fig. 3c). To further determine the inhibitory effects of Pri on the NEK7-NLRP3 interaction, we tested whether Pri directly inhibited the NLRP3-NEK7 interaction. We found that Pri inhibited the NEK7-NLRP3 interaction in HEK-293T cells, which suggested that Pri directly disturbed the NEK7-NLRP3 interaction (Fig. 3d). However, Pri did not prevent the NLRP3-NLRP3 interaction in HEK-293T cells, which suggested that Pri did not affect NLRP3 oligomerization (Fig. 3e). In agreement with this result, we found that Pri had no effect on endogenous NLRP3 oligomerization by SDD-AGE immunoblotting (Supplementary Fig. 1a). In addition, Pri did not suppress the ATPase activity of NLRP3 (Supplementary Fig. 1b), which was essential for NLRP3 oligomerization. Moreover, Pri could not block the NLRP3-ASC interaction in HEK-293T cells (Fig. 3f). Collectively, these results indicate that Pri inhibits NLRP3 inflammasome activation via direct blockage of the NEK7-NLRP3 interaction.

a LPS-primed BMDMs were treated with Pri for 30 min and then stimulated with nigericin for 30 min. Western blotting analysis of ASCs in cross-linked pellets and lysates (input) was performed. Protein levels were quantified, and GAPDH was used as the loading control. b LPS-primed BMDMs stimulated with nigericin in the presence or absence of Pri. Endogenous immunoprecipitation (IP) and Western blotting analysis of the interaction of NLRP3 and NEK7. Protein levels were quantified, and GAPDH was used as the loading control. c LPS-primed BMDMs stimulated with nigericin in the presence or absence of Pri. Endogenous immunoprecipitation (IP) and Western blotting analysis of the interaction of NLRP3 and ASC. Protein levels were quantified, and GAPDH was used as the loading control. d Exogenous immunoprecipitation and Western blotting analysis of the interaction of VSV-NLRP3 and Flag-NEK7 in HEK-293T cells. Protein levels were quantified, and GAPDH was used as the loading control. e Exogenous immunoprecipitation and Western blotting analysis of the interaction of VSV-NLRP3 and Flag-NLRP3 in HEK-293T cells. Protein levels were quantified, and GAPDH was used as the loading control. f Exogenous immunoprecipitation and Western blotting analysis of the interaction of VSV-NLRP3 and Flag-ASC in HEK-293T cells. Protein levels were quantified, and GAPDH was used as the loading control. Data are representative of three independent experiments. **P < 0.01 and ***P < 0.001.
Pri inhibits NLRP3 inflammasome activation via a Michael acceptor
Previous results demonstrated that targeting NLRP3 is important for the inhibition of the NEK7-NLRP3 interaction [20, 21, 24]. To investigate the possibility of a Pri interaction with NLRP3, we analyzed the binding properties of Pri to the NLRP3 inflammasome. A washing experiment indicated that Pri inhibited nigericin-induced IL-1β secretion and caspase-1 activation after washout, which suggests that the inhibitory effects of Pri are irreversible (Fig. 4a, b). Pri contains an α, β-unsaturated carbonyl group, which may function as a Michael acceptor by reacting with the cysteine thiols of target proteins. To address whether this moiety of Pri was essential for its inhibitory effects on NLRP3 inflammasome activation, Pri was preincubated with dithiothreitol (DTT) or glutathione (GSH) to reduce the disulfide bonds. Notably, DTT and GSH completely blocked the Pri-induced inhibition of NLRP3 inflammasome activation (Fig. 4c, d). To further confirm whether the carbon–carbon double bond of Pri was responsible for NLRP3 inflammasome inhibition, we detected the effects of wilforol A, which is an altered quinone methide of Pri that is obtainable via reduction of the carbon–carbon double bond (Fig. 4e). In contrast, wilforol A did not suppress nigericin-induced IL-1β secretion, which suggests that wilforol A had no effect on NLRP3 inflammasome activation. These results indicate that the Michael acceptor of Pri is responsible for the inhibition of NLRP3 inflammasome activation.

