
Abstract
Diabetic nephropathy (DN) is one of the most common causes of end-stage renal disease worldwide. ω3-Fatty acids (ω3FAs) were found to attenuate kidney inflammation, glomerulosclerosis, and albuminuria in experimental and clinical studies of DN. As G protein-coupled receptor 120 (GPR120) was firstly identified as the receptor of ω3FAs, we here investigated the function of GPR120 in DN. We first examined the renal biopsies of DN patients, and found that GPR120 expression was negatively correlated with the progression of DN. Immunofluorescence staining analysis revealed that GPR120 protein was mainly located in the podocytes of the glomerulus. A potent and selective GPR120 agonist TUG-891 (35 mg · kg−1 · d−1, ig) was administered to db/db mice for 4 weeks. We showed that TUG-891 administration significantly improved urinary albumin excretion, protected against podocyte injury, and reduced collagen deposition in the glomerulus. In db/db mice, TUG-891 administration significantly inhibited the mRNA and protein expression of fibronectin, collagen IV, α-SMA, TGF-β1, and IL-6, and downregulated the phosphorylation of Smad3 and STAT3 to alleviate glomerulosclerosis. Similar results were observed in high-glucose-treated MPC5 podocytes in the presence of TUG-891 (10 μM). Furthermore, we showed that TUG-891 effectively upregulated GPR120 expression, and suppressed TAK1-binding protein-1 expression as well as the phosphorylation of TAK1, IKKβ, NF-κB p65, JNK, and p38 MAPK in db/db mice and high-glucose-treated MPC5 podocytes. Knockdown of GPR120 in MPC5 podocytes caused the opposite effects of TUG-891. In summary, our results highlight that activation of GPR120 in podocytes ameliorates renal inflammation and fibrosis to protect against DN.
Similar content being viewed by others

Introduction
Diabetic nephropathy (DN) is one of the most common causes of end-stage renal disease worldwide [1]. The pathogenetic mechanisms of diabetic kidney damage are poorly understood, and therefore, there is a lack of distinct molecular targets. Currently, there are no preventive or curative therapies for DN, and the disease is progressive; complete recovery is unlikely. However, chronic inflammation and podocyte injury lead to albuminuria and renal fibrosis in the progression of DN [1,2,3,4]. ω3-Fatty acids (ω3FAs), the major ingredients in fish oil, exert anti-inflammatory effects and are beneficial in preventing albuminuria, glomerulosclerosis, and tubulointerstitial fibrosis, according to most DN studies [4,5,6,7,8]. Notably, ω3FAs could act as ligands of G protein-coupled receptor 120 (GPR120) [9]. GPR120 is implicated in crucial homeostatic processes, including adipogenesis, inflammation, and insulin sensitivity, as well as the secretion of hormones, prompting many studies to focus on GPR120 in the realm of drug discovery [10, 11]. However, the detailed role of GPR120 in DN has not yet been well demonstrated.
Activation of GPR120 has been reported to exhibit potent anti-inflammatory effects and improve systemic insulin sensitivity in diabetic and/or obese mice [9, 12,13,14]. GPR120 agonism could also suppress IKKβ/NF-κB and JNK/c-Jun signaling in macrophages to alleviate inflammation in diabetic mice [9]. Activated GPR120 binds to the scaffold protein β-arrestin2 [15]. Then, the GPR120–β-arrestin2 complex is internalized into the cytoplasm and binds to TAK1-binding protein-1 (TAB1), which in turn blocks the TAB1/TAK1 association to inhibit the generation of proinflammatory cytokines [9, 16]. Therefore, GPR120 indeed plays crucial roles in diabetes-related inflammatory responses, and agonism of GPR120 represents a promising therapeutic approach [17, 18].
Since the efficacy of ω3FAs in fish oils for sustained GPR120 agonism is low, a high-affinity agonist that would be of potential clinical benefit is required [13]. TUG-891, a potent, selective, and commercially available GPR120 agonist, has been developed and confirmed to promote β-arrestin recruitment via GPR120 [11, 15, 19]. Administration of TUG-891 effectively ameliorated inflammatory disturbances and improved glucose homeostasis by enhancing insulin sensitivity in adipose tissue following chronic sleep fragmentation [20]. Similarly, activation of brown adipocytes with the GPR120 agonist TUG-891 was a promising strategy to increase lipid combustion and reduce obesity [21]. In our previous study, TUG-891 also decreased the number of TUNEL-positive tubular epithelial cells in mice with cisplatin-induced acute kidney injury and reduced the apoptosis rates of cisplatin-stimulated HK-2 cells by repressing ER stress and associated apoptosis, as shown by flow cytometry [22]. This finding prompted us to investigate the favorable function of TUG-891 against DN. In this study, we examined GPR120 expression in the kidneys of DN patients to further determine whether TUG-891 treatment alleviated kidney fibrosis and inflammation by activating GPR120.
Materials and methods
Preparation of human kidney samples
The use of renal biopsies for this study was approved by the Ethics Committees of West China Hospital of Sichuan University (2016-273). Written informed consents and renal tissues were obtained from patients with biopsy-verified DN. The severity of DN (renal biopsy DN stage and estimated glomerular filtration rate) was obtained from a standard pathology report and laboratory test results from West China Hospital. Normal renal tissues were obtained from nephrectomy specimens for the treatment of renal clear cell carcinoma, and the opposite poles of tumors were used as controls.
