
Abstract
Cerebral edema is a pathological hallmark of various central nervous system (CNS) insults, including traumatic brain injury (TBI) and excitotoxic injury such as stroke. Due to the rigidity of the skull, edema-induced increase of intracranial fluid significantly complicates severe CNS injuries by raising intracranial pressure and compromising perfusion. Mortality due to cerebral edema is high. With mortality rates up to 80% in severe cases of stroke, it is the leading cause of death within the first week. Similarly, cerebral edema is devastating for patients of TBI, accounting for up to 50% mortality. Currently, the available treatments for cerebral edema include hypothermia, osmotherapy, and surgery. However, these treatments only address the symptoms and often elicit adverse side effects, potentially in part due to non-specificity. There is an urgent need to identify effective pharmacological treatments for cerebral edema. Currently, ion channels represent the third-largest target class for drug development, but their roles in cerebral edema remain ill-defined. The present review aims to provide an overview of the proposed roles of ion channels and transporters (including aquaporins, SUR1-TRPM4, chloride channels, glucose transporters, and proton-sensitive channels) in mediating cerebral edema in acute ischemic stroke and TBI. We also focus on the pharmacological inhibitors for each target and potential therapeutic strategies that may be further pursued for the treatment of cerebral edema.
Similar content being viewed by others
Introduction
Cerebral edema complicates severe central nervous system (CNS) injuries, and is considered one of the pathological hallmarks of traumatic brain injury (TBI) and excitotoxic injury such as stroke [1,2,3]. Cerebral edema is the leading cause of death within the first week of stroke, with up to 80% mortality in severe cases [4, 5]. It also accounts for up to 50% mortality in TBI [1]. Given the rigidity of the skull, any increase in intracranial fluid due to edema can significantly raise intracranial pressure (ICP) and compromise perfusion [6]. Physiological ICP ranges between 5 and 15 mmHg, however acute brain injury may elevate this value to over 20 mmHg. If left unchecked, this elevated ICP can compress the surrounding brain tissue and lead to ischemia, further accumulation of the edema fluid, brain herniation, and eventually death [6, 7]. Patients who survive from cerebral edema often experience long-term consequences such as chronic encephalopathy, and vascular dementia [8]. Currently available treatments for cerebral edema include hypothermia and osmotherapy. The most commonly used osmotic agents are mannitol and hypertonic saline. Both are osmotic diuretics and can temporarily reduce ICP by generating an osmotic gradient across the blood–brain barrier (BBB) that draws excess water into the vasculature [9]. The potential adverse effects of these osmotic agents include acute renal failure, hypotension, hemolysis, and congestive heart failure [10]. They may also provoke a rebound elevation in ICP [10]. Surgical procedures, including drainage of cerebrospinal fluid (CSF) and decompressive craniotomy, are also frequently performed [7, 11, 12]. Unfortunately, these current treatments are only symptomatic, nonspecific, and often cause significant complications [13]. As there is no effective therapeutic intervention or drug for cerebral edema, continued pharmaceutical research in this field including the discovery and screening of pharmacological agents is of critical importance.
First described by Klatzo et al. in 1967, cerebral edema may be classified as cytotoxic or vasogenic based on the status of the BBB [2] (Fig. 1). Cytotoxic edema, also known as oncotic cell swelling, manifests minutes after acute brain injury. As a premorbid process following the activation of ion channels, cells experience an acute influx of cations, mainly Na+. Cation influx, in turn, drives Cl− influx [14, 15]. These resultant changes in osmotic gradients immediately translate to water influx from the interstitial compartment to the intracellular compartment, causing cells to swell. Cell blebbing, thus, is a key characteristic of cytotoxic edema. The process of cytotoxic edema takes place in all cell types in the brain, including neurons, glial cells, endothelial cells, and is particularly prominent in astrocytes.

In cytotoxic edema (also known as oncotic cell swelling), cells experience an acute influx of solutes (black dots), mainly Na+ and Cl−. Changes in osmotic gradients translate to water influx (blue arrow) from the interstitial compartment to the intracellular compartment. Cytotoxic edema is particularly prominent in astrocytes. Ionic edema is defined as brain swelling due to water influx from an external fluid source in the presence of an intact BBB. Solute and water influx are mediated by ion channels and transporters of endothelial cells. Upregulation of ion channels and transporters also occurs in astrocytes. Astrocyte swelling may lead to the release of excitatory amino acids (EAAs). Vasogenic edema includes the breakdown of the BBB and the extravasation of serum proteins such as albumin and immunoglobulin G (IgG). The transport of solutes and proteinaceous fluid may occur directly (large blue arrow) or through pinocytic vesicles. Multiple factors, including metalloproteinase (MMP), substance P, vascular endothelial growth factor (VEGF), thrombin, and pro-inflammatory cytokines such as tumor necrosis factor (TNF), interleukins (IL)-6, and IL-1β are involved. Together, they mediate neuroinflammation and further degradation of tight junctions, exacerbating cerebral edema. Astrocytes and endothelial cells may experience oncotic cell death and retraction, respectively. Figure created with BioRender.com.
Since cell volume regulation is often compromised in cases of brain injury, sustained unregulated ion channel activation and subsequent water influx often lead to oncotic cell death [3, 16]. It should be noted that while cytotoxic edema resembles a redistribution of water, it does not increase brain water content or cause brain swelling by itself. Rather, it initiates ionic and vasogenic edema, which lead to brain swelling. As the influx of Na+, Cl–, and water persists, the ionic constituents and the volume of the interstitial compartment start to deplete. This is due to the fact that the intracellular compartment is much larger than the interstitial compartment, which only comprises ~12% of the total volume [14, 15]. Cytotoxic edema thereby generates a new Na+ gradient, that can potentially serve as a driving force for the subsequent formation of ionic edema [17].
Ionic edema was only recently classified as a distinct subtype of cerebral edema. It is defined as brain swelling due to water influx from an external fluid source in the presence of an intact BBB [16]. The development of ionic edema is an extracellular process, and is thought to be mediated by ion channels and transporters of endothelial cells. It is essentially a two-step process, during which solutes and water are first transported into endothelial cells via the luminal membrane, and subsequently transported out via the abluminal membrane [14, 18]. As endothelial cells are key components of the neurovascular unit, dysregulation of these cells may cause an early transient leakage of the BBB. Acute cerebrovascular insults that generate cytotoxic and ionic edema early-on (<24 h), are also known to develop secondary vasogenic edema after 2–4 days. In fact, this biphasic edema response is often seen in ischemic stroke and TBI [8]. A very recent study suggested that the immediate source of Na+ and water-mediated ionic edema in acute brain injuries could also arise from CSF, rather than being entirely from the vascular compartment [19]. CSF is thought to transport along the penetrating perivascular spaces into the brain parenchyma, and this process is facilitated by aquaporins expressed at the perivascular astrocytic endfeet. This glymphatic system may have a major role in developing ionic edema.
Vasogenic edema, but not ionic edema, includes the breakdown of the BBB and the extravasation of serum proteins such as albumin and immunoglobulin G from the capillaries into the interstitium [14]. BBB hyperpermeability not only favors the influx of salt and proteinaceous fluid, either directly or through pinocytic vesicles, but also promotes the infiltration of circulating leukocytes, which has been speculated to promote the clearance of cellular debris after injuries [20]. Aside from oncotic cell death in endothelial cells and astrocytes, which normally maintain BBB integrity, multiple proteins have been proposed to be upregulated to facilitate vasogenic edema and BBB breakdown. These proteins include matrix metalloproteinase (MMP), substance P, vascular endothelial growth factor, thrombin, and pro-inflammatory cytokines such as tumor necrosis factor, interleukins (IL)-6, and IL-1β [21,22,23,24,25,26]. Together, they mediate neuroinflammation and the downregulation of tight junction and/or basement membrane proteins. Vasogenic edema can further increase the interstitial oncotic pressure due to proteineous fluid influx, which can potentially occlude small vessels and cause local hypoperfusion. In turn, these processes may positively feedback to upregulate ion channels and/or transporters, which can potentially exacerbate cytotoxic edema.
Clinically, patients who suffer from acute ischemic stroke or TBI are likely to experience cytotoxic edema at the injury core, with ionic and vasogenic edema playing a more profound role in the larger penumbra [16, 27]. In TBI, brain edema typically results from ischemia, which is considered a secondary injury of TBI. However, it should be noted that brain edema can develop despite sufficient cerebral blood flow. Therefore, the initiation and propagation of edema in TBI may also arise directly from mitochondrial impairment induced by the primary mechanical insult, irrespective of cerebral perfusion [27].
Excitotoxicity plays a central role in mediating cell swelling and neuronal death. In both acute ischemic stroke and TBI, acute glutamate release triggers the accumulation of intracellular Na+ and Ca2+ [28, 29]. Elevated intracellular Na+ concentration is initially compensated by the Na+/K+ ATPase pumps. Under prolonged ischemic conditions however, oxygen and glucose are depleted and the ensuing ATP depletion renders the Na+/K+ ATPase pumps inactive [15]. As previously discussed, the altered Na+ gradient can immediately lead to Cl− and water influx and causes neuronal swelling. Studies also indicate that the glutamate-mediated neuronal death in ischemic stroke and TBI is dependent on Na+ and Cl− influx. Moreover, extracellular K+ is elevated significantly from 2.7–3.5 mM to 50–80 mM during acute brain injuries [30]. This increase promotes the rapid uptake of K+ by surrounding astrocytes to reduce K+ imbalance [30]. However, the uptake of K+, along with Cl−, can also lead to cytotoxic edema in astrocytes, as KCl accumulates and draws water into the cells [30]. In neurons, overactivation of the glutamate receptors, namely N-methyl-D-aspartic acid (NMDA) and α-amino 3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors, further contributes to Ca2+ imbalance and subsequent neuronal cell death [31]. The increased intracellular Ca2+ is immediately sequestered by various Ca2+ buffers, such as the mitochondria. However, prolonged Ca2+ uptake by the mitochondria affects the mitochondrial membrane potential, leading to impaired ATP synthesis and reactive oxygen species production. The excessive Ca2+ is then pumped back into the cytoplasm, and Ca2+ overload triggers the activation of various downstream excitotoxic cascades [31]. The overall ionic imbalance and its related events can work both independently to initiate BBB dysfunction and synergistically to propagate each other’s actions and exacerbate BBB breakdown.
Ion channels play important roles in both glutamatergic and non-glutamatergic mechanisms in cerebral ischemia and stroke. Although glutamate receptor-triggered excitotoxicity is traditionally thought to be the central mechanism in acute brain injuries including stroke, recently, more attention has been drawn to non-glutamatergic mechanisms as the clinical trials of anti-excitotoxic therapies have not reached the expected benefits in stroke patients [31,32,33]. These alternative non-glutamatergic mechanisms can also lead to ionic imbalance and eventually neuronal cell death. Various ion channels have been implicated in cerebral ischemia and stroke. These ion channels include neuronal ATP-sensitive potassium (KATP (Kir6.2/SUR1)) channels, transient receptor potential (TRP) channels, acid-sensing ion channels (ASICs), hemichannels, volume-regulated anion channels (VRACs), and other ion exchangers and nonselective cation channels [34,35,36,37,38,39,40] (Fig. 2). The critical roles of ion channels in stroke-related complications such as cerebral edema have also been investigated and confirmed in many studies.

Arrows indicate the direction of transport. Note that KCC2 and NCX are more likely to operate in their reverse modes under conditions of moderate and severe injury, and ClC direction of rectification varies between family members. The respective expression of these channels and transporters depends on the cell type and stages of cerebral edema. Figure created with BioRender.com.