a, b LPS-primed BMDMs were treated with various concentrations of Pri for 15 min, washed three times, and then stimulated with nigericin for 30 min. Western blotting analysis of cleaved IL-1β and caspase-1 (p20) in SN and pro-IL-1β and pro-caspase-1 in input (a). Protein levels were quantified, and GAPDH was used as the loading control. Supernatants were analyzed using ELISA for IL-1β (b). c, d LPS-primed BMDMs were pretreated with DTT (0.05 mM), GSH (0.2 mM), Pri, or their mixture (Pri and DTT/GSH were preincubated for 30 min) and then stimulated with nigericin for 30 min. Western blotting analysis of cleaved IL-1β and caspase-1 (p20) in SN and pro-IL-1β and pro-caspase-1 in input (c). Protein levels were quantified, and GAPDH was used as the loading control. Supernatants were analyzed using ELISA for IL-1β (d). e Chemical structure of wilforol A. f LPS-primed BMDMs were treated with various concentrations of wilforol A (0.1, 0.5, 1.0, and 2.0 μM) for 30 min and then stimulated with nigericin for 30 min. Supernatants were analyzed using ELISA for IL-1β. Data are expressed as the means ± SEM (n = 6) from three independent experiments (b, d, f) or are representative of three independent experiments (a, c). ***P < 0.001.
Pri suppresses LPS-induced systemic inflammation and MSU-induced peritonitis via inhibition of NLRP3 inflammasome activation
To address whether Pri suppresses inflammatory responses via inhibition of the NLRP3 inflammasome in vivo, we examined the effects of Pri on NLRP3 inflammasome activation in LPS-induced systemic inflammation. The results showed that Pri and Dex significantly decreased the levels of serum IL-1β and IL-18 compared to the LPS group in wild-type (WT) mice (Fig. 5a, b). However, Pri treatment had a limited effect on IL-1β and IL-18 in Nlrp3-/- mice (Fig. 5c, d). Pri also showed a milder effect on serum TNF-α production, which was inflammasome independent (Fig. 5e).

a, b WT mice were intraperitoneally injected with LPS (20 mg/kg body weight) in the presence or absence of Pri (500 μg/kg) and Dex (1.0 mg/kg). IL-1β (a) and IL-18 (b) were measured in serum by ELISA. c–e Nlrp3+/+ and Nlrp3−/− mice were intraperitoneally injected with LPS (20 mg/kg) in the presence or absence of Pri (500 μg/kg). IL-1β (c), IL-18 (d), and TNF-α (e) were measured in serum by ELISA. f, g WT mice were intraperitoneally injected with MSU crystals (1 mg per mouse) in the presence or absence of Pri (500 μg/kg) and Dex (1.0 mg/kg). IL-1β was measured in the serum (f) and peritoneal cavity (g) by ELISA. h, i. Nlrp3+/+ and Nlrp3−/− mice were intraperitoneally injected with MSU crystals (1 mg per mouse) in the presence or absence of Pri (500 μg/kg). IL-1β was measured in the serum (h) and peritoneal cavity (i) by ELISA. j Neutrophil numbers in the peritoneal cavity from Nlrp3+/+ or Nlrp3−/− mice intraperitoneally injected with MSU crystals (1 mg per mouse) in the presence or absence of Pri (500 μg/kg). Data are expressed as the mean and SEM (n = 6) from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Increasing evidence suggests that the NLRP3 inflammasome is involved in MSU-induced peritonitis and results in higher IL-1β production and neutrophil influx in the abdominal cavity [9]. We investigated the inhibitory effects of Pri on NLRP3 inflammasome activation in the MSU-induced peritonitis model. Our results showed that treatment with both Pri and Dex significantly alleviated IL-1β production in serum and lavage fluid after intraperitoneal injection of MSU (Fig. 5f, g) but not in Nlrp3−/− mice (Fig. 5h, i). Moreover, Pri treatment suppressed the total number of neutrophils triggered by MSU (Fig. 5j). Taken together, these results indicate that Pri prevents inflammation via inhibition of the NLRP3 inflammasome in vivo.