Animal model
The animal experimental protocols were approved by the Ethics Committee for Experimental Research and Animal Care and Use Ethics Committee of Sichuan University (2020061A), and the animal care and experimental procedures were performed in accordance with The Guide for the Care and Use of Laboratory Animals from National Research Council (US) Committee. Male C57BLKS/J db/db and db/m mice (8 weeks old) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). The mice were housed in a temperature-controlled room (23 ± 2 °C) under a 12-h light/dark cycle with free access to food and water. After 2 weeks of adaptation to the housing conditions, the C57BLKS/J db/db and db/m mice were randomly divided into three groups (n = 6 in each group): control (db/m mice), model (db/db mice), and TUG-891 treatment (db/db mice) groups. TUG-891 (≥98% purity; Sigma-Aldrich, Shanghai, China) was administered at a dose of 35 mg/kg body weight once a day by gavage to db/db mice. Equal volumes of 1% dimethyl sulfoxide vehicle dissolved in 0.9% normal saline were administered once daily to db/m control and db/db model mice. All mice were sacrificed after 4 weeks of treatment.
Cell culture and siRNA transfection
Conditionally immortalized podocytes (MPC5) were established as previously described [23] and purchased from the ATCC agency Shanghai Limai Biological Engineering Co., Ltd (Shanghai, China). The cells were cultured in RPMI-1640 (SH30809.01B, HyClone, Beijing, China) containing 10% fetal bovine serum (SH30084.03, HyClone, Beijing, China) and 1% penicillin-streptomycin solution (SV30010, HyClone, Beijing, China) at 37 °C in a humidified atmosphere of 5% CO2/95% air. All experiments were performed with cells that were passaged four to ten times. For experiments that were performed in six-well plates, the cells were seeded at a density of 1 × 105 cells/well. Cells were serum-starved in medium containing 1% serum for 24 h, followed by treatment with medium containing normal glucose (11 mM), high mannitol (11 mM glucose + 20 mM mannitol), high glucose (30 mM), or high glucose (30 mM) plus TUG-891 (10 μM) for 24 h as indicated.
GPR120 siRNAs and scrambled siRNA were mixed with transfection reagent (riboFECTTM CP transfection kit (166T); RiboBio, Guangzhou, China) according to the manufacturer’s instructions. The sequences of the siRNAs were as follows:
GPR120 siRNA629, sense 5′-GCACAUUGGAUUGGCCCAATT-3′, and antisense 5′-UUGGGCCAAUCCAAUGUGCTT-3′; GPR120 siRNA746, sense 5′-UCACGAAAGCAUCGCGGAATT-3′, and antisense 5′-UUCCGCGAUGCUUUCGUGATT-3′; and nonspecific scramble siRNA, sense 5′-UUCUCCGAACGUGUCACGUTT-3′, and antisense 5′-ACGUGACACGUUCGGAGAATT-3′ (GenePharma, Shanghai, China). MPC5 cells were seeded in six-well plates and transfected with a final concentration of 50 nM GPR120 and scramble siRNAs using riboFECTTM CP reagent in RPMI-1640 (SH30809.01B) containing 10% fetal bovine serum (SH30084.03, HyClone, Beijing, China) without penicillin-streptomycin for 24 h, and then the medium was changed to growth medium for additional incubation. After transfection, the cells were treated with high-glucose (HG) media (30 mM) for another 24 h to evaluate the effects of the siRNAs on MPC5 cells.
Blood and urine examination
Random urine was collected by metabolic cages, and the urine albumin concentration was measured by a mouse urine albumin enzyme-linked immunosorbent assay kit (Mlbio, Shanghai, China). Serum and urinary creatinine, serum glucose, blood urea nitrogen (BUN), cholesterol, low-density lipoprotein cholesterol (LDL-C), and triglyceride levels were measured using a biochemical automatic analyzer (BS-240, Mindray, Shenzhen, China).
Histological examination
Kidney tissues were fixed in 10% phosphate-buffered formalin, cut into 4 μm sections and then subjected to hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), and Masson staining for morphological analysis. The stained sections were viewed by light microscopy at magnifications of ×200 and ×400. PAS staining was used for the semiquantitative analysis of glomerulosclerosis. The degree of glomerulosclerosis was defined as thickening of the basement membrane and mesangium. Twenty glomeruli in each kidney were graded in terms of the severity of glomerular damage (0, normal; 1, slight glomerular damage, mesangial matrix expansion, and/or hyalinosis with focal adhesion involving 25% of the glomerulus; 2, sclerosis of 26%–50% of the glomerulus; 3, sclerosis of 51%–75% of the glomerulus; and 4, sclerosis of 75% of the glomerulus). Quantitative analysis of Masson staining was performed using ImageJ 6.0 (National Institutes of Health, Bethesda, MD, USA). Histological analysis was performed in a blinded manner.
Immunohistochemistry (IHC) staining analysis
Kidney tissues that were fixed in 10% phosphate-buffered formalin, embedded in paraffin, and cut into 4-μm-thick sections were used for immunohistochemical staining. Heat-induced epitope retrieval was performed on dewaxed sections in citrate buffer (pH 6.0) at 95 °C for 40 min. Endogenous peroxidase activity in tissues was blocked with 3% H2O2. Then, the slides were incubated with the primary antibodies anti-GPR120, anti-collagen IV, and anti-α-SMA (Abcam, Cambridge, MA, USA) diluted 1:200 in PBS in a humidified chamber at 4 °C overnight. Then, the slides were incubated with horseradish peroxidase-incubated secondary antibodies for 45 min, and the immunoreactivity was revealed by diaminobenzidine after the slides were washed. Nuclear staining was performed with hematoxylin. The slides were observed under a light microscope, and quantitative analysis of the staining was performed by ImageJ 6.0.