To date, we still lack effective pharmacological agents in attenuating cerebral edema formation and progression despite significant mortality and clinical complications. A crucial step in developing novel and effective therapies is to first understand the molecular and cellular mechanisms of cerebral edema. Ion channels currently represent the third-largest target class in drug development and are critical mediators throughout the stages of cerebral edema as well as stroke. Thus, pharmacological agents that target or modulate ion channel functions have the therapeutic potential to attenuate cerebral edema in acute brain injuries (Table 1). This review aims to (1) summarize the proposed roles of ion channels in mediating cerebral edema in acute ischemic stroke and TBI, as well as the current progress and challenges in discovering ion channel inhibitors; and (2) discuss the potential pharmacological strategies in the drug development for cerebral edema.
Aquaporin (AQP)
AQPs are specialized cell membrane proteins that are permeable to water molecules [41]. Each member of the AQP family has a molecular weight of ~30 kDa and consists of six transmembrane (TM) helices, two opposing short helices, and intracellular carboxyl (C) and amino (N) termini [41]. The two short helices contain the signature triplet amino acid NPA (Asn-Pro-Ala) motif, which facilitates the movement of water [41].
AQPs are organized in homotetramers, where each AQP has four water channels that function independently. This organization also gives rise to a central pore, of which ions and/or gases can flow through depending on the specific AQP subtype [41]. Water flows through AQPs bidirectionally depending on the osmotic gradients. Among the 13 mammalian AQP members, AQP1, -4, and -9 are the most abundant subtypes expressed in the brain [41].
AQP1 is mainly expressed in epithelial cells of the choroid plexus and contributes to CSF production and regulation. Attributing to its ability to transport glycerol in addition to water, AQP9 is mainly involved in energy metabolism [42]. Moreover, AQP4 is the most abundant water channel found throughout the brain and is intensively studied in the context of acute brain injuries.
AQP4 is highly expressed at astrocytic endfeet, where it closely contacts the cerebral vascular compartment at the BBB [41, 42]. In acute brain injuries, however, astrocytic AQP4 expression can reorganize from being highly polarized at endfeet to widely distributed throughout cell bodies and the fine astrocytic processes [43, 44]. Interestingly, most of the neurons in the CNS do not express functional AQPs, suggesting the water influx in neurons is mediated by other transport mechanisms [45, 46]. A low level of AQP4 expression has been detected in endothelial cells, as well as ependymal cells lining the ventricles.
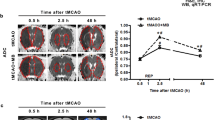
Both TBI and ischemic stroke yield distinct patterns of AQP4 activation [47,48,49,50]. In mouse models of transient middle cerebral artery occlusion (tMCAO), AQP4 expression was found to be rapidly upregulated in astrocytes immediately after the onset of stroke, and the upregulation was correlated temporally with the extent of edema [50]. This correlation was only observed in AQP4, but not AQP1 or AQP9. In addition, a second peak of maximal brain swelling at 48-h post-ischemia coincided with the second peak of AQP4 expression [50]. AQP4 mRNA was also maximally upregulated and corresponded to brain swelling in the peri-infarcted cortex 3 days after focal MCAO in rats, as monitored by MRI [48]. Consistent with these findings, AQP4−/− mice showed a significant reduction in mortality, infarction, and brain edema following both transient and permanent MCAO (pMCAO), with the BBB remaining intact [51,52,53]. AQP4−/− mice also exhibited improvement in long-term neurobehavioural outcomes [51,52,53]. Similar protective effects of AQP4 deficiency were reported even in a severe, global ischemia model [54].
In contrast, reductions in AQP4 expression at the perivascular astrocytic endfeet have been demonstrated in rat and mouse models of tMCAO [43, 55]. AQP4 mRNA was downregulated at 24 h after pMCAO [56], and AQP4−/− mice showed higher mortality, post-ischemic inflammation, and brain atrophy following tMCAO [57]. These contradictory findings may be explained by the severity of the injury, prompting the hypothesis that under severe insults, damaged brain cells are not able to sufficiently synthesize new AQP4 proteins.
In various models of acute TBI, genetic ablation of AQP4 in mice reduced ICP, cell death, water accumulation, astrogliosis, and lesion volume [58, 59]. Consistent with these findings, small-interfering RNA (siRNA) targeting AQP4 resulted in reduced edema formation, neuroinflammation, and neuronal cell death after TBI in rats [60]. The downregulation of AQP4 expression in response to experimental brain trauma has also been reported [49, 61]. A significant decrease in AQP4 expression, correlating with increased brain water content, was reported 48-h post injury in a rat model of TBI [61]. In another study, both AQP4 immunolabelling and AQP4 mRNA were reduced in the edematous cortex along with impaired BBB integrity [49].
Although few human studies have been conducted, AQP4 expression was found to be elevated in brain specimens of patients with ischemic stroke [62]. A more recent postmortem study however, has suggested that the changes in perivascular AQP4 expression are regional-specific, and significantly differ between gray and white matter [55]. CSF from patients with severe TBI showed a significantly elevated level of AQP4 and brain specimens of TBI patients revealed an increase in AQP4 expression from 15 h to 8 days post injury [63, 64].
Aside from injury severity, conflicting results in AQP4 expression following acute brain injuries can also be explained by its dual function, namely the early-on deleterious role in edema formation and the later beneficial role in edema resolution [65]. In fact, increased AQP4 expression was observed to be associated with edema resolution, measured over time using MRI [47, 50, 60]. Moreover, this increase in AQP4 was mainly distributed at the perivascular astrocytic endfeet and the glia limitans, which suggests the potential role of AQP4 in facilitating edematous fluid elimination through the subarachnoid space [47, 50]. The dual function of AQP4, therefore, suggests that AQP4 inhibitors may be beneficial in reducing cerebral edema if administrated at early stages after the brain injury. In addition, it cannot be excluded that some astrocyte function could be altered in AQP4−/− mice, for example, the ability of glutamate reuptake could be affected due to a reduced level of glutamate transporter GLT-1 [66]. Furthermore, altered K+ reuptake by astrocytes has also been reported in AQP4−/− mice, which may contribute to the neuroprotection observed in AQP4−/− animals [65, 67].
Heavy metals, such as mercury and zinc have been known to directly bind to AQP subtypes and inhibit AQP-mediated water transport in vitro. However, the therapeutic potential of using heavy metals in managing cerebral edema is largely limited due to their nonspecific and potentially toxic properties. In the past decade, targeting AQP4 by small molecule inhibitors has become the main focus of pharmacological research, and several small molecule inhibitors have been identified. Studies by Huber et al. identified a series of arylsulfonamides, antiepileptics, and related small molecules that can inhibit AQP4-mediated water transport in hAQP4-expressing Xenopus oocytes [68]. Among these compounds, the carbonic anhydrase inhibitor Acetazolamide (AZA), which is commonly used for reducing intraocular pressure in glaucoma, exerts the most potent and dose-dependent inhibition (IC50 = 0.86 μM) against AQP4 [68]. This inhibitory effect of AZA on AQP4-mediated water conductance has been both confirmed and completely refuted in other in vitro studies [69, 70]. A very recent study has shown the in vivo protective effects of AZA against cerebral edema in a mouse model of TBI. Treatment of AZA prevents the reorganization of AQP4 and attenuates cytotoxic edema in both the cortex and hippocampus [71]. A novel AQP4 inhibitor TGN-020 was later identified by Huber et al. with an IC50 of 3 μM. Administration of TGN-020, both pre- and post-MCAO, reduced cerebral edema, cortical infarction, gliosis, and albumin effusion in rodents [72, 73]. Crystal structures of rAQP4 bound to AZA and TGN-020 have been revealed by in silico studies [68, 74]. The residues of rAQP4 responsible for the binding sites of these two compounds are quite conserved to hAQP4, suggesting their potential inhibitory effects on hAQP4 [68, 74]. However, their effects on AQP4 are not selective, as ACA and TGN-020 may additionally inhibit AQP1.
The phenylbenzamides AER-270 (IC50 = 0.42 μM) and its phosphonate prodrug derivative AER-271, are small molecule AQP4 inhibitors that have been most recently identified. AER-271 has a >5000-fold enhanced water solubility, and can be converted by phosphatases to AER-270 in vivo. A very recent study showed that AER-271 selectively and effectively reduced AQP4-mediated cerebral edema and improved neurological outcome in rodent models of both tMCAO and water intoxication [75]. However, despite of having a trend in reducing ICP and contusion volume, AER-271 seems to be ineffective in reducing cerebral edema in various TBI models [76, 77]. Currently, a Phase-I clinical trial of AER-271 has been completed by Aeromics Inc. OH, USA. A Phase-II trial evaluating AER-271 in cerebral edema prevention in acute ischemic stroke was supposed to begin in 2019. Updates regarding the trial progress have yet to be released.
The loop diuretics bumetanide and its derivatives have also been proposed to inhibit AQP4. Bumetanide has been shown to reduce brain edema following tMCAO via reducing AQP4 expression [78, 79]. However, bumetanide also exerts inhibitory effects on the sodium–potassium–chloride cotransporter (NKCC), the potassium-chloride cotransporter (KCC), and AQP1 of the choroid plexus epithelium, thus its beneficial effects on edema could be due to inhibition of these other targets [79, 80]. Other compounds including phorbol myristate acetate, a PKC activator, and vasopressin 1A receptor antagonist, SR49059, have also been suggested to reduce ischemic and trauma-induced edema by modulating AQP4, yet the complete mechanisms of their actions remain unclear [81,82,83]. The specific monoclonal AQP4 antibody Aquaporumab has been shown in vivo to reduce neuromyelitis optica, an autoimmune demyelinating disease, but it has yet to be evaluated in ischemic stroke or TBI [84]. A more recent in vivo study has revealed that targeting AQP4 subcellular localization, rather than directly targeting the channel activity, significantly reduced cell-surface expression of AQP4 and AQP4-mediated CNS edema, which offers an alternative approach for anti-edematous therapy [85].
To conclude, the variations in AQP4 expression observed in different in vivo studies depend on the degree of injury severity, the pathological models, and the dual nature of AQP4 function. Small molecule AQP4 inhibitors, with defined target-specific and time-specific pharmacological properties, may serve as a potential treatment for cerebral edema. The therapeutic effects of AER-271 are yet to be evaluated in clinical trials. Regardless, many reports have served as “proof of principle” studies, highlighting the important role of AQP4 in cerebral edema. The discovery of selective AQP4 inhibitor(s) will be a critical goal in drug development.
SUR1-TRPM4
The ion channel complex sulfonylurea receptor 1—TRP melastatin 4 (SUR1-TRPM4) was first discovered in reactive astrocytes obtained from the hypoxic core by Chen et al. [86]. This ion channel complex is formed by two subunits, namely, the regulatory subunit SUR1 and the pore-forming subunit TRPM4. SURs belong to the superfamily of ATP-binding cassette (ABC) transporters, which use the energy derived from ATP hydrolysis to mediate a variety of cellular functions [87]. SUR1 and SUR2 are unique among ABC transporters in that they serve as ion channel regulators [87]. Under physiological conditions, SUR1 is expressed in neurons, but not in astrocytes or endothelial cells.
Contrary to what is seen under physiological conditions, brain tissue isolated from rat ischemic core shows an upregulation of SUR1 protein and mRNA in neurons, astrocytes, and endothelial cells [88]. Upregulation of SUR1 was also observed in rat cortical impact TBI model, with a doubling of the SUR1 gene ABCC8 6-h post TBI [89]. Clinically, SUR1 expression is elevated in the CSF of TBI patients and is undetectable in healthy controls. SUR1 expression in these patients correlates with an increased risk of cerebral edema [90]. Single nucleotide polymorphisms of ABCC8 have also been associated with a higher risk of edema development in TBI patients [91]. Although SUR1 acts as the regulatory subunit of both ATP-sensitive K+ (KATP) channels and the nonselective cation channel NCCa-ATP, the upregulation of SUR1 and resultant cerebral edema in acute brain injuries were proposed to be associated with its regulation of NCCa-ATP, but not of KATP [88, 89]. This is further confirmed in a very recent study using highly specific antisense oligodeoxynucleotides [92].