Pri prevents HFD-induced metabolic disorders by suppressing NLRP3 inflammasome activation
Previous studies have demonstrated that NLRP3 inflammasome activation was involved in HFD-induced metabolic disorders [10, 11]. Therefore, we assessed the possible inhibitory effects of Pri on HFD-induced metabolic disorders. WT mice were challenged with a HFD for 12 weeks and then treated daily with Pri (100 μg/kg) for another 6 weeks. HFD mice treated with Pri showed less weight gain and food intake (Fig. 6a, b). To evaluate the safety of Pri in vivo, we detected its toxicity after treatment. No notable differences in blood biochemical parameters were observed between the vehicle and Pri-treated groups (Supplementary Fig. 2), which indicates that the efficacy of Pri was not due to a toxic response. After 6 weeks, Pri treatment reduced the fasting glucose concentration (Fig. 6c). Pri also improved insulin sensitivity and glucose tolerance (Fig. 6d, e). We then evaluated the effects of Pri on the metabolic parameters of healthy mice. C57BL/6J mice that fed a normal diet (chow) were treated daily with Pri (100 μg/kg) for 6 weeks. Pri treatment exhibited no significant effects on these metabolic parameters (Fig. 6f–h). Notably, histological analyses indicated that Pri reduced HFD-induced massive hepatic steatosis and intracellular lipid accumulation in the liver (Fig. 6i).

a, b The HFD-induced diabetic mouse model, body weights, and food intake of 12-week HFD-fed WT mice during 6 weeks of vehicle or Pri treatment (100 μg/kg). c Fasting blood glucose concentrations of 12-week HFD-fed WT mice treated with vehicle or Pri (100 μg/kg) for 6 weeks. d, e The insulin tolerance test (ITT) (d) and glucose tolerance test (GTT) (e) were performed after 12-week HFD-fed WT mice, which were treated with vehicle or Pri (100 μg/kg) for 6 weeks. f–h Body weights (f), food intake (g), and fasting plasma glucose levels (h) of C57BL/6J mice fed a normal diet with or without intraperitoneal administration of Pri daily at a dose of 100 μg/kg for 6 weeks. i Representative hematoxylin and eosin (H&E) and Oil Red O staining of liver sections of 12-week HFD-fed WT mice treated with vehicle or Pri (100 μg/kg) for 6 weeks. j–l Twelve-week HFD-fed WT mice treated with vehicle or Pri for 6 weeks. IL-1β in the serum (j) or adipose tissue and liver (k) was measured by ELISA. Western blotting analysis of caspase-1 (p20) in adipose tissue (l). The caspase-1 p20 protein was quantified, and GAPDH was used as the loading control. Data are expressed as the means ± SEM (n = 6) from three independent experiments or are representative of three independent experiments. **P < 0.01, ***P < 0.001.
To further determine whether Pri reversed HFD-induced metabolic disorders via suppression of NLRP3 inflammasome activation, we detected IL-1β production and caspase-1 activation. As expected, Pri treatment reduced IL-1β production in the serum, adipose, and liver tissues (Fig. 6j, k). Pri also suppressed HFD-induced caspase-1 activation (Fig. 6l).