Immunofluorescence (IF) staining analysis
Renal specimens were embedded in OCT compound, cut into 4 μm sections on a cryostat and stored at −80 °C until use. Nonspecific binding sites were blocked with PBS containing 5% bovine serum at room temperature for 1 h. For staining, we incubated the frozen specimens with the following primary antibodies diluted to 1:200 at 4 °C overnight: anti-GPR120 (Santa Cruz, Dallas, TX, USA) and anti-nephrin (Affinity Biosciences, Cincinnati, OH, USA). The corresponding secondary antibodies (1:500 dilution; Jackson ImmunoResearch, West Grove, PA, USA) were used to display the fluorescent signals and were applied for 1 h after the sections were washed with PBS. Then, the samples were stained with DAPI (D8200, Solarbio, Beijing, China) and mounted with cover clips. Pictures were exported from ZEN 2012 microscopy software (blue edition) and analyzed by ImageJ. Slides that contained MPC5 cells were stained with anti-GPR120 (1:200, Abcam, Cambridge, MA, USA) and anti-β-arrestin2 (1:200, ProteinTech Group, Wuhan, Hubei, China). Images were first collected and segmented based on the Hoechst fluorescence. A multistep algorithm was developed to quantitatively analyze biomarker expression in podocytes by using Harmony software (Perkin Elmer, Shanghai, China).
Electron microscopy
Kidney tissues were fixed in cold 2.5% glutaraldehyde for 72 h at 4 °C and then washed with PBS (0.2 mol/L, pH 7.4) for 2 h. Samples were further incubated for 2 h at room temperature with 1% osmic acid. After being dehydrated with ethanol and washed with epoxypropane, all samples were embedded in EPON 812 overnight at room temperature. Ultrathin sections (40–60 nm) were cut (EM UC61rt, Leica, Wetzlar, Germany), stained with uranyl acetate/lead citrate, and viewed on a Hitachi H7650 microscope (Calgary, AB, Canada).
Western blot analysis
Kidney tissue and MPC5 podocytes were homogenized in RIPA buffer containing 4% cocktail proteinase inhibitors to isolate proteins. Equal amounts of protein were separated by SDS-polyacrylamide gels and then transferred onto a PVDF membrane (Bio-Rad, Hercules, CA, USA). After being blocked with 5% nonfat milk, the membranes were incubated with primary antibodies (Supplementary Table S1) overnight at 4 °C, followed by incubation with the following secondary antibodies for 1 h at room temperature: horseradish peroxidase-labeled goat anti-rabbit IgG (HA1001, 1:2000 dilution; HuaBio, Hangzhou, China) or goat anti-mouse IgG (HA1006, 1:2000 dilution; HuaBio, Hangzhou, China). Finally, the proteins were developed with an enhanced chemiluminescence reagent (Millipore Corporation, Boston, MA, USA). Immunoblots were visualized using an Odyssey infrared imaging system (Bio-Rad, ChemiDoc MP, mANUSC, Bio-Rad Laboratories Inc., Hercules, CA, USA) and quantified using ImageJ 6.0 (National Institutes of Health, Bethesda, MD, USA). All Western blotting experiments were performed in triplicate.
RNA isolation and quantitative real-time PCR
Total RNA was extracted from kidney and spleen tissues using a total RNA extraction kit (Foregene, Chengdu, China) according to the protocols and was reverse transcribed into cDNA using a PrimeScriptTM RT reagent kit (Takara, Kusatsu, Japan). The mRNA concentration was measured using a Scan Drop 100 (Analytik Jena, Thuringia, Germany) instrument. Quantitative real-time PCR was performed by using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) in a PCR system (CFX Connect; Bio-Rad, Hercules, CA, USA). The primers used for target mRNA detection are listed in Supplementary Table S2. Relative gene expression was normalized to that of β-actin by comparison with the control groups using CFX Manager™ Software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
The data are presented as the mean ± SD. Differences between two groups were analyzed by unpaired two-tailed t-tests. ANOVA and Tukey’s post hoc tests were applied for comparisons between more than two groups by using GraphPad Prism version 6 (GraphPad Software, La Jolla, CA, USA). A P value less than 0.05 was considered significant.
Results
GPR120 expression was decreased in renal biopsies of DN patients
To explore the role of GPR120 in human DN, we compared the expression of GPR120 in diabetic kidney tissues (n = 18) and nondiabetic controls (n = 6) by IHC and IF staining (Fig. 1). The clinical characteristics of these patients are shown in Table 1. The IHC and IF analyses showed that the intensity of GPR120 staining was negatively correlated with the progression of the disease. The levels of GPR120 in the glomerulus were significantly higher in nondiabetic control kidneys than in DN patient kidneys (Fig. 1a–d). The colocalization of GPR120 and the podocyte marker nephrin was also examined in the glomeruli of kidney tissues obtained from both control and early or advanced DN patients (Fig. 1e, f). The double IF staining results revealed that the coexpression of GPR120 and nephrin significantly decreased in the glomeruli of human diabetic kidneys compared to those of nondiabetic control kidneys, which also revealed that the decreased GPR120 expression was in the podocytes of patients with DN according to the progression of the disease. These experimental findings illustrated the existence and participation of GPR120 in DN.

Representative immunohistochemical (IHC) staining (a) and immunofluorescence (IF) staining (c) of GPR120 in nondiabetic healthy control subjects and DN patients. Quantitative IHC (b) and IF (d) analysis of GPR120 expression in renal glomeruli from control (n = 6), early DN (n = 10), and advanced DN (n = 8) specimens was performed using ImageJ 6.0 software. e Double IF staining of GPR120 and Nephrin in glomeruli from control and DN patients. f Colocalization analysis (Pearson’s r and overlap r coefficient) of GPR120 and nephrin was performed using ImageJ 6.0 software. The results showed that GPR120 was expressed in the podocytes of normal human kidneys. In contrast, reduced expression of GPR120 was observed in the podocytes of patients with DN. Magnification: ×200. The data are represented as the mean ± SD. ***P < 0.001 versus the control. ###P < 0.001 versus early DN.