TRPM4 is a Ca2+-activated, nonselective, monovalent cation channel. TRPM4 alone is a functional ion channel, whereas SUR1 cannot translocate to the cell membrane without binding to a pore-forming subunit [93]. Reciprocally, SUR1 doubles TRPM4’s affinity for Ca2+-calmodulin, and its sensitivity to intracellular Ca2+ when they co-assemble as heteromers. Unlike most of the ion channels and transporters implicated in cerebral edema, SUR1-TRPM4 channels are expressed de novo only after acute CNS injuries [94]. The expression of functional SUR1-TRPM4 occurs approximately 3-h post MCAO in the ischemic core, and 8-h post MCAO in the penumbra [88].
ATP depletion or a nanomolar change of Ca2+ concentration rapidly activates SUR1-TRPM4 [86, 94]. In acute brain injuries, activation of SUR1-TRPM4 by increased intracellular Ca2+ initially reduces the driving force of Ca2+ influx [94]. Sustained SUR1-TRPM4 activation under conditions of severe ATP depletion, however, can lead to Na+ influx and oncotic cell death.
A recent study demonstrated that SUR1-TRPM4 can physically assemble with astrocytic AQP4 to form a novel heteromultimeric water/ion channel complex [95]. Primary astrocytes in vitro showed that TRPM4 and AQP4 were closely co-localized and synergistically mediated a fast, high-capacity TM water influx that drove astrocytic swelling [95]. The same study also validated their finding in vivo in a mouse model of brain edema, and speculated that the co-assembly of AQP4 and SUR1-TRPM4 may further amplify the role of TRPM4 as a negative regulator of Ca2+ influx [95]. Nonetheless, future studies are required to demonstrate the functional implications of SUR1-TRPM4-AQP4 complex in the context of acute brain injuries.
Sulfonylureas, such as glibenclamide and tolbutamide, are potent inhibitors of SUR-regulated channels, including KATP and NCCa-ATP. SUR1-TRPM4 can be inhibited by glibenclamide (EC50 = 48 nM) with high affinity and specificity. Glibenclamide administration in vitro effectively prevented SUR1-TRPM4-mediated Na+ influx, cytotoxic edema, and oncotic cell death [86]. Furthermore, in vivo effects of glibenclamide on brain edema have been widely studied in different rat models of ischemic stroke, including thromboembolic stroke, pMCAO, and tMCAO models [96]. In all of these studies, glibenclamide blocked the ischemia-induced upregulation of NCCa-ATP channels and improved stroke outcomes. In regard to TBI, rats treated with glibenclamide showed attenuated BBB disruption, brain water content, and contusion size [97].
Clinically, the FDA-approved, antidiabetic medication glibenclamide (aka. glyburide) appeared promising in both Phase-I- and Phase-II trials (Remedy Pharmaceuticals) [98, 99]. Intravenous administration of glibenclamide at non-hypoglycemic doses attenuated water diffusivity and cerebral edema in stroke patients with large hemispheric infarction [99]. Patients who underwent glibenclamide therapy (GAMES-RP) also show improved survival and long-term clinical outcomes, with no identified serious adverse effects [98, 100]. These protective effects of glibenclamide were proposed to be associated with a reduced level of MMP-9 [98]. A randomized, placebo-controlled, double-blind, Phase-IIa trial (Remedy Pharmaceuticals, Inc., NY, USA) is currently underway to test the effects of glibenclamide in patients with moderate-to-severe TBI.
Together these findings indicate that the SUR1-TRPM4 channel is critically involved in cerebral edema formation. SUR1-TRPM4 inhibitors are feasible to safely reduce brain swelling and improve clinical outcomes following acute ischemic stroke and TBI. The time interval between the onset of injury and the transcriptional upregulation of SUR1-TRPM4 also extends the therapeutic window of glibenclamide treatment. Genetic variability in ABCC8 may provide valuable information on the prognosis and treatment of brain edema. Although the pharmacological aspect of this direction seems more advanced compared to AQP4 pharmacology, the therapeutic relevance of SUR1-TRPM4 channel inhibitors awaits validation from the above-mentioned clinical trials.
Our lab has shown that KATP (Kir6.2/SUR1) channels are neuroprotective through potentially inducing cellular hyperpolarization during ischemia and hypoxic preconditioning (HPC) [35, 101]. Previously, we demonstrated that pretreatment with tolbutamide in neonatal mice abolished the effects of HPC-induced neuroprotection and worsened neurodegeneration following hypoxic-ischemic insults. Contrary, pretreating with a KATP activator, diazoxide, enhanced neuroprotection [101]. Our lab also showed that administering tolbutamide to mice subjected to pMCAO resulted in exacerbated brain damage and poorer behavioral outcomes. Opposing effects including reduced brain damage and improved behavioral outcomes were obtained using the KATP activator diazoxide on the pMCAO model [102]. Furthermore, a subsequent meta-analysis illustrated that patients with type-2 diabetes mellitus (T2DM) had a greater odds ratio for stroke morbidity when treated with sulfonylureas [102]. A possible explanation is that anti-type-2 diabetes medication sulfonylureas may have interfered with the neuroprotective role of the KATP channels in mediating membrane hyperpolarization during ischemia as well as HPC. Taken together, sulfonylureas may have nonspecific effects on KATP (Kir6.2/SUR1), as well as NCCa-ATP channels; further investigations are needed to elucidate their individual effects when specific blockers for each channel have been identified.
Chloride channels
Cl− is the most abundant anion in humans under physiological conditions. The intracellular chloride level, [Cl−]i, in neurons is tightly regulated to ensure appropriate electrical responses to the inhibitory neurotransmitters γ-aminobutyric acid (GABA) and glycine. Specifically, low [Cl−]i stimulates Cl− influx through GABAA and glycine receptors, resulting in hyperpolarization. High [Cl−]i, in contrast, stimulates Cl− efflux, resulting in depolarization [103]. Regulation of [Cl−]i is also essential to maintain normal cell volume. This is achieved either through regulatory volume increase (RVI) or regulatory volume decrease (RVD), the processes by which osmotically shrunken or swollen cells are restored to their original volumes [103]. The putative candidates for [Cl−]i regulation in the context of cerebral edema are the NKCC, KCC, VRAC, TMEM16A, cystic fibrosis (CF) transmembrane conductance regulator (CFTR), and voltage-gated chloride channels (ClCs).
NKCC
First described by Geck et al., NKCCs are membrane proteins that maintain cell volume by RVI [104]. Two isoforms of NKCCs are currently characterized; NKCC1 is found in most mammalian cells whereas NKCC2 is exclusively found in the kidney [105]. In the CNS, the NKCC1 isoform (132 kDa) is expressed in neurons, astrocytes, endothelial cells, and epithelial cells of the choroid plexus [105]. Under physiological conditions, cell shrinkage triggers NKCC1 activation and the electroneutral influx of 1 Na+, 1 K+, and 2 Cl−, coupled with an isosmotic water flow [104]. NKCC1 is considered a secondary-active transporter, and its activity is driven by TM Na+ and K+ gradients as established by Na+/K+ ATPase [105].
The overall structure of NKCC1 is a dimer, consisting of 12 TM domains with intracellular N- and C-termini [106]. NKCC1 activation is associated with phosphorylation at its N-terminal domain and its activity can be stimulated by various factors that are well-implicated in acute brain injuries [107]. These factors include high extracellular K+, excitatory amino acids (EAAs), hypoxia, aglycemia, and arginine vasopressin [108, 109]. As previously mentioned, extracellular K+ is rapidly elevated in the early stages of acute ischemic stroke, reaching 20 times its original concentration [30]. The elevated [K+]o creates a large driving force, and stimulates NKCC1 activity in neurons and the surrounding astrocytes to facilitate rapid K+ uptake [105]. Since K+ returns to the cells with Na+, Cl−, and water, the net effect is an increase in intracellular osmolarity and cytotoxic edema [103, 110]. In fact, both neurons and astrocytes of NKCC1−/− mice exhibited reduced intracellular Na+ accumulation, and cytotoxic edema after oxygen-glucose deprivation (OGD) [110]. In astrocytes, K+-induced NKCC1 activation also leads to the release of EAAs such as glutamate, which in turn stimulates NKCC1 activity [111]. NKCC1−/− astrocytes showed decreased EAAs release and astrocytic swelling compared to wild-type astrocytes in response to either elevated [K+]o or OGD [110, 112]. This K+-induced NKCC1 activation in astrocytes can be completely abolished by either removal of extracellular Ca2+ or blocking the L-type voltage-dependent Ca2+ channels, suggesting that the activation of NKCC1 under conditions of high [K+]o is mediated through Ca2+-dependent pathways [108].
Unlike astrocytes and neurons, where NKCC1 is primarily responsible for cytotoxic edema, NKCC1 activation in BBB endothelial cells mediates ionic and vasogenic edema following injuries [113]. Specifically, the luminal-facing NKCC1 facilitates the influx of Na+, which is then pumped out through the abluminal-facing Na+/K+ ATPase [113]. Consistent with in vitro studies, the expression of NKCC1 is upregulated and correlated with cell swelling and brain edema in both ischemic stroke and TBI models in rats [80, 114,115,116]. In addition, both NKCC1−/− and NKCC1+/− mice showed less brain damage following tMCAO, with a ~45% and ~30% reduction, respectively, in infarction volume [110]. Brain edema was also significantly attenuated in both NKCC1−/− and NKCC1+/− mice [110]. Together, these studies suggest that targeting NKCC1 may be beneficial in reducing cerebral edema following TBI and acute ischemic stroke.
The FDA-approved loop diuretic bumetanide is a potent inhibitor of NKCC1 and NKCC2. Low doses of bumetanide can specifically inhibit NKCC1, without producing the diuretic effects associated with NKCC2 inhibition. In animal studies, intravenous administration of bumetanide significantly reduced infarction volume and brain edema in rats after pMCAO [80]. Bumetanide is also protective against TBI in terms of reducing brain contusion and brain edema [115]. These effects of bumetanide are potentially mediated though anti-neuroinflammatory pathways, including the inhibition of TRPV4 and phosphorylation of MEK, ERK, and Akt [115].
Since bumetanide contributes to [Cl−]i and GABAergic regulation, it has been shown to have promising therapeutic effects in neurological disorders related to impaired GABAergic signaling in clinical trials. These neurological disorders include temporal lobe epilepsy, autism spectrum disorder, and Parkinson’s disease. Clinical trials using bumetanide to treat ischemic and TBI-related edema have not yet been performed.
It is worth mentioning that bumetanide does not readily cross the BBB under physiological conditions. This is due to its high ionization (>99%) and plasma protein binding (>95%) properties [117, 118]. The activation of NKCC1 in endothelial cells, in fact, promotes the uptake of systemically administered bumetanide [80, 105]. To improve the brain bioavailability of bumetanide, a series of prodrugs with higher lipophilicity have been recently identified. Compared with other prodrugs, administration of the ester prodrug BUM5 obtained a higher brain concentration of bumetanide [119]. BUM5 was also thought to be more efficacious than bumetanide in rodent models of epilepsy [119, 120]. A novel NKCC1 inhibitor STS66, with high BBB penetration, has been proposed very recently to have a higher efficacy than BUM5 and bumetanide in reducing infarction and cerebral edema after ischemic stroke in mice [121]. Together, these findings suggest that compounds that specifically target NKCC1 with a better BBB penetration property will hold more therapeutic potential in treating brain diseases related to altered Cl− level.