To address whether Pri reversed HFD-induced metabolic disorders dependent on the NLRP3 inflammasome, Nlrp3+/+ and Nlrp3−/− mice were fed a HFD for 12 weeks and then received Pri (100 μg/kg) daily for 6 weeks. Pri treatment reversed the weight gain, reduced the fasting blood glucose level and improved insulin sensitivity and glucose tolerance in HFD-fed Nlrp3+/+ mice (Fig. 7a–d). However, these effects were not observed in HFD-fed Nlrp3−/− mice after Pri treatment (Fig. 7a, b, e, f). Notably, Pri treatment reduced serum IL-1β production, adipose tissue, and cleaved caspase-1 activation, but it exerted minimal effects on HFD-fed Nlrp3−/− mice (Fig. 7g–i). Taken together, our results demonstrate that Pri reverses HFD-induced metabolic disorders via suppression of NLRP3 inflammasome activation.

a, b In the HFD-induced diabetic mouse model, the body weights and food intake of 12-week HFD-fed Nlrp3+/+ and Nlrp3−/− mice during 6 weeks of vehicle or Pri treatment were measured. Insulin tolerance test (ITT) (c, e) and glucose tolerance test (GTT) (d, f) performed in 12-week HFD-fed Nlrp3+/+ (c, d) and Nlrp3−/− (e, f) mice treated with vehicle or Pri for 6 weeks. g–i Twelve-week HFD-fed WT and Nlrp3−/− mice treated with vehicle or Pri for 6 weeks. IL-1β in the serum (g) and liver (h) was measured by ELISA. Western blotting analysis of caspase-1 (p20) in adipose tissue as indicated (i). The caspase-1 p20 protein was quantified, and GAPDH was used as the loading control. Data are expressed as the means ± SEM (n = 6) from three independent experiments or are representative of three independent experiments. ***P < 0.001.
Discussion
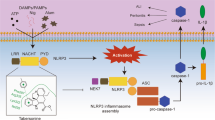
We demonstrated that Pri, an active ingredient of the traditional Chinese medicinal herbs Celastraceae and Hippocrateaceae, selectively inhibited NLRP3 inflammasome activation in vitro and in vivo (Fig. 8). Pri may be a promising agent for novel therapeutics against NLRP3-related inflammatory diseases.

Schematic representation of the mechanism of action of Pri.
Although Pri shows potential biological activity, including anti-autoimmune arthritis and anti-inflammatory activity [29, 30, 41], the mechanism and target of Pri are not well understood. Our results demonstrated that Pri was a potent inhibitor of the NLRP3 inflammasome. Pri treatment significantly prevented systemic inflammation, peritonitis, and type-2 diabetes in vivo. Notably, the beneficial effects of Pri on inflammation-associated diseases were absent in Nlrp3−/− mice, which suggests that the anti-inflammatory activities of Pri depended on the NLRP3 inflammasome. Moreover, Pri had no obvious side effects. Therefore, Pri may provide an improved therapeutic strategy for NLRP3-related inflammatory diseases.
Our results demonstrated that Pri specifically inhibited the NLRP3 inflammasome, but it had no effects on other inflammasomes. The multiple proteins in the NLRP3 inflammasome include NEK7, NLRP3, ASC, and caspase-1, which contribute to NLRP3 inflammasome activation [37,38,39,40]. Our results indicated that Pri suppressed the interaction between NEK7 and NLRP3, which interfered with the assembly and activation of the NLRP3 inflammasome. These results demonstrated that the NEK7-NLRP3 interaction may be targeted by small molecules to inhibit NLRP3 inflammasome activation. How Pri blocked the NEK7-NLRP3 interaction was an important question. Previous results showed that the targeting nucleotide-binding and oligomerization (NACHT) domain of NLRP3 was required for the physical interaction between NEK7 and NLRP3 [24]. Pri contains an electrophilic α, β-unsaturated carbonyl moiety that tends to react with a variety of electron donors. The cysteine thiol groups that are ubiquitously present in cellular proteins may be covalently modified. Because the NLRP3 NACHT domain contains cysteine residues, it is possible that Pri reacted covalently with the thiols of the cysteine residues in the NLRP3 NACHT domain. The present study revealed that the thiol-reducing agents DTT and GSH reversed Pri-induced NLRP3 inflammasome inactivation. Consistent with this finding, Pri suppressed NLRP3 inflammasome activation, but wilforol A, which lacks an α, β-unsaturated carbonyl moiety, was incapable of inhibiting NLRP3 inflammasome activation. Therefore, our findings suggest that Pri may form a covalent bond with NLRP3 to inhibit NLRP3 inflammasome activation. The binding site of Pri to NLRP3 protein needs to be further identified.