Activation of GPR120 by TUG-891 alleviated renal damage in db/db mice
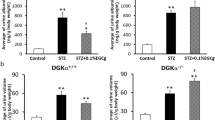
To evaluate whether GPR120 agonism could protect against DN, we treated 10-week-old db/db mice with the GPR120 agonist TUG-891 for 4 weeks. The results showed that TUG-891 treatment slightly reduced body weight, food intake, plasma glucose, serum cholesterol, and serum LDL-C in db/db diabetic mice. TUG-891 did not affect BUN or serum triglycerides in db/db mice (Table 2). Importantly, the urine albumin/creatinine ratio was significantly improved in TUG-891-treated db/db mice (Fig. 2d). Histological analysis indicated that TUG-891 markedly decreased glomerulosclerosis in db/db mice compared with control mice (Fig. 2a, b, e). Electron microscopy analysis revealed that TUG-891 ameliorated podocyte injury, including the attenuation of glomerular basement membrane thickness and foot process effacement in db/db mice (Fig. 2c, f, g). These results highlighted that TUG-891 exerted a renoprotective effect against DN.

Representative H&E (a) and PAS (b) staining of kidneys from db/m (n = 6), db/db (n = 6), and TUG-891-treated db/db (n = 6) mice. Magnification: ×400. c Representative electron microscopy images of the glomerulus (magnification: ×15,000). Arrows: Foot process effacement, fusion of podocytes, and glomerular basement membrane thickness. d Analysis of the urine albumin/creatinine ratio in each group. e Glomerular sclerosis index of 20 randomly selected glomeruli per mouse in each group. f Quantification of glomerular basement membrane (GBM) thickness and g the numbers of podocyte foot processes in the glomeruli of the different groups. **P < 0.01, ***P < 0.001 compared with db/m mice; #P < 0.05, ###P < 0.001 compared with db/db mice.
Activation of GPR120 by TUG-891 ameliorated renal fibrosis and inflammation in db/db mice
To determine the protective mechanism of TUG-891 in DN, we further examined renal fibrosis and inflammation in db/db mice. Our data showed that TUG-891 clearly reduced collagen deposition in the glomeruli of kidney tissues compared with that of untreated db/db mice, as indicated by Masson’s trichrome staining (Fig. 3a, b). TUG-891-mediated decreases in the expression levels of collagen IV and the myofibroblast marker α-SMA were also observed by IHC staining (Fig. 3c–f). The Western blot (Fig. 4a, c) and real-time PCR (Fig. 4b) results showed that TUG-891 reduced the protein and mRNA expression of fibrotic fibronectin, collagen IV, α-SMA, and TGF-β1 in diabetic kidneys. Similar results showed that the increase of protein and mRNA of the proinflammatory cytokine IL-6 were abrogated by TUG-891 treatment in db/db mice (Fig. 4a–c). In addition, the phosphorylation of Smad3, JAK2, and STAT3, which result from the activation of TGF-β1, IL-6, and Smad2, were significantly reduced by TUG-891 (Fig. 4d, e). These data indicated that TUG-891 ameliorated kidney fibrosis and inflammation in db/db mice.

Representative images (a) and quantification (b) of collagen disposition (%) in Masson-stained kidney sections. Magnification: ×400. IHC staining for c collagen IV and e α-SMA in db/m, db/db, and TUG-891-treated db/db mice. d, f Quantification of the areas (%) of collagen IV- and α-SMA-positive cells in the glomeruli. *P < 0.05, **P < 0.01, ***P < 0.001 compared with db/m mice; #P < 0.05; ##P < 0.01, ###P < 0.001 compared with db/db mice.

Protein (a) and mRNA (b) expression of fibronectin, collagen IV, α-SMA, TGF-β1, and IL-6 in the kidneys of mice was determined by Western blotting and real-time qPCR. c Quantitative analysis of fibronectin, collagen IV, α-SMA, TGF-β1, and IL-6 protein expression. Protein expression (d) and quantitative analysis (e) of Smad2, phospho-Smad3/Smad3, phospho-JAK2/JAK2, and phospho-STAT3/STAT3. *P < 0.05, **P < 0.01, ***P < 0.001 compared with db/m mice; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with db/db mice. All Western blot analyses were performed in two randomized mice from each group, and the experiments were repeated in triplicate.
GPR120 agonism activated the β-arrestin2/TAB1 pathway to suppress TAK1 and downstream signaling in the kidneys of db/db mice
As noted previously, TUG-891 acted as a potent and selective agonist of GPR120, but it showed only limited selectivity compared with that of mouse GPR40/free fatty acid receptor 1, complicating its use in vivo in this species [19]. Hence, we investigated the expression of GPR120 and GPR40 in the kidneys and spleens of db/db mice by real-time PCR (Supplementary Fig. S1). According to a previous report, Gpr120 was expressed in male C57BL/6 wild-type mouse kidneys and spleens [9]. Our data confirmed that Gpr120 expression was much higher than Gpr40 expression in the kidney tissues of db/db mice, suggesting that TUG-891 reduced renal damage in DN by activating GPR120 instead of GPR40. Furthermore, the double IF staining results indicated the colocalization of nephrin and GPR120 in the glomeruli of kidney tissues, which revealed the decreased expression of GPR120 in podocytes of db/db mice with urine albumin (Fig. 5a). Administration of TUG-891 restored the diabetes-induced decrease in renal GPR120 protein and mRNA expression compared with that of db/db mice (Fig. 5a–d).