Note that the function of NKCC1 requires ATP indirectly, and thus is mostly involved in early stages of edema development, during which the activity of the Na+/K+ ATPase is still adequate, and the ion gradients are not completely collapsed. In this context, it might be valuable to couple an inhibitor of NKCC1 with a drug that targets the later stages, when ATP is depleted and NKCC1 activity is compromised. The SUR1-TRPM4 channels, as mentioned above, are only transcribed and expressed under injury conditions, suggesting that they may play a more predominant role in the later stages of cerebral edema. Thus, using a combined regimen of bumetanide plus glibenclamide may therefore offer greater clinical outcomes.
KCC
KCCs are another group of cotransporters that are critical for Cl− homeostasis. KCCs were first discovered in red blood cells, where they are entirely responsible for RVD [122]. Four different subtypes of KCCs (KCC1-4) have been identified. Early work described the association between human malignancy and upregulated KCC expression and activity; namely, augmented transport rates were found to accompany increased KCC1, -3, and -4 mRNA levels in cervical cancer cells [123]. This enhanced osmotic sensitivity, supported by KCC-mediated Cl− transport, is consistent with the ability of glioma cells to thrive in the edematous environment they often present with. In the D54-MG cell line as well as in glioma cells from acute patient biopsies, 30%–40% of Cl− efflux during RVD occurs through KCC1 and -3a, primarily within the initial stages of volume regulation. KCC-specific inhibitor, [(dihydroindenyl)oxy] alkanoic acid (DIOA), was observed to inhibit the hypotonic-activated Cl− current and resultantly slow the RVD [124].
Although the KCC family have been detected in glioma, cervical cancer cells and recently, lens epithelial cells, the KCC2 isoform (123.6 kDa) is generally thought to be neuronal-specific [125, 126]. Unlike other subtypes, which are activated only after osmotic stress, KCC2 is distinct in its activation under isotonic conditions.
Structurally, KCC2 consists of 12 TM domains with intracellular N- and C-termini [127]. The C-terminus contains multiple phosphorylation sites, which are critical for KCC2 regulation [127]. Under physiological conditions, KCC2 maintains Cl− homeostasis and cell volume by extruding Cl− along with K+ in the stoichiometry of 1:1 [127, 128]. The direction of ion transport via KCC2 is reversible and is largely driven by the electrochemical gradient of K+ [127, 128]. By maintaining low [Cl−]i, KCC2 ensures the hyperpolarizing inhibitory effects of GABA and glycine by keeping the reversal potential of Cl− below the membrane potential (ECl < Vm) in mature neurons [129].
Impaired KCC2 activity and Cl− imbalance have been associated with several neurological conditions including traumatic and ischemic injuries, epilepsy, neuropathic pain, and schizophrenia [130,131,132]. In excitotoxic injuries, the large increase in extracellular K+ causes a significant reduction in KCC2-mediated Cl− extrusion. In extremely high [K+]o conditions, KCC2-mediated transport of K+ and Cl− may reverse and promote edema formation by facilitating Cl−, K+, and water influx [103].
More importantly, impaired Cl− extrusion could lead to a positive shift in EGABA. The resultant reduction in GABAergic and glycinergic inhibition may initially promote mechanisms of plasticity and recovery, resembling developmental features in the immature brain [128]. However, [Cl−]i overload, as generated in severe injuries, can lead to a depolarizing shift in EGABA. Subsequent Cl− efflux through GABAAR could, instead, generate a large depolarizing GABA current [103, 128]. This switch of GABA from inhibitory to excitatory contributes to hyperexcitability [128]. Thus, it is not surprising that the two common clinical sequelae of ischemic stroke, namely edema and seizures, are related to impaired KCC2 activity and expression as well as the dysregulation of [Cl−]i [103].
Altered KCC2 expression and Cl− homeostasis have been reported in experimental models of ischemic stroke and TBI. Specifically, hippocampal slices subjected to OGD showed a progressive downregulation of KCC2 with the accumulation of [Cl–]i [133]. In a mouse tMCAO model, Jaenisch et al. found a significant postischemic downregulation of KCC2 protein in neurons, and this downregulation was more pronounced in severe (120 min) compared to mild (60 min) ischemia [134]. In a fluid-percussion model of TBI, the expression and function of KCC2 were downregulated, leading to impaired GABAergic inhibition [135]. A recent study using the controlled cortical impact model of TBI in rats also showed decreased mRNA and protein expression of KCC2 in the injured cortex and these changes were associated with cerebral edema and functional deficits [136].
Although not yet well understood, BDNF/TrkB and glutamate-dependent pathways have been proposed to contribute to the downregulation and/or inactivation of KCC2 in acute brain injuries. A recent study using dissociated rat neurons demonstrated that the glutamate-mediated activation of NMDARs and Ca2+ influx led to dephosphorylation of KCC2 at Ser940, causing a significant loss of KCC2 function [137]. NMDAR activation also leads to KCC2 cleavage by the calcium-activated protease calpain [138]. Conversely, inhibiting dephosphorylation of Ser940 or using a calpain inhibitor reduced the glutamate-induced downregulation of KCC2 and improved the maintenance of hyperpolarizing GABAergic inhibition [137]. Multiple studies have also proposed that BDNF is one of the upstream regulators of KCC2; BDNF downregulates KCC2 expression through TrkB-mediated signaling in mature neurons while upregulating KCC2 in immature neurons or injured neurons [129, 139,140,141]. This up-regulatory effect of BDNF on KCC2 in injured neurons seems to be dependent on GABAA-mediated depolarization, and is also speculated to promote mechanisms of repair and recovery [141]. Melatonin has been implicated in restoring KCC2 expression, attenuating brain edema, cell death, and neurological deficits after TBI in rats [136]. These neuroprotective effects of melatonin were mediated partially through the BDNF/ERK pathway, although the detailed mechanisms remain undefined [136].
The contribution of the KCC2 to cell swelling and the mechanisms of KCC2 regulation in acute brain injuries have not yet been elucidated as understanding these mechanisms has proven challenging. KCC2−/− mice die shortly after birth due to respiratory failure or spontaneous generalized seizures [142, 143]. Hypomorphic KCC2-deficient mice also exhibit increased seizure susceptibility and anxiety-like behaviors [144]. Furthermore, compounds like furosemide and bumetanide have been shown to inhibit KCC, however, their inhibitory effects are more potent toward NKCC1 than KCC, especially at low doses [117].
More recent inhibitors of KCC include DIOA, ML077, and VU0463271 [145,146,147]. Among these, VU0463271 has been identified to block KCC2 with the highest potency (IC50 = 61 nM) and display >100-fold selectivity toward KCC vs. NKCC1 [147]. Administration of VU0463271 led to a depolarizing shift in EGABA and hyperexcitability [146]. Consistent with this, silencing KCC2 using shRNA increased [Cl−]i and neuronal susceptibility to excitotoxicity [145]. The effects of KCC2 knockdown or VU0463271 in ischemic stroke and TBI have not yet been addressed. Nevertheless, considering the dual function of KCC2 in both cell volume and GABAergic signaling regulation, pharmacological agents that rescue or activate, rather than inhibit KCC2 function may be a potential therapeutic strategy for attenuating brain edema and hyperexcitability in various neurological disorders.
VRAC
VRACs or volume-sensitive outwardly rectifying anion channels (VSORs) are widely expressed, Cl−-permeable anion channels. VRACs maintain cell volume through RVD, and mediate the swelling-induced Cl− current ICl,swell [148, 149]. Under physiological conditions, RVD via VRACs is accomplished by the efflux of Cl− coupled with K+ and subsequent water efflux, leading to cell shrinkage [149]. Other than cell volume regulation, ICl,swell has been implicated in various cellular functions such as synaptic transmission, cell proliferation, migration, and apoptosis [149].
VRAC is ubiquitously expressed in mammalian tissues. VRAC currents were first reported in 1988 by Cahalan and Lewis in human T lymphocytes and Hazama and Okada in human intestine epithelial cells [150, 151]. Both groups showed that application of a hypotonic solution activated the outward rectifying ICl,swell while the inhibition of ICl,swell coincided with a reduced RVD [150, 151]. In the CNS, VRAC or VRAC-like currents have been reported in neurons, astrocytes, and microglia [149]. Despite decades of extensive research, the molecular nature of VRAC remains ill-defined. Recent studies identified using genome-wide RNAi screening have identified the leucine-rich repeat-containing 8 (LRRC8) proteins as molecular components of VRAC. VRAC was proposed to be a hetero-hexametric complex (~800 kDa) composed of LRRC8A (also known as SWELL1) with at least one other LRRC8 paralogue (LRRC8B-E) [152, 153]. These LRRC8 paralogues form assemblies with distinct biophysical properties. More recent studies suggest that LRRC8A can also assemble into homo-hexametric channels [154, 155]. In fact, knockdown of LRRC8A using siRNA abolished ICl,swell, suggesting that the LRRC8A is indispensable for VRAC-mediated conductance [153]. Point mutations in LRRC8A also caused a significant change in its anion selectivity [152]. In addition to Cl−, VRACs are permeable to other anions such as I− and amino acids, including glutamate, aspartate, and taurine, and other small osmolytes. Besides cell swelling, VRAC activation can be induced isovolumetrically by various cellular factors. Low intracellular ionic strength, for example, directly activates VRAC [156]. Under isotonic conditions, VRAC activation can also be initiated by Na+-overload via glutamate receptors and ATP [157, 158].
In acute ischemic stroke and TBI, overactivation of VRACs due to persistent depolarization results in Cl− influx, rather than efflux as in RVD [148]. Using the OGD model, Zhang et al. showed a progressive increase in ICl,swell amplitude over the period of post-OGD reperfusion due to VRAC activation [157]. The same study also revealed that OGD-induced ICl,swell was significantly attenuated by inhibitors of both AMPAR (NBQX) and NMDAR (AP-5), suggesting that the activation of VRAC and the induction of ICl,swell require excessive Na+-loading via these glutamate receptors [157].
Upon activation, VRACs can mediate the release of EAAs such as glutamate and aspartate, and can modulate neuronal excitability by changing Cl− permeability independent of the GABAA receptors [149, 159]. An important pathophysiological implication in ischemic or traumatic injuries is that astrocytes at the site of injury rapidly swell, activate VRACs and release EAAs [148, 160]. Although the permeability to glutamate is not as high as to Cl−, VRAC-mediated EAA release is sufficient to evoke depolarization and increase [Ca2+]i in adjacent neurons, exacerbating excitotoxic cell death [160]. Consistently, a recent in vivo study demonstrated that astrocyte-specific LRRC8A knockout mice were protected after tMCAO, with reduced glutamate-dependent neuronal excitability [158].
Studies showed that ATP released from damaged or swollen neurons and astrocytes significantly increased VRAC-mediated EAA release from astrocytes [161]. Since the activation of VRACs in astrocytes is dependent on exogenous ATP, the VRAC-mediated EAA release was more profound in the penumbra, where neuronal intracellular ATP level remains high [159].
VRAC inhibitors, including tamoxifen and the more selective ethacrynic acid derivative, DCPIB, have shown neuroprotection against ischemic stroke [162, 163]. DCPIB in vitro decreased neuronal intracellular Cl− concentration induced by low ionic strength and abolished OGD-induced cell death [38, 157]. DCPIB also significantly reduced infarction volume and improved neurobehavioral deficits in vivo in a rat tMCAO model [162]. These neuroprotective effects were elicited through the inhibition of VRAC-mediated EAA release and Cl− influx [159, 162]. DCPIB has been previously defined as a potent and selective VRAC inhibitor and does not inhibit other Cl− channels (CFTR, ClC, CaCC) at concentration ranges used for VRAC. A more recent study, however, suggested that DCPIB may also target other glutamate transporters pathways, such as GLT-1 and connexin hemichannel in glial cells [163]. Therefore, the in vivo effect of DCPIB on reducing EAA release may not be solely attributed to its action on VRACs. Further research on characterizing the specific mechanisms of DCPIB action is required. Nevertheless, these studies concluded that targeting VRAC may provide a new therapeutic approach to cerebral edema following acute brain injuries.