In conclusion, our results demonstrated that Pri exhibited selective and potent inhibitory activity against NLRP3 inflammasome activation in vitro and in vivo, which resulted in preventive and therapeutic effects against NLRP3 inflammasome-associated diseases. The results revealed the mechanism of Pri inhibition of NLRP3 inflammasome activation and provide a potential pharmacological therapeutic strategy for the treatment of NLRP3-associated diseases.
References
Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35.
Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13:148–59.
Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res. 2020;30:315–27.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22.
Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013;23:201–12.
Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59:1192–9.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88.
Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1 beta in type 2 diabetes. Nat Immunol. 2010;11:897–904.
Zhang X, Xu A, Lv J, Zhang Q, Ran Y, Wei C, et al. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur J Med Chem. 2020;185:111822.
Yan YQ, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154–63.
Yan YQ, Jiang W, Liu L, Wang XQ, Ding C, Tian ZG, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160:62–73.
Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–9.
Guo C, Xie S, Chi Z, Wang D. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802–16.
Ahn H, Kim J, Jeung EB, Lee GS. Dimethyl sulfoxide inhibits NLRP3 inflammasome activation. Immunobiology. 2014;219:315–22.
Coll RC, Robertson AAB, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187:61–70.
Jiang H, He HB, Chen Y, Huang W, Cheng JB, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214:3219–38.
Huang Y, Rong R. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. 2018;10:e8689.
Guo WJ, Sun Y, Liu W, Wu XX, Guo LL, Cai PF, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10:972–85.
Wang ZL, Xu G, Gao Y, Zhan XY, Qin N, Fu SB, et al. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm Sin B. 2019;9:734–44.
He HB, Jiang H, Chen Y, Deng X, Jiang W, Zhou R. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9:2550.
Petronelli A, Pannitteri G, Testa U. Triterpenoids as new promising anticancer drugs. Anticancer Drugs. 2009;20:880–92.
Luo DQ, Wang H, Tian X, Shao HJ, Liu JK. Antifungal properties of pristimerin and celastrol isolated from Celastrus hypoleucus. Pest Manag Sci. 2005;61:85–90.
Yousef BA, Hassan HM, Zhang LY, Jiang ZZ. Anticancer potential and molecular targets of pristimerin: a mini-review. Curr Cancer Drug Targets 2017;17:100–8.
Zhao Q, Liu Y, Zhong J, Bi Y, Ren Z, Li X, et al. Pristimerin induces apoptosis and autophagy via activation of ROS/ASK1/JNK pathway in human breast cancer in vitro and in vivo. Cell Death Discov. 2019;5:125.
El-Agamy DS, Shaaban AA, Almaramhy HH, Elkablawy S, Elkablawy MA. Pristimerin as a novel hepatoprotective agent against experimental autoimmune hepatitis. Front Pharmacol. 2018;9:292.
Zhao Q, Bi Y, Zhong J, Ren ZT, Liu YX, Jia JJ, et al. Pristimerin suppresses colorectal cancer through inhibiting inflammatory responses and Wnt/beta-catenin signaling. Toxicol Appl Pharmacol. 2020;386:114813.
Kim HJ, Park GM, Kim JK. Anti-inflammatory effect of pristimerin on lipopolysaccharide-induced inflammatory responses in murine macrophages. Arch Pharm Res. 2013;36:495–500.