a Double IF staining of nephrin and GPR120 in renal tissues from db/m-, db/db-, and TUG-891-treated db/db mice. Magnification: ×400. Protein expression (b) and quantitative Western blot (c), and real-time qPCR (d) analysis of GPR120 in db/m-, db/db-, and TUG-891-treated kidneys of db/db mice. The results showed that GPR120 was expressed in the podocytes of mouse kidneys, and increased expression of GPR120 was observed after TUG-891 treatment. *P < 0.05, ***P < 0.001 compared with db/m mice; ##P < 0.01, ###P < 0.001 compared with db/db mice. All Western blot analyses were performed in two randomized mice from each group, and the experiments were repeated in triplicate.
Furthermore, activation of GPR120 by TUG-891 treatment increased β-arrestin2 expression, which was then internalized into the cytoplasm to bind TAB1, which in turn blocked TAB1/TAK1 association. Interestingly, TAB1 expression and phosphorylated TAK1 were significantly decreased after the application of TUG-891 in db/db mice. The expression of TAK1 downstream effectors, such as phosphorylated IKKβ, NF-κB p65 JNK and p38 mitogen-activated protein kinase (MAPK), was also suppressed by TUG-891 (Fig. 6a, b). Overall, TUG-891 interacted with GPR120, activated the β-arrestin2/TAB1 pathway and subsequently repressed TAK1 phosphorylation and downstream proinflammatory/fibrotic signaling, which in turn attenuated DN damage.

Representative Western blot images (a) and quantitative analyses (b) of TAB1, phospho-TAK1/TAK1, phospho-IKKβ/IKKβ, phospho-NF-κB/NF-κB, phospho-JNK/JNK, phospho-p38/p38, and β-actin levels. *P < 0.05, **P < 0.01, ***P < 0.001 compared with db/m mice; #P < 0.05, ###P < 0.001 compared with db/db mice. All Western blot analyses were performed in two randomized mice from each group, and the experiments were repeated in triplicate.
GPR120 agonism activated the β-arrestin2/TAB1 pathway to inhibit fibrosis and inflammation in podocytes
Our in vivo study confirmed that the administration of TUG-891 increased intrarenal GPR120 expression in podocytes in db/db kidneys. To clarify the in vitro role of GPR120 in podocytes, we cultured murine podocytes (MPC5 cells) in HG media for 24 h. Western blot analysis (Fig. 7a, b) showed an increasing trend in fibronectin, collagen IV, α-SMA, TGF-β1, and IL-6 expression in HG-induced MPC5 cells, and the addition of TUG-891 effectively suppressed these increases in protein expression. Moreover, consistent with the in vivo study, TAB1, as well as phosphorylated TAK1, IKKβ, NF-κB p65, JNK, and p38 MAPK, were also reduced after TUG-891 treatment (Fig. 7c, d). Notably, IF analysis showed that TUG-891 increased GPR120 and β-arrestin2 levels in HG-stimulated MPC5 cells (Fig. 8a–d). Furthermore, MPC5 cells were transfected with GPR120 siRNA, which therefore suppressed GPR120 expression (Fig. 9a, c, d). After transfection, MPC5 cells cultured in HG medium showed increased expression of fibronectin, TGF-β1 and IL-6. Interestingly, although the amount of GPR120 was reduced, β-arrestin2 and TAB1 were elevated, and TAK1, p38 MAPK, and downstream pathways were significantly activated via phosphorylation (Fig. 9b, e, f). These alterations indicated that TUG-891 activated the GPR120/β-arrestin2/TAB1 pathway, blocked the association of TAB1/TAK1, and repressed the activation of TAK1 downstream signaling, thus ameliorating fibrosis and inflammation (Fig. 7e). However, knockdown of GPR120 had the opposite effect on HG-induced podocyte injury.

Representative Western blot images (a) and quantitative analyses (b) of fibronectin, collagen IV, α-SMA, TGF-β1 and IL-6, and β-actin levels in MPC5 cells. Representative Western blot images (d) and quantitative analyses (c) of TAB1, phospho-TAK1, phospho-IKKβ, phospho-NF-κB/NF-κB, phospho-JNK/JNK, phospho-p38/p38, and β-actin levels in MPC5 cells. e Schematic diagram of the mechanism of the GPR120 agonist TUG-891 in DN. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the NG group; #P < 0.05; ##P < 0.01, ###P < 0.001 compared with the HG group. All Western blot analyses were performed in triplicate.

Representative images of IF staining and quantitative analyses of GPR120 (a, b) and β-Arrestin2 (c, d) levels. ***P < 0.001 compared with the NG group; #P < 0.05; ###P < 0.001 compared with the HG group.

The knockdown efficiencies of two siRNA sequences targeting GPR120 (siGPR120) were evaluated by (a, c) Western blotting and (d) real-time PCR in MPC5 cells. Cells were transfected with scramble siRNA or GPR120 siRNA and treated with 30 mM HG for 24 h. Representative Western blot images (b) and quantitative analyses (e, f) of fibronectin, TGF-β1 and IL-6, GPR120, β-arrestin2, TAB1, phospho-Smad3/Smad3, phospho-STAT3/STAT3, phospho-TAK1/TAK1, phospho-IKKβ/IKKβ, phospho-NF-κB/NF-κB, phospho-JNK/JNK, phospho-p38/p38, and β-actin levels in MPC5 cells. *P < 0.05, **P < 0.01, ***P < 0.001 compared with scramble siRNA. All Western blot analyses were performed in triplicate.