TMEM16A
TMEM16A, the first of ten members of the Anoctamin or TM protein 16 family, was first reported in 2008 by three independent groups to confer Ca2+-activated Cl− currents [164,165,166]. TMEM16A and close paralog TMEM16B, have since been identified to function as calcium-activated chloride channels (CaCCs) with differing activation and deactivation kinetics and [Ca2+] activation thresholds that suggest a prominent role for the latter in native olfactory sensory neuron CaCC activity [167]. Meanwhile, TMEM16A expression coincides with that of endogenous CaCCs including secretory epithelial cells, airway and vascular smooth muscle cells, sensory neurons, and various endothelial cell types [164]. Accordingly, under physiologic conditions, anion permeation via TMEM16A regulates mucus secretion, smooth muscle contraction, gut mobility, and RVD [164, 168].
Homology modeling of fungal homolog, nhTMEM16, and recent single-particle cryoEM of TMEM16A revealed the homodimeric structure consisting of 10 TM domains, two independently activated ion conduction pores as well as highly conserved Ca2+ binding sites and pore-lining residues critical for Cl− selectivity and channel gating [169,170,171]. As a CaCC, TMEM16A exhibits both Vm and [Ca2+]i gating properties; at low [Ca2+]i the current is largely voltage-dependent and outwardly rectifying, however as [Ca2+]i increases, Ca2+ alone may induce a strong Cl− current characterized by a linear I–V relationship [168, 172]. Extracellular Cl− has also been suggested to stabilize the open conformation, and allosterically modulate Ca2+ sensitivity of TMEM16A [172].
In endothelial cells, pathological- or agonist-induced high cytosolic Ca2+ activates TMEM16A, leading to Cl− efflux and cell volume modulation [173]. However, isovolumetric conditions may also activate CaCCs, implicating TMEM16A in the cell shrinkage that initiates apoptotic volume decrease [174]. Interestingly, Almaça et al. showed that through an ATP-purinergic P2Y receptor binding mechanism and ERK1/2 activation, TMEM16A contributes to swelling-activated Cl− conductance. siRNA-TMEM16A was found to suppress ICl,swell and RVD in three separate cell lines under increased extracellular hypotonicity, while TMEM16A overexpression augmented swelling-activated Cl− currents. Furthermore, TMEM16A inhibitor, CACCinh-A01, was observed to significantly reduce ICl,swell to an extent comparable to both tamoxifen and nonspecific Cl− channel blocker, DIDS. ICl,swell was also nearly abolished in epithelial cells of TMEM16A−/− mice [175].
Although significant progress in understanding the molecular and structural underpinnings of TMEM16A function has been made, the precise interaction mechanisms of modulators have yet to be clearly addressed [168]. Much of recent literature has explored the therapeutic effects of TMEM16A inhibitors on cancer cell lines given the role of TMEM16A-activated signal transduction on cancer cell proliferation and migration – TMEM16A overexpression is commonly accounted for by gene amplification in malignant tumors, and cell-specific signaling pathways in non-tumor cells under pathological conditions [174, 176]. In addition, TMEM16A dysfunction has been associated with CF, hypertension, and more recently, stroke [176].
TMEM16A was recently proposed to contribute to ischemic damage after being identified in cardiac vascular endothelial cells of mice; TMEM16A-mediated ICl(Ca) was found to be potentiated under hypoxic conditions via increased protein expression levels, and enhanced channel sensitivity to [Ca2+]i and membrane depolarization [173]. Moreover, TMEM16A was shown to play a role in regulating BBB permeability following ischemic stroke. tMCAO of adult mice resulted in a marked upregulation of TMEM16A, with particular abundance in brain epithelial cells. CaCCinh-A01, TMEM16A antagonist, attenuated brain infarction and functional deficits, and preserved BBB integrity by preventing the loss of tight junction proteins, and downregulating ICAM-1 as well as neutrophil adhesion and infiltration [177]. In vitro, silencing of TMEM16A was also found to rescue OGD-induced transendothelial permeability by suppressing the NF-κB-ICAM-1 signaling pathway [177]. It is important to note that T16ainh-A01, another well-documented TMEM16A inhibitor, did not yield the same protective effects; CaCCinh-A01 is distinct in its ability to both inhibit channel activity as well as reduce TMEM16A proteasomal turnover, while T16ainh-A01 has no effect on channel expression levels [178]. Moreover, MONNA is another compound that exerts inhibitory effects on TMEM16A-mediated ICl(Ca). While ample evidence supports the antagonizing effects of these small molecule inhibitors, recent literature has demonstrated that T16ainh-A01, CaCCinh-A01, and MONNA were still able to induce vasorelaxation in rodent resistance vessels under Cl−-free conditions, thereby challenging their selectivity for TMEM16A Cl− conductance [179].
While the role of TMEM16A in cerebral edema following acute brain injury remains elusive, its contribution to both ICl,swell and ICl(Ca) combined with the effects of TMEM16A inhibition on preventing BBB leakage has profound implications for acute stroke patients and warrant further research [180].
CFTR
CFTR is a cAMP-activated Cl−-selective channel belonging to the superfamily of ABC transporters. It is unique amongst other ABCs in that it functions as an ion channel [181]. CFTR is a large protein (180 kDa), consisting of 12 TM domains, 2 cytosolic nucleotide-binding domains (NBDs), and a regulatory (R) domain [181]. The NBDs are responsible for ATP hydrolysis, and phosphorylation of the R domain is required for channel opening, which leads to bidirectional Cl− transport down its electrochemical gradient [181]. Besides Cl−, CFTR is permeable to bicarbonate, thiocyanate, and other anions.
Although the brain is not traditionally viewed as an affected organ in CF, patients with CF who underwent lung transplants are associated with a higher risk of developing neurologic complications, such as seizure and stroke, among other lung transplant recipients [182, 183]. In the CNS, CFTR is mainly expressed in neurons, but not in glial cells [181]. A study by Zhang et al. showed that CFTR acted to prevent neuronal apoptosis following cerebral ischemia-reperfusion in mice [184]. Stimulating CFTR with its activator forskolin suppressed apoptosis whereas silencing CFTR by siRNA enhanced apoptosis [184]. These effects of CFTR were thought to be mediated by regulating oxidative stress and glutathione in mitochondria-dependent pathways.
The role of CFTR in edema formation is primarily studied in the lung, where CFTR regulates the secretion and absorption of alveolar fluid. Specifically, CFTR expression was upregulated in pulmonary edema, whereas inhibition of CFTR decreased alveolar fluid secretion [185]. Consistent with this, CFTR−/− mice were protected from pulmonary edema [185]. In the CNS, the upregulation of CFTR mRNA and protein has been associated with infection-induced cerebral edema through unknown mechanism [186]. A very recent study has also established a strong relationship between cerebral artery CFTR expression and impaired cerebrovascular reactivity as well as perfusion in mouse models of heart failure and subarachnoid hemorrhage [187].
High-affinity inhibitors as well as potentiators and correctors of CFTR have been largely identified via high-throughput screening in the last two decades. The CFTR potentiator VX-770/ivacaftor has been shown to be highly effective in restoring channel activity and has been approved for treating patients with a variety of CFTR mutations [188]. To rescue the specific F508del mutation, CFTR correctors, such as VX-809/lumacaftor, have been discovered to improve folding and stability of CFTR at the cell surface [188].
Regarding CFTR inhibitors, the thiazolidinone CFTRinh-172 (IC50 = 300 nM) and the glycine hydrazide GlyH101 (IC50 = 5 μM), both identified by Verkman and colleagues in the early 2000s, are mostly studied [189, 190]. Both CFTRinh-172 and GlyH101 have been shown to significantly reduce alveolar Cl− and fluid secretion in different animal models of pulmonary edema [185, 191]. CFTRinh-172 has also been implicated in blocking toxin-induced intestinal Cl− and fluid secretion [190, 192]. A third class of CFTR inhibitors, the pyrimido-pyrrolo-quinaxolinediones, including PPQ-102 (IC50 = 90 nM) and its related compound BPO-27 (IC50 = 8 nM) were later discovered by the same group, with improved potency, solubility, and stability [193].
Recent comparative studies, however, suggested that GlyH101 and PPQ-102 are not CFTR-selective. At concentration ranges used to inhibit CFTR, the VRAC- and CaCC-mediated Cl− conductance can also be inhibited by these two compounds [194, 195]. The most-selective CFTR inhibitor to date is CFTRinh-172 as it does not affect CaCC and only exerts minor inhibition on VRAC conductance even with high concentrations [194].
While CFTR is capable of driving edema formation, further research is required to investigate its functional role in the context of cerebral edema following acute brain injuries. The effects of selective and potent CFTR inhibitors in managing cerebral edema also remain to be evaluated.
ClCs
Nine members of the ClC family, facilitating voltage-dependent Cl− transport, have been identified. While the ClC family share a dimeric structure and Cl− gradient dependency, ClC-1, -2, -Ka, and -Kb function as plasma membrane Cl− channels, while ClC-3 to -7 operate as Cl−/H+ exchangers localized to intracellular compartments [196, 197]. Several members of the ClC family have been proposed as viable candidates for cell volume regulation and osmotic homeostasis, primarily ClC-2 and -3.
ClC-2 are widely expressed in mammalian tissue and activated upon hyperpolarization and mild decreases in extracellular pH levels [196]. Although the inwardly rectifying Cl− conductance and anion selectivity characteristic of ClC-2 are distinct from typical ICl,swell, ClC-2 was demonstrated to contribute to volume-sensitive changes of hepatoma tissue culture (HTC) cells. Microinjection of HTC cells with ClC-2 antibodies inhibited volume-activated currents and delayed hypotonic-induced cell volume recovery [198]. Furthermore, administration of DIDS, to which ClC-2 is largely resistant, was found to block ~40% of volume-activated currents, implicating the involvement of ClC-2 in the remaining 60% [198]. The presumptive role of ClC-2 in regulating cerebral ion homeostasis is furthermore illustrated by the proposition of CLCN2, encoding ClC-2, as a candidate gene for leukodystrophy. ClC-2−/− mice were found to display CNS myelin vacuolation accompanied by enhanced microglial activation and impaired BBB, hypothesized to result from disrupted ion homeostasis of the astrocytic/oligodendrocytic network [199]. The pathology of ClC-2−/− mice resembles human leukodystrophy and although controversial, evidence does suggest that CLCN2 mutations are associated with leukoencephalopathies characterized by intramyelinic edema [200]. An observational analytical study found that CLCN2 mutations leading to ClC-2 loss-of-function occurred in 6/7 patients that presented with such disease criteria. Interestingly, healthy controls exhibited abundant ClC-2 membrane expression in the panglial syncytium, a network of glial cells specialized for osmotic homeostasis in myelinated axons [200].