Hui B, Yao X, Zhou QH, Wu ZY, Sheng P, Zhang LP. Pristimerin, a natural anti-tumor triterpenoid, inhibits LPS-induced TNF-alpha and IL-8 production through down-regulation of ROS-related classical NF-kappa B pathway in THP-1 cells. Int Immunopharmacol. 2014;21:501–8.
Dirsch VM, Kiemer AK, Wagner H, Vollmar AM. The triterpenoid quinonemethide pristimerin inhibits induction of inducible nitric oxide synthase in murine macrophages. Eur J Pharmacol. 1997;336:211–7.
Hui B, Zhang LP, Zhou QH, Hui L. Pristimerin inhibits LPS-triggered neurotoxicity in BV-2 microglia cells through modulating IRAK1/TRAF6/TAK1-mediated NF-kappa B and AP-1 signaling pathways in vitro. Neurotox Res. 2018;33:268–83.
Zhao Q, Yu XJ, Zhang HW, Liu YB, Zhang XX, Wu XX, et al. RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in Fadd-deficient mice. Cell Rep. 2017;19:798–808.
Yu XJ, Zhao Q, Zhang XX, Zhang HW, Liu YB, Wu XX, et al. Celastrol ameliorates inflammation through inhibition of NLRP3 inflammasome activation. Oncotarget. 2017;8:67300–14.
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–206.
Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun. 2016;7:11929.
He Y, Zeng MY, Yang DH, Motro B, Nunez G. Nek7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–7.
Shi HX, Wang Y, Li XH, Zhan XM, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol. 2016;17:250–8.
Tong L, Nanjundaiah SM, Venkatesha SH, Astry B, Yu H, Moudgil KD. Pristimerin, a naturally occurring triterpenoid, protects against autoimmune arthritis by modulating the cellular and soluble immune mediators of inflammation and tissue damage. Clin Immunol. 2014;155:220–30.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (81902852 and 81502548), the Natural Science Foundation of Hubei Provincial Department of Education (D20182101), the Foundation of Health Commission of Hubei Province (WJ2019M053), China Postdoctoral Science Foundation (2020M67022), the Biomedical Research Foundation, Hubei University of Medicine (HBMUPI201809), the Foundation for Innovative Research Team of Institute of Medicine and Nursing, Hubei University of Medicine (2017YHKT01), Faculty Development Grants from Hubei University of Medicine (2018QDJZR06), the Open Project of Hubei Key Laboratory of Embryonic Stem Cell Research (2020ESOF008) and Wudang Local Chinese Medicine Research of Hubei University of Medicine (WDCM2019006) and Innovative Research Program for Graduates of Hubei University of Medicine (YC2019010, YC2020005). We sincerely thank Prof. Rong-bin Zhou (University of Science and Technology of China, Hefei, China) for providing the NLRP3, NEK7, and ASC plasmids. We extremely appreciate Prof. Yu Li (Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai, China) for providing NLRP3 knockout (Nlrp3−/−) mice.
Author information
Authors and Affiliations
Contributions
QZ and XJY conceived and planned the experiments; QZ, YB, JG, YXL, JZ, LRP, YT, and XJY performed the experiment and analyzed the data. QZ and XJY wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhao, Q., Bi, Y., Guo, J. et al. Pristimerin protects against inflammation and metabolic disorder in mice through inhibition of NLRP3 inflammasome activation. Acta Pharmacol Sin 42, 975–986 (2021). https://doi.org/10.1038/s41401-020-00527-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00527-x
Keywords
This article is cited by
-
The role of inflammasomes in human diseases and their potential as therapeutic targets
Signal Transduction and Targeted Therapy (2024)
-
Acetylation is required for full activation of the NLRP3 inflammasome
Nature Communications (2023)
-
NEK7: a new target for the treatment of multiple tumors and chronic inflammatory diseases
Inflammopharmacology (2022)

{kind=link}
{kind=link}