Discussion
In this study, we first demonstrated that GPR120 was involved in the pathology of DN, and its expression was negatively correlated with the progression of renal biopsy-verified DN patients. Moreover, GPR120 was confirmed to be expressed in glomerular podocytes in the kidneys of DN patients and db/db mice. Furthermore, oral administration of the GPR120 agonist TUG-891 significantly reduced urinary albumin excretion, improved podocyte injury, and reduced collagen deposition in the glomerulus. Mechanistically, TUG-891 activated the GPR120/β-arrestin2/TAB1 pathway and suppressed the phosphorylation of TAK1, IKKβ, NF-κB p65, JNK, and p38 MAPK to alleviate fibrosis and inflammation in the kidneys of diabetic db/db mice and HG-stimulated MPC5 podocytes.
Renal fibrosis and inflammation, which are fueled by the local synthesis of profibrotic factors (for example, TGF-β1) and cytokines (such as IL-6), play important roles in the development and progression of DN [2, 3]. Glomerular inflammatory infiltration and progressive glomerulosclerosis gradually impair glomerular filtration and accelerate podocyte demise and urinary albumin excretion in diabetic kidneys [1]. In previous studies, ω3FAs attenuated inflammatory infiltration into the glomeruli by inhibiting the secretion of cytokines and the resultant fibrosis and sclerosis [4]. ω3FA supplementation protected against podocyte injury in experimental DN and attenuated albuminuria both in experimental and human DN [7, 24, 25]. However, little is known about the role of the ω3FA receptor GPR120 in the kidneys in DN. Interestingly, the potent, selective and commercially available GPR120 agonist TUG-891 effectively alleviated albuminuria and glomerulosclerosis and exerted a similar renoprotective effect as that of ω3FAs. Our findings also highlighted that GPR120 agonism by TUG-891 restored diabetic renal injury and may be a promising therapeutic strategy.
After first being identified as an orphan GPCR in human genome databases in 2003 [26], GPR120 was found to be mainly expressed in the lung, adipose tissue, gastrointestinal tract, pancreas, pituitary gland, islets of Langerhans, and macrophages in humans and mice [12]. Our study showed that GPR120 was expressed in normal glomeruli and that GPR120 deposition was decreased with the severity of DN in renal biopsies obtained from patients. This evidence suggests a strong, negative correlation between GPR120 signaling and the progression of DN. Since glomerular podocyte injury is a major cause of albuminuria and glomerulosclerosis in diabetic patients, and ω3FAs could attenuate podocyte injury [24], we further explored the role of the GPR120 agonist TUG-891 in podocytes in diabetic kidneys. In our study, GPR120 and nephrin were coexpressed in the kidneys of humans and db/db mice, which further confirmed GPR120 expression in podocytes. Moreover, TUG-891 treatment restored the diabetes-induced downregulation of GPR120 in db/db mice and in murine podocytes. This observation indicated that the increase in GPR120 was consistent with the ω3FA-mediated increase in GPR120 expression in the rat colon [27]. Taken together, our results suggest the potential protective role of the GPR120 agonist TUG-891 in diabetic glomerular and podocyte injury.
Several studies have reported that GPR120 agonism is involved in preventing the occurrence and development of metabolic disorders such as obesity and diabetes [9, 12, 13, 28]. GPR120 activation directly or indirectly inhibited inflammation, modulated hormone secretion in the digestive system, attenuated insulin resistance, and regulated lipid and/or glucose metabolism in adipose, liver, and muscle tissues, which may help prevent obesity and diabetes. The pathological mechanism of DN is always accompanied by chronic, low-grade tissue inflammation and metabolic disorders in the kidneys [1], whereas the favorable effect of GPR120 activation has not been previously detected in renal tissue. A recent study demonstrated that autologous transplantation of peritoneal macrophages programmed by GPR120 agonism was an effective treatment for delaying renal interstitial fibrosis following UUO surgery [29]. In this study, TUG-891 inhibited the expression of fibrotic markers such as fibronectin, collagen IV, α-SMA, and TGF-β1 and the inflammatory cytokine IL-6 in the kidneys of db/db mice and in HG-stimulated podocytes. Furthermore, TUG-891 treatment significantly decreased the phosphorylation of Smad3 and STAT3 to reduce collagen deposition. These results demonstrated that GPR120 agonism reduced kidney fibrosis and inflammation to exert therapeutic effects on DN.
GPR120 activation has been shown to produce anti-inflammatory effects in a broad range of cells, including macrophages (RAW 264.7 cells), mature adipocytes, Kupffer cells, microglia, and hypothalamic neurons [12]. The mechanism involves activated GPR120 binding to the scaffold protein β-arrestin2. Then, the GPR120–β-arrestin2 complex is internalized into the cytoplasm and binds to TAB1, which in turn blocks the TAB1/TAK1 association and inhibits TAK1 phosphorylation [9, 16]. TAK1 activation is an important signaling step for stimulating the IKKβ/NF-κB and MAPK pathways and mediating the profibrotic effects of the Smad-independent TGF-β signaling pathway [30]. Therefore, this repression could inhibit the downstream proinflammatory and fibrosis cascades, including the induction of MCP-1, TNF-α, IL-6, COX-2, collagens, and other products in the development of DN [30, 31].