Several Cl− currents including ICl,swell, have been attributed to ClC-3 [201, 202]. Early studies found that use of ClC-3 antisense oligonucleotide or ClC-3 antibody inhibited VRAC currents in multiple cell types, thereby identifying ClC-3 as a molecular component of volume-regulated cation channel (VRCC) regulation [201, 203]. As a VRCC, ClC-3 has been associated with proliferation and apoptosis in rodent vascular smooth muscle cells, both of which are associated with cerebrovascular remodeling in cardiovascular disease such as stroke [204]. However, the contribution of ClC-3 to ICl,swell has remained inconsistent partially due to the lack of a selective inhibitor [202]. Nevertheless, ClC-3 along with ClC-2, -5, -6, and -7 were recently proposed as the repertoire of Cl− channels contributing to RVD in human glioma cells; under changing osmolarities, glioma cells can notably regulate their volume, at times recovering to a volume lower than baseline levels [124]. Five members of the ClC family were identified as the candidates accounting for 60%–70% of the Cl− conductance during RVD. 5-nitro-2(3-phenylpropylamino)benzoic acid was shown to act synergistically with Cd2+ to inhibit ClC-mediated conductance, which corresponds with the respective sensitivities of ClC-3 and ClC-2 to these inhibitors [124].
The ClC family, particularly ClC-2 and -3, are notable candidate proteins for regulating ion homeostasis and swelling-induced RVD, and incite future study into their potential role in cerebral edema following acute injury such as stroke and TBI.
NCX
The sodium–calcium exchanger (NCX) is a bidirectional ion channel that maintains intracellular [Na+] and [Ca2+] homeostasis. All three mammalian isoforms of NCX (NCX1, NCX2, and NCX3) are expressed in neurons and glial cells, and are associated with various cellular functions such as neurotransmitter release and myelin formation [205]. Structurally, eukaryotic NCX consists of 10 TM domains with a large intracellular regulatory loop between TM5 and TM6 [206]. This loop includes two regulatory Ca2+-binding domains, CBD1 and CBD2, which enable the dynamic regulation of [Ca2+]i [206, 207].
The modes of ion transport via NCX depend on the electrochemical gradients of Na+ and Ca2+. Under physiological conditions, NCX operates in its forward mode. It maintains a low level of intracellular Ca2+ by extruding one Ca2+ in exchange with three Na+, with an electrogenic current [208]. Changes in [Na+]i are then balanced through Na+/K+ ATPase. In this forward mode, either an increase in [Na+]i or a decrease in [Ca2+]i leads to NCX inactivation and decreased NCX current [209]. The reverse mode of NCX, which mediates Na+ efflux and Ca2+ influx, has been associated with CNS injuries. In addition to the 3Na+:1Ca2+ transport ratio, two minor modes of NCX transport have also been proposed to operate at low rates [210].
In acute ischemic stroke, the mode of operation of NCX differs at the ischemic core and the penumbra. NCX operates in its forward mode to extrude Ca2+ in the ischemic penumbra, where ATPase activity remains high and is still able to counterbalance the influx of Na+ [211]. In contrast, ATPase activity is often compromised at the ischemic core. Na+ overload, in addition to mediating cytotoxic edema, increases Em. When Em exceeds ENCX, NCX will shift to its reverse mode [212]. This reverse mode of NCX operation has also been shown in vitro after severe, but not mild or moderate TBI [213]. Reversal of NCX can result in two opposing effects in the injury core; it increases Ca2+-dependent cytotoxicity and decreases Na+-dependent cell swelling [212, 214]. However, the net effect and the precise mechanism of NCX working in its reverse mode are still debatable. The expression and operation mode of the three NCX subtypes in permanent vs. transient ischemic injury also seems to be varied, which adds another level of complexity to the current understanding. Both NCX3−/− and NCX2−/− mice showed increased infarction volume in tMCAO models, suggesting their potentially beneficial role in ischemia/reperfusion states, during which they are likely to operate in the forward mode to reduce intracellular Ca2+ [215, 216]. The beneficial effect of NCX, however, has not been observed in NCX3−/− mice subjected to pMCAO [217].
A variety of compounds have been identified to inhibit NCX activity [211]. Among these, KB-R7943 and SEA0400 have been studied in experimental models of stroke [218, 219]. Compared with KB-R7943, SEA0400 is more potent and selective since many other Ca2+-dependent pathways can also be altered by KB-R7943 [219, 220]. SEA0400 is thought to inhibit the reverse mode of NCX, and primarily inhibits NCX1, with a low affinity for NCX2 and no inhibitory effect on NCX3 [221].
Whereas NCX1 might be protective in pMCAO, pharmacological studies using SEA0400 suggested that NCX1, unlike NCX2 and NCX3, may aggravate brain injury in ischemia/reperfusion states [214, 222]. Specifically, SEA0400 inhibited the Na+-dependent Ca2+ uptake in vitro and reduced infarction volume in vivo after tMCAO [219]. SEA0400 also attenuated vasogenic edema and BBB disruption in rats after radiofrequency lesion [222]. These findings are challenged by a more recent study by Molinaro et al., where selective and conditional NCX1 knockout animals showed increased neuronal damage after tMCAO [223]. Consistently, both overexpression of NCX1 and administration of neurounina-1, a novel compound that increases NCX1 and NCX2 activities, reduced infarction [223, 224].
To date, the understanding of NCX function in mediating brain edema in acute brain injuries is limited, and subtype-specific NCX inhibitors are not yet available. Knockout studies, however, provide a “proof of principle” that inhibition of NCX2 and NCX3 might be beneficial in ischemic/reperfusion injuries. Considering the dynamic modes of NCX operation in brain injuries and the variable roles carried out by each NCX subtype, targeting NCX using pharmacological agents should consider controlling the site and timing of administration.
Glucose transporters (GLUTs)
Glucose is the major source of energy for the brain, and a continuous supply of glucose is critical for maintaining neuronal function. Delivery of glucose from the blood to the brain is mediated by specific GLUTs expressed in BBB endothelial cells, neurons, and glial cells. Once transported into the brain, glucose can be directly utilized by the neurons or converted into lactate by the astrocytes before importing into the neurons [225, 226].
GLUTs can be classified into two families, namely, the Na+-independent or facilitated GLUTs, and the Na+-dependent or Na+-glucose cotransporters (SGLT) [227]. The GLUT family consists of 12 TMDs and facilitates the transport of glucose down its concentration gradient. Among the 14 members of GLUT, GLUT1 and GLUT3 are the major isotypes found in CNS [228]. The glycosylated form (55 kDa) of GLUT1 is highly expressed on both luminal and abluminal membranes of the endothelial cells, being the predominant GLUT at the BBB [227, 229]. The less glycosylated form of GLUT1 (45 kDa) and GLUT3 is respectively expressed in astrocytes and neurons [227]. The SGLT family consists of 14 TMDs [228]. There are 12 known human SGLT (SLC5) gene family members, 6 of them (SGLT1 to -6) have been identified to transport or sense monosaccharides. SGLT1 and SGLT2 are the most studied isotypes with a particular focus on their roles in mediating intestinal and renal glucose reabsorption [228]. In contrast to GLUTs, SGLTs transport glucose against its concentration gradient along with Na+. The stoichiometric ratios of Na+ to glucose through SGLT1 and SGLT2 are 2:1 and 1:1, respectively [228]. In the CNS, SGLT1 is mainly expressed in neurons. Functional expression of SGLT2 has been detected in several regions of the rat brain, however, the precise distribution of SGLT2 and other SGLTs is currently unknown [230]. Although the presence of SGLT in BBB endothelial cells has been speculated in vitro, in vivo studies suggested an absence of SGLT-mediated glucose transport at the BBB under physiological conditions [230,231,232].
Enhanced gene or protein expression of both GLUT1 and SGLT1 at the BBB has been implicated in experimental models of ischemic stroke and TBI [231, 233, 234]. Consistent with these findings, brain imaging studies have suggested elevated glucose uptake and glucose hypermetabolism during ischemic stroke, especially in peri-infarcted regions [234, 235]. Due to the high abundance of GLUTs at the BBB, they are thought to represent a major route for transendothelial water flux during ionic edema, as they have the ability to passively transport water, analogously to the AQPs [13, 14]. In fact, mice pretreated with SGLT inhibitor, phlorizin, showed reduced infarction volume and brain edema after pMCAO [231]. Downregulating SGLT1 was also shown to be beneficial in TBI in terms of reducing cerebral edema and contusion volume [233]. These protective effects of SGLT inhibition are potentially due to reduced Na+ and water transport through the endothelial cell, as well as reduced acidosis and free radical production from the anaerobic metabolism of glucose [231].
In human studies, high blood glucose level has been described as an important predictor for early cerebral edema in patients with ischemic stroke, although the reasons for this phenomenon remained unknown [236, 237]. A recent postmortem study that examined 15 TBI cases found that the expression of SGLT1 and SGLT2 was significantly higher in contusional tissue, compared to that of the contralateral uninjured tissue or healthy brain tissue [238]. SGLT1 and SGLT2 in contusional regions also exhibit a biphasic pattern of increase that peaked at 72-h post injury, which may correlate with delayed secondary injuries after TBI [238].
Collectively, these findings suggest a combined role for both SGLT and GLUT in mediating glucose and water transport at the BBB under ischemic conditions [231]. To date, pharmacological inhibitors of GLUTs are extensively studied in T2DM, and multiple SGLT inhibitors have been approved by the FDA in treating diabetes. Our current understanding of brain glucose transport in acute brain injuries, however, is still limited, thereby warranting future investigations to elucidate the pathophysiological roles brain GLUTs play.
Proton-sensitive channels/exchangers
ASIC
ASICs are a class of ligand-gated, voltage-insensitive cation channels that belong to the epithelial sodium channel/degenerin family. ASICs are widely expressed in the CNS and activated by extracellular acidosis. Six subunits of ASIC (ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3, and ASIC4) have been identified, with ASIC1a having the highest proton sensitivity. Each subunit consists of 2 TM domains, a cysteine-rich extracellular loop, and intracellular N- and C-termini. The subunits may assemble into homomer or heterotrimeric complexes, with the homotrimeric ASIC1a and heteromeric ASIC2b/1a predominantly expressed [239, 240]. ASICs are generally more permeable to Na+, and to a lesser degree to Ca2+ and K+ [241]. Under physiological conditions, ASICs are noted for their roles in synaptic plasticity, nociception, and mechanoception [242, 243]. ASICs have also been noted in ischemic stroke, multiple sclerosis, and epilepsy [13].
Extracellular acidosis in both ischemic stroke and TBI worsens secondary brain damage. With normal extracellular pH maintained around 7.3, ischemia and TBI typically induce a reduction of pH to 6.3 and 6.7, respectively, or even lower depending on the severity of the insult [243]. The build up of lactic acid rising from 1 to 15 mM due to anaerobic metabolism drives the activation of ASICs via the ASIC1a subunit [244]. ASICs-mediated Na+ influx leads to cell swelling and depolarization [13]. In severe acidosis conditions, hyperactive ASICs can also contribute to excessive Ca2+, aggravating the neuronal injury in a glutamate receptor-independent manner [37].
While ASICs have not been explicitly found to affect fluid buildup in the brain following acute brain injuries, inhibiting ASIC1 has been noted to be neuroprotective following cerebral ischemia [13]. Compounds such as nonspecific inhibitor amiloride or ASIC1a-specific inhibitor psalmotoxin-1 (PcTx1) reduced acidosis and OGD-induced neuronal cell death in vitro, while potentiating ASICs activity has been shown to exacerbate acidosis-mediated neuronal injury [37, 245, 246]. ASICs inhibitors or knockout of ASIC1a were also found to substantially reduce infarction volume after tMCAO [37, 247, 248]. In experimental models of TBI, the severity of functional deficits was reduced in ASIC1−/− mice compared to wild-type mice [242]. Even in the presence of cerebral acidosis in the brain, ASIC1−/− mimicked the effects of neutralization with HCO3− [242]. In fact, amiloride has been suggested to exert more protective effects in attenuating hippocampal damage than other more specific NCX or sodium hydrogen exchanger (NHE) inhibitors in a rat TBI model [249]. Taken together, the inhibition of ASIC subunits in the CNS may prove to be a therapeutic tool in addressing the acidosis-mediated Ca2+ overload that exacerbates the effects of secondary injury.