In the present study, we found that TUG-891-treated murine podocytes increased intracellular β-arrestin2 levels, which may increase the association of β-arrestin2 and TAB1 and then block the interaction of TAB1 with TAK1. Interestingly, TAB1 and MAPK p38 expression levels were both decreased in vitro and in vivo after the addition of TUG-891. As p38 MAPK is a critical mediator of kidney inflammatory and fibrotic disease and activation of the atypical p38 MAPK pathway requires the direct interaction of TAB1 and p38 MAPK, the downregulation of TAB1 may enable the selective inhibition of p38 MAPK signaling in DN [32,33,34]. Moreover, the levels of phosphorylated TAK1 and downstream proinflammatory signaling pathways (IKKβ/NF-κB and JNK) were increased in db/db mice and murine podocytes, and fewer phosphorylated proteins associated with the above effectors were found after TUG-891 treatment. It has been described that TAB1 and TAK1 cooperate to mediate TGF-β-induced p38 MAPK activation and the stimulation of collagens in DN-related renal fibrosis [34]. Accumulating evidence indicates that TGF-β1-induced TAK1 activation triggers downstream signaling pathways, such as the JNK, p38 MAPK, or NF-κB cascades, and regulates the expression of extracellular matrix proteins, including collagens and fibronectin, and inflammatory cytokines [31, 35,36,37]. In addition, TGF-β1-induced Smad activation is also indispensable for TGF-β1-induced collagen expression through crosstalk with the TAK1–MKK3–p38 MAPK signaling axis [35], which could further increase renal fibrosis in DN. Overall, activation of the GPR120/β-arrestin2/TAB1 signaling pathway by TUG-891 is an attractive strategy to inhibit the downstream profibrotic and proinflammatory pathways of TAK1, such as p38 MAPK, JNK, and IKKβ/NF-κB, in the treatment of DN.
Several studies have reported that GPR120 activation improved hepatic insulin sensitization and enhanced glucose uptake in adipose tissue via Gq/11/phosphoinositide 3-kinase signaling [9, 13, 14, 17]. However, our study only showed slight changes in fasting blood glucose levels in TUG-891-treated db/db mice. Moreover, it has been demonstrated that the direct activation of central GPR120 could reduce appetite by inhibiting neuropeptide Y neurons and suppressing the rewarding effects of palatable food [38]. In our study, food intake was decreased by TUG-891 treatment compared with that of untreated db/db mice, but this difference was not significant. These nonsignificant differences may be because the treatment duration (4 weeks) is not long enough for TUG-891 to cause marked improvements in the blood glucose level or food intake. In addition to improving glucose uptake, GPR120 activation has a beneficial effect on hepatic lipid metabolism, decreasing liver triglycerides and diacylglycerols [9]. Our results revealed that serum LDL but not triglycerides was decreased in TUG-891-treated mice. Nevertheless, while these favorable results illustrated that TUG-891 may have beneficial effects in regulating lipid and/or glucose metabolism, TUG-891 attenuated diabetic renal injury mainly by repressing fibrosis and inflammation in DN.
In general, our study revealed that GPR120 expression was decreased in human diabetic kidneys according to the progression of DN. The GPR120 agonist TUG-891 ameliorated podocyte injury, albuminuria, and glomerulosclerosis in db/db mice by activating the intracellular GPR120/β-arrestin2/TAB1 pathway, which in turn inhibited the downstream profibrotic and proinflammatory pathways associated with TAK1. Our results suggest that activation of GPR120 may provide a novel approach for the treatment of DN.
References
Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. 2017;12:2032–45.
Brosius FC 3rd. New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev Endocr Metab Disord. 2008;9:245–54.
Schena FP, Gesualdo L. Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol. 2005;16:S30–33.
Shapiro H, Theilla M, Attal-Singer J, Singer P. Effects of polyunsaturated fatty acid consumption in diabetic nephropathy. Nat Rev Nephrol. 2011;7:110–21.
Syren ML, Turolo S, Marangoni F, Milani GP, Edefonti A, Montini G, et al. The polyunsaturated fatty acid balance in kidney health and disease: a review. Clin Nutr. 2018;37:1829–39.
Elajami TK, Alfaddagh A, Lakshminarayan D, Soliman M, Chandnani M, Welty FK. Eicosapentaenoic and docosahexaenoic acids attenuate progression of albuminuria in patients with type 2 diabetes mellitus and coronary artery disease. J Am Heart Assoc. 2017;6:e004740.
Garman JH, Mulroney S, Manigrasso M, Flynn E, Maric C. Omega-3 fatty acid rich diet prevents diabetic renal disease. Am J Physiol Ren Physiol. 2009;296:F306–16.
Yanagida K, Hla T. A dark side to omega-3 fatty acids. Nature. 2017;552:180–1.
Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–98.
Kimura I, Ichimura A, Ohue-Kitano R, Igarashi M. Free fatty acid receptors in health and disease. Physiol Rev. 2019;100:171–210.
Milligan G, Alvarez-Curto E, Hudson BD, Prihandoko R, Tobin AB. FFA4/GPR120: pharmacology and therapeutic opportunities. Trends Pharmacol Sci. 2017;38:809–21.
Im DS. FFA4 (GPR120) as a fatty acid sensor involved in appetite control, insulin sensitivity and inflammation regulation. Mol Asp Med. 2018;64:92–108.
Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med. 2014;20:942–7.
Talukdar S, Olefsky JM, Osborn O. Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol Sci. 2011;32:543–50.
Li A, Yang D, Zhu M, Tsai KC, Xiao KH, Yu X, et al. Discovery of novel FFA4 (GPR120) receptor agonists with beta-arrestin2-biased characteristics. Future Med Chem. 2015;7:2429–37.
Ren Z, Chen L, Wang Y, Wei X, Zeng S, Zheng Y, et al. Activation of the omega-3 fatty acid receptor GPR120 protects against focal cerebral ischemic injury by preventing inflammation and apoptosis in mice. J Immunol. 2019;202:747–59.
Zhang D, Leung PS. Potential roles of GPR120 and its agonists in the management of diabetes. Drug Des Devel Ther. 2014;8:1013–27.
Dranse HJ, Kelly ME, Hudson BD. Drugs or diet? Developing novel therapeutic strategies targeting the free fatty acid family of GPCRs. Br J Pharmacol. 2013;170:696–711.
Hudson BD, Shimpukade B, Mackenzie AE, Butcher AJ, Pediani JD, Christiansen E, et al. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol Pharmacol. 2013;84:710–25.