NHE
NHEs are another major group of pH-regulated channels. Under physiological conditions, NHEs regulate pHi by the extrusion of 1 H+ in exchange of 1 Na+ [243]. In addition to pH regulation, NHEs also mediate RVI in osmotically shrunken cells by driving Na+ and water influx [243]. Structurally, NHEs consist of 12 TM segments, with intracellular N- and C-termini. Among the ten identified NHEs, NHE1 is the most abundantly expressed isoform in the CNS and is expressed in neurons, astrocytes, the luminal membrane of the BBB endothelial cells, and microglia [243].
Enhanced NHE1 activity has been associated with cerebral edema in ischemic stroke. In neurons and astrocytes, NHE1 activation contributes to Na+ overload and cytotoxic edema after OGD [250, 251]. This NHE1-mediated swelling is significantly reduced in NHE1−/− astrocytes [250]. A more recent study showed that selective knockout of astrocytic NHE1 reduced cerebral edema, infarction volume, and functional deficits after tMCAO [252]. NHE1−/− mice also exhibited reduced astrogliosis and BBB breakdown, related to a reduced MMP-9 level [252]. NHE1 activity in BBB endothelial cells can also be stimulated by ischemic conditions to facilitate the influx of Na+ from the vasculature [253].
Two classes of pharmacological agents are known to inhibit NHE activity and have shown beneficial effects in experimental models of ischemic stroke [243]. The amiloride and its analogs dimethylamiloride and ethylisopropylamiloride inhibit NHEs activity in a nonselective manner [243]. However, as previously mentioned, amiloride is a nonspecific inhibitor as it also exerts inhibitory effects on ASICs and NCX [243]. Guanidine derivatives, including HOE-694, HOE-642 (cariporide), EMD-85131 (eniporide), and SM-20220, contribute to the second, and more selective class of NHE inhibitors [243]. Among all the guanidine derivatives, HOE-642 is the most selective and potent inhibitor of NHE1 and has demonstrated neuroprotection in reducing Na+ uptake and cerebral edema in both pMCAO and tMCAO models [243, 252, 253]. Furthermore, animals treated with a combination of HOE-642 and the NKCC1 inhibitor bumetanide, showed further reduction in cerebral edema and infarction volume in the initial hours following stroke onset compared to those treated with HOE-642 or bumetanide alone [253]. Based on current findings, NHE1 seems to be a promising target for attenuating cerebral edema in ischemic stroke. The involvement of NHE1 in TBI and the roles of other brain NHEs constitute an important direction for future studies.
Summary
Brain edema is a complicated pathophysiological process that involves a variety of ion channels and transporters. Ion conduction of the same ion channel or transporter may also be altered at different stages of the edema. To date, the understanding of edema formation and propagation is limited. The discovery of effective anti-edematous drugs is challenging due to the dynamic nature of the process. As each ion channel and transporter has its distinct role in mediating brain edema, combinational therapeutic approaches targeting multiple ion channels or transporters may maximize the efficacy of anti-edematous therapy. The discovery of selective ion channel inhibitors thus constitutes a critical direction for future studies. Furthermore, continued investigations of the cellular and molecular mechanisms related to ion channel-mediated edema in acute brain injuries are necessary in order to yield novel and effective therapeutic strategies.
References
Donkin JJ, Vink R. Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol. 2010;23:293–9.
Klatzo I. Pathophysiological aspects of brain edema. Acta Neuropathol. 1987;72:236–9.
Rungta RL, Choi HB, Tyson JR, Malik A, Dissing-Olesen L, Lin PJC, et al. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. 2015;161:610–21.
Berrouschot J, Sterker M, Bettin S, Köster J, Schneider D. Mortality of space-occupying (‘malignant’) middle cerebral artery infarction under conservative intensive care. Intensive Care Med. 1998;24:620–3.
Hacke W, Schwab S, Horn M, Spranger M, De Georgia M, von Kummer R. ‘Malignant’ middle cerebral artery territory infarction: clinical course and prognostic signs. Arch Neurol. 1996;53:309–15.
Hill MD, Hachinski V. Stroke treatment: time is brain. Lancet. 1998;352:SIII10–4.
Ayata C, Ropper AH. Ischaemic brain oedema. J Clin Neurosci. 2002;9:113–24.
Thrane AS, Thrane VR, Nedergaard M. Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014;37:620–8.
Hinson HE, Stein D, Sheth KN. Hypertonic saline and mannitol therapy in critical care neurology. J Intensive Care Med. 2013;28:3–11.
Ziai WC, Toung TJK, Bhardwaj A. Hypertonic saline: first-line therapy for cerebral edema? J Neurol Sci. 2007;261:157–66.
Polin RS, Shaffrey ME, Bogaev CA, Tisdale N, Germanson T, Bocchicchio B, et al. Decompressive bifrontal craniectomy in the treatment of severe refractory posttraumatic cerebral edema. Neurosurgery. 1997;41:84–92.
Moritz ML, Ayus JC. Hyperosmolar therapy for raised intracranial pressure. N Engl J Med. 2012;367:2555–6.
Jha RM, Kochanek PM, Simard JM. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology. 2019;145:230–46.
Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36:513–38.
Liang D, Bhatta S, Gerzanich V, Simard JM. Cytotoxic edema: mechanisms of pathological cell swelling. Neurosurg Focus. 2007;22:E2.
Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–68.
Wang Y, Hu W, Perez-Trepichio AD, Ng TC, Furlan AJ, Majors AW, et al. Brain tissue sodium is a ticking clock telling time after arterial occlusion in rat focal cerebral ischemia. Stroke. 2000;31:1386–91.
Ito U, Ohno K, Nakamura R, Suganuma F, Inaba Y. Brain edema during ischemia and after restoration of blood flow: Measurement of water, sodium, potassium content and plasma protein permeability. Stroke. 1979;10:542–7.
Mestre H, Du T, Sweeney AM, Liu G, Samson AJ, Peng W, et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science. 2020;367:eaax7171.
Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327–34.
Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:2010–30.
Gerstner ER, Duda DG, di Tomaso E, Ryg PA, Loeffler JS, Sorensen AG, et al. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6:229–36.
Kovács Z, Ikezaki K, Samoto K, Inamura T, Fukui M. VEGF and flt: expression time kinetics in rat brain infarct. Stroke. 1996;27:1865–72.
Aslam M, Ahmad N, Srivastava R, Hemmer B. TNF-alpha induced NFκB signaling and p65 (RelA) overexpression repress Cldn5 promoter in mouse brain endothelial cells. Cytokine. 2012;57:269–75.
Masada T, Hua Y, Xi G, Yang GY, Hoff JT, Keep RF. Attenuation of intracerebral hemorrhage and thrombin-induced brain edema by overexpression of interleukin-1 receptor antagonist. J Neurosurg. 2009;95:680–6.
Donkin JJ, Turner RJ, Hassan I, Vink R. Substance P in traumatic brain injury. Prog Brain Res. 2007;161:97–109.
Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22:E1.
Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–88.
Guerriero RM, Giza CC, Rotenberg A. Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep. 2015;15:27. https://doi.org/10.1007/s11910-015-0545-1.
Leis JA, Bekar LK, Walz W. Potassium homeostasis in the ischemic brain. Glia. 2005;50:407–16.
Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–75.
Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:1–7.
Tymianski M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci. 2011;14:1369–73.
Sun HS, Feng ZP, Barber PA, Buchan AM, French RJ. Kir6.2-containing ATP-sensitive potassium channels protect cortical neurons from ischemic/anoxic injury in vitro and in vivo. Neuroscience. 2007;144:1509–15.
Sun HS, Feng ZP, Miki T, Seino S, French RJ. Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol. 2006;95:2590–601.
Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci. 2009;12:1300–7.
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98.
Wong R, Abussaud A, Leung JW, Xu BF, Li FY, Huang S, et al. Blockade of the swelling-induced chloride current attenuates the mouse neonatal hypoxic-ischemic brain injury in vivo. Acta Pharmacol Sin. 2018;39:858–65.
Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–7.
Thompson RJ, Jackson MF, Olah ME, Rungta RL, Hines DJ, Beazely MA, et al. Activation of pannexin-1 hemichannels augments aberrant bursting in the hippocampus. Science. 2008;322:1555–9.
Yang B. Aquaporins. Advances in experimental medicine and biology book series 969. Dordrecht: Springer Nature; 2017.
Amiry-Moghaddam M, Ottersen OP. The molecular basis of water transport in the brain. Nat Rev Neurosci. 2003;4:991–1001.
Frydenlund DS, Bhardwaj A, Otsuka T, Mylonakou MN, Yasumura T, Davidson KGV, et al. Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc Natl Acad Sci USA. 2006;103:13532–6.
Steiner E, Enzmann GU, Lin S, Ghavampour S, Hannocks MJ, Zuber B, et al. Loss of astrocyte polarization upon transient focal brain ischemia as a possible mechanism to counteract early edema formation. Glia. 2012;60:646–59.
Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, et al. Aquaporin water channels—from atomic structure to clinical medicine. J Physiol. 2002;542:3–16.
Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex. 2007;17:787–802.
Fukuda AM, Pop V, Spagnoli D, Ashwal S, Obenaus A, Badaut J. Delayed increase of astrocytic aquaporin 4 after juvenile traumatic brain injury: possible role in edema resolution? Neuroscience. 2012;222:366–78.
Taniguchi M, Yamashita T, Kumura E, Tamatani M, Kobayashi A, Yokawa T, et al. Induction of aquaporin-4 water channel mRNA after focal cerebral ischemia in rat. Brain Res Mol Brain Res. 2000;78:131–7.
Ke C, Poon WS, Ng HK, Pang JCS, Chan Y. Heterogeneous responses of aquaporin-4 in oedema formation in a replicated severe traumatic brain injury model in rats. Neurosci Lett. 2001;301:21–4.
de Castro Ribeiro M, Hirt L, Bogousslavsky J, Regli L, Badaut J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J Neurosci Res. 2006;83:1231–40.
Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–63.
Yao X, Derugin N, Manley GT, Verkman AS. Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci Lett. 2015;584:368–72.
Hirt L, Fukuda AM, Ambadipudi K, Rashid F, Binder D, Verkman A, et al. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J Cereb Blood Flow Metab. 2017;37:277–90.
Akdemir G, Ratelade J, Asavapanumas N, Verkman AS. Neuroprotective effect of aquaporin-4 deficiency in a mouse model of severe global cerebral ischemia produced by transient 4-vessel occlusion. Neurosci Lett. 2014;574:70–5.
Stokum JA, Mehta RI, Ivanova S, Yu E, Gerzanich V, Simard JM. Heterogeneity of aquaporin-4 localization and expression after focal cerebral ischemia underlies differences in white versus grey matter swelling. Acta Neuropathol Commun. 2015;6:61.
Sato S, Umenishi F, Inamasu G, Sato M, Ishikawa M, Nishizawa M, et al. Expression of water channel mRNA following cerebral ischemia. Acta Neurochir Suppl. 2000;76:239–41.
Shi WZ, Qi LL, Fang SH, Lu YB, Zhang WP, Wei EQ. Aggravated chronic brain injury after focal cerebral ischemia in aquaporin-4-deficient mice. Neurosci Lett. 2012;520:121–5.
Liang F, Luo C, Xu G, Su F, He X, Long S, et al. Deletion of aquaporin-4 is neuroprotective during the acute stage of micro traumatic brain injury in mice. Neurosci Lett. 2015;598:29–35.