Gozal D, Qiao Z, Almendros I, Zheng J, Khalyfa A, Shimpukade B, et al. Treatment with TUG891, a free fatty acid receptor 4 agonist, restores adipose tissue metabolic dysfunction following chronic sleep fragmentation in mice. Int J Obes. 2016;40:1143–9.
Schilperoort M, van Dam AD, Hoeke G, Shabalina IG. The GPR120 agonist TUG-891 promotes metabolic health by stimulating mitochondrial respiration in brown fat. EMBO Mol Med. 2018;10:e8047.
Huang Z, Guo F, Xia Z, Liang Y, Lei S, Tan Z, et al. Activation of GPR120 by TUG891 ameliorated cisplatin-induced acute kidney injury via repressing ER stress and apoptosis. Biomed Pharmacother. 2020;126:110056.
Mundel P, Reiser J, Zúñiga Mejía Borja A, Pavenstädt H, Davidson GR, Kriz W, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–58.
Sieber J, Lindenmeyer MT, Kampe K, Campbell KN, Cohen CD, Hopfer H, et al. Regulation of podocyte survival and endoplasmic reticulum stress by fatty acids. Am J Physiol Ren Physiol. 2010;299:F821–9.
Soleimani A, Taghizadeh M, Bahmani F, Badroj N, Asemi Z. Metabolic response to omega-3 fatty acid supplementation in patients with diabetic nephropathy: a randomized, double-blind, placebo-controlled trial. Clin Nutr. 2017;36:79–84.
Fredriksson R, Hoglund PJ, Gloriam DE, Lagerstrom MC, Schioth HB. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003;554:381–8.
Cheshmehkani A, Senatorov IS, Kandi P, Singh M, Britt A, Hayslett R, et al. Fish oil and flax seed oil supplemented diets increase FFAR4 expression in the rat colon. Inflamm Res. 2015;64:809–15.
Datilo MN, Sant’Ana MR, Formigari GP, Rodrigues PB, de Moura LP, da Silva ASR, et al. Omega-3 from flaxseed oil protects obese mice against diabetic retinopathy through GPR120 receptor. Sci Rep. 2018;8:14318.
Wang L, Ren X, Tian XF, Cheng XL, Zhao YY, Li QY, et al. Protective effects of GPR120 agonist-programmed macrophages on renal interstitial fibrosis in unilateral ureteral obstruction (UUO) rats. Biomed Pharmacother. 2019;117:109172.
Xu X, Fan Z, Qi X, Shao Y, Wu Y. The role of TGF-beta-activated kinase 1 in db/db mice and high glucose-induced macrophage. Int Immunopharmacol. 2016;38:120–31.
Kim SI, Choi ME. TGF-beta-activated kinase-1: new insights into the mechanism of TGF-beta signaling and kidney disease. Kidney Res Clin Pr. 2012;31:94–105.
Burton JC, Grimsey NJ. Ubiquitination as a key regulator of endosomal signaling by GPCRs. Front Cell Dev Biol. 2019;7:43.
Lim AK, Nikolic-Paterson DJ, Ma FY, Ozols E, Thomas MC, Flavell RA, et al. Role of MKK3-p38 MAPK signalling in the development of type 2 diabetes and renal injury in obese db/db mice. Diabetologia. 2009;52:347–58.
Kim SI, Kwak JH, Zachariah M, He Y, Wang L, Choi ME. TGF-beta-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-beta1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Ren Physiol. 2007;292:F1471–8.
Choi ME, Ding Y, Kim SI. TGF-beta signaling via TAK1 pathway: role in kidney fibrosis. Semin Nephrol. 2012;32:244–52.
Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell Tissue Res. 2012;347:11–20.
Hocevar BA, Prunier C, Howe PH. Disabled-2 (Dab2) mediates transforming growth factor beta (TGFbeta)-stimulated fibronectin synthesis through TGFbeta-activated kinase 1 and activation of the JNK pathway. J Biol Chem. 2005;280:25920–7.
Auguste S, Fisette A, Fernandes MF, Hryhorczuk C, Poitout V, Alquier T, et al. Central agonism of GPR120 acutely inhibits food intake and food reward and chronically suppresses anxiety-like behavior in mice. Int J Neuropsychopharmacol. 2016;19:pyw014.
Acknowledgements
The study was supported by the National Key Research and Development Program (2016YFC1305403) and the 135 Project for Disciplines of Excellence from West China Hospital of Sichuan University.
Author information
Authors and Affiliations
Contributions
LM and PF participated in the research design. TTW, LTY, FG, SBT, LC, and RSH conducted the experiments. TTW and FG contributed new reagents or analytical tools. TTW and LTY performed the data analysis. TTW and LM wrote or contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Wei, Tt., Yang, Lt., Guo, F. et al. Activation of GPR120 in podocytes ameliorates kidney fibrosis and inflammation in diabetic nephropathy. Acta Pharmacol Sin 42, 252–263 (2021). https://doi.org/10.1038/s41401-020-00520-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00520-4
Keywords
This article is cited by
-
Interference of periostin attenuates pathological changes, proinflammatory markers and renal fibrosis in diabetic kidney injury
Genes & Genomics (2023)
-
GPR120 modulates epileptic seizure and neuroinflammation mediated by NLRP3 inflammasome
Journal of Neuroinflammation (2022)
-
FFAR4 improves the senescence of tubular epithelial cells by AMPK/SirT3 signaling in acute kidney injury
Signal Transduction and Targeted Therapy (2022)
-
Mechanism of miR-365 in regulating BDNF-TrkB signal axis of HFD/STZ induced diabetic nephropathy fibrosis and renal function
International Urology and Nephrology (2021)