Yao X, Uchida K, Papadopoulos MC, Zador Z, Manley GT, Verkman AS. Mildly reduced brain swelling and improved neurological outcome in aquaporin-4 knockout mice following controlled cortical impact brain injury. J Neurotrauma. 2015;32:1458–64.
Fukuda AM, Adami A, Pop V, Bellone JA, Coats JS, Hartman RE, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab. 2013;33:1621–32.
Kiening KL, van Landeghem FKH, Schreiber S, Thomale UW, von Deimling A, Unterberg AW, et al. Decreased hemispheric Aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci Lett. 2002;324:105–8.
Aoki K, Uchihara T, Tsuchiya K, Nakamura A, Ikeda K, Wakayama Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003;106:121–4.
Hu H, Yao HT, Zhang WP, Zhang L, Ding W, Zhang SH, et al. Increased expression of aquaporin-4 in human traumatic brain injury and brain tumors. J Zhejiang Univ Sci B. 2005;6:33–7.
Lo Pizzo M, Schiera G, Di Liegro I, Di Liegro CM, Pál J, Czeiter E, et al. Aquaporin-4 distribution in control and stressed astrocytes in culture and in the cerebrospinal fluid of patients with traumatic brain injuries. Neurol Sci. 2013;34:1309–14.
Verkman AS, Binder DK, Bloch O, Auguste K, Papadopoulos MC. Three distinct roles of aquaporin-4 in brain function revealed by knockout mice. Biochim Biophys Acta. 2006;1758:1085–93.
Li YK, Wang F, Wang W, Luo Y, Wu PF, Xiao JL, et al. Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology. 2012;37:1867–78.
Zhang H, Verkman AS. Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci. 2008;37:1–10.
Huber VJ, Tsujita M, Nakada T. Identification of Aquaporin 4 inhibitors using in vitro and in silico methods. Bioorg Med Chem. 2009;17:411–7.
Tanimura Y, Hiroaki Y, Fujiyoshi Y. Acetazolamide reversibly inhibits water conduction by aquaporin-4. J Struct Biol. 2009;166:16–21.
Yang B, Zhang H, Verkman AS. Lack of aquaporin-4 water transport inhibition by antiepileptics and arylsulfonamides. Bioorg Med Chem. 2008;16:7489–93.
Glober NK, Sprague S, Ahmad S, Mayfield KG, Fletcher LM, Digicaylioglue MH, et al. Acetazolamide treatment prevents redistribution of astrocyte aquaporin 4 after murine traumatic brain injury. Neurosci J. 2019;2019:2831501.
Igarashi H, Huber VJ, Tsujita M, Nakada T. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol Sci. 2011;32:113–6.
Pirici I, Balsanu TA, Bogdan C, Margaritescu C, Divan T, Vitalie V, et al. Inhibition of aquaporin-4 improves the outcome of ischaemic stroke and modulates brain paravascular drainage pathways. Int J Mol Sci. 2017;19:46.
Kamegawa A, Hiroaki Y, Tani K, Fujiyoshi Y. Two-dimensional crystal structure of aquaporin-4 bound to the inhibitor acetazolamide. MicroscOPY. 2016;65:177–84.
Farr GW, Hall CH, Farr SM, Wade R, Detzel JM, Adams AG, et al. Functionalized phenylbenzamides inhibit aquaporin-4 reducing Cerebral edema and improving outcome in two models of CNS injury. Neuroscience. 2019;404:484–98.
Wallisch J, Jha R, Vagni V, Feldman K, Dixon C, Farr G, et al. Effect of the novel aquaporin-4 antagonist AER-271 in combined TBI plus hemorrhagic shock in mice. Crit Care Med. 2015;43:6–7.
Kochanek PM, Bramlett HM, Dixon CE, Dietrich WD, Mondello S, Wang KKW, et al. Operation brain trauma therapy: 2016 update. Mil Med. 2018;183:303–12.
Migliati ER, Amiry-Moghaddam M, Froehner SC, Adams ME, Ottersen OP, Bhardwaj A. Na+-K+-2Cl- cotransport inhibitor attenuates cerebral edema following experimental stroke via the perivascular pool of aquaporin-4. Neurocrit Care. 2010;13:123–31.
Migliati E, Meurice N, DuBois P, Fang JS, Somasekharan S, Eeckett B, et al. Inhibition of aquaporin-1 and aquaporin-4 water permeability by a derivative of the loop diuretic bumetanide acting at an internal pore-occluding binding site. Mol Pharmacol. 2009;76:105–12.
O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE. Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab. 2004;24:1046–56.
Fazzina G, Amorini AM, Marmarou CR, Fukui S, Okuno K, Dunbar JG, et al. The protein kinase C activator phorbol myristate acetate decreases brain edema by aquaporin 4 downregulation after middle cerebral artery occlusion in the rat. J Neurotrauma. 2010;27:453–61.
Okuno K, Taya K, Marmariu CR, Ozisik P, Fazzina G, Kleindienst A, et al. The modulation of aquaporin-4 by using PKC-activator (phorbol myristate acetate) and V1a receptor antagonist (SR49059) following middle cerebral artery occlusion/reperfusion in the rat. Acta Neurochir Suppl. 2008;102:431–6.
Marmarou CR, Liang X, Abidi NH, Parveen S, Taya K, Henderson SC, et al. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014;1581:89–102.
Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11:535–44.
Kitchen P, Salman MM, Halsey AM, Clarke-Bland C, MacDonald JA, Ishida H, et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell. 2020;181:784–99.
Chen M, Simard JM. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J Neurosci. 2001;21:6512–21.
Aittoniemi J, Fotinou C, Craig TJ, de Wet H, Proks P, Ashcroft FM. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos Trans R Soc Lond B Biol Sci. 2009;364:257–67.
Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanoca S, Melnitchenko L, et al. Newly expressed SUR1-regulated NCCa-ATPchannel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12:433–40.
Patel AD, Gerzanich V, Geng Z, Simard JM. Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. J Neuropathol Exp Neurol. 2010;69:1177–90.
Jha RM, Puccio AM, Chou SHY, Chang CCH, Wallisch JS, Molyneaux BJ, et al. Sulfonylurea receptor-1: a novel biomarker for cerebral edema in severe traumatic brain injury. Crit Care Med. 2017;45:e255–64.
Jha RM, Puccio AM, Okonkwo DO, Zusman BE, Park SY, Wallisch J, et al. ABCC8 single nucleotide polymorphisms are associated with cerebral edema in severe TBI. Neurocrit Care. 2017;26:213–24.
Woo SK, Tsymbalyuk N, Tsymbalyuk O, Ivanova S, Gerzanich V, Simard JM. SUR1-TRPM4 channels, not KATP, mediate brain swelling following cerebral ischemia. Neurosci Lett. 2020;718:134729.
Zerangue N, Schwappach B, Yuh NJ, Lily YJ. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–48.
Simard JM, Woo SK, Schwartzbauer GT, Gerzanich V. Sulfonylurea receptor 1 in central nervous system injury: A focused review. J Cereb Blood Flow Metab. 2012;32:1699–717.
Stokum JA, Kwon MS, Woo SK, Tsymbalyuk O, Vennekens R, Gerzanich V, et al. SUR1-TRPM4 and AQP4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia. 2018;66:108–25.
Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, Gerzanich V. Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke. Stroke. 2009;40:604–9.
Zweckberger K, Hackenberg K, Jung CS, Hertle DN, Kiening KL, Unterberg AW, et al. Glibenclamide reduces secondary brain damage after experimental traumatic brain injury. Neuroscience. 2014;272:199–206.
Sheth KN, Elm JJ, Molyneaux BJ, Hinson H, Beslow LA, Sze GK, et al. Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016;15:1160–9.
Kimberly WT, Battey TWK, Pham L, Wu O, Yoo AJ, Furie KL, et al. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit Care. 2014;20:193–201.
Sheth KN, Petersen NH, Cheung K, Elm JJ, Hinson HE, Molyneaux BJ, et al. Long-term outcomes in patients aged ≥70 years with intravenous glyburide from the Phase II GAMES-RP study of large hemispheric infarction an exploratory analysis. Stroke. 2018;49:1457–63.
Sun HS, Xu B, Chen W, Xiao A, Turvola E, Alibraham A, et al. Neuronal KATP channels mediate hypoxic preconditioning and reduce subsequent neonatal hypoxic-ischemic brain injury. Exp Neurol. 2015;263:161–71.
Liu R, Wang H, Xu B, Chen W, Turlova E, Dong N, et al. Cerebrovascular safety of sulfonylureas: the role of KATP channels in neuroprotection and the risk of stroke in patients with type 2 diabetes. Diabetes. 2016;65:2795–809.
Glykys J, Dzhala V, Egawa K, Kahle KT, Delpire E, Staley K. Chloride dysregulation, seizures, and cerebral edema: a relationship with therapeutic potential. Trends Neurosci. 2017;40:276–94.
Geck P, Pietrzyk C, Burckhardt BC, Pfeifferl B, Heinz E. Electrically silent cotransport of Na+, K+ and Cl− in ehrlich cells. Biochim Biophys Acta. 1980;600:432–47.
Chen H, Sun D. The role of Na–K–Cl co–transporter in cerebral ischemia. Neurol Res. 2005;27:280–6.
Chew TA, Orlando BJ, Zhang J, Latorraca NR, Wang A, Hollingsworth SA, et al. Structure and mechanism of the cation–chloride cotransporter NKCC1. Nature. 2019;572:488–92.
Pacheco-Alvarez D, San Cristóbal P, Meade P, Moreno E, Vazquez N, Díaz A, et al. The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;281:28755–63.
Su G, Haworth RA, Dempsey RJ, Sun D. Regulation of Na+-K+-Cl− cotransporter in primary astrocytes by dibutyryl cAMP and high [K+](o). Am J Physiol Cell Physiol. 2000;279:C1710–21.
Huang WD, Pan J, Xu M, Su W, Lu YQ, Chen ZJ, et al. Changes and effects of plasma arginine vasopressin in traumatic brain injury. J Endocrinol Invest. 2008;31:996–1000.
Chen H, Luo J, Kintner DB, Shull GE, Sun D. Na+-dependent chloride transporter (NKCC1)-null mice exhibit less gray and white matter damage after focal cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:54–66.
Su G, Kintner DB, Sun D. Contribution of Na+-K+-Cl− cotransporter to high-[K+] o-induced swelling and EAA release in astrocytes. Am J Physiol Cell Physiol. 2002;282:C1136–46.
Su G, Kintner DB, Flagella M, Shull GE, Sun D. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. 2002;282:C1147–60.
Foroutan S, Brillault J, Forbush B, O’Donnell ME. Moderate-to-severe ischemic conditions increase activity and phosphorylation of the cerebral microvascular endothelial cell Na+-K+-Cl− cotransporter. Am J Physiol Cell Physiol. 2005;289:C1492–501.
Yan Y, Dempsey RJ, Sun D. Na+-K+-Cl− cotransporter in rat focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:711–21.
Lu KT, Huang TC, Tsai YH, Yang YL. TRPV4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J Neurochem. 2017;140:718–27.
Lu KT, Wu CY, Yen HH, Peng JHF, Wang CL, Yang YL. Bumetanide administration attenuated traumatic brain injury through IL-1 overexpression. Neurol Res. 2007;29:404–9.
Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74.
Hampel P, Römermann K, MacAulay N, Löscher W. Azosemide is more potent than bumetanide and various other loop diuretics to inhibit the sodium-potassium-chloride-cotransporter human variants hNKCC1A and hNKCC1B. Sci Rep. 2018;8:9877.
Töllner K, Brandt C, Töpfer M, Brunhofer G, Erker T, Gabriel M, et al. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann Neurol. 2014;75:550–62.
Erker T, Brandt C, Töllner K, Schreppel P, Twele F, Schidlitzki A, et al. The bumetanide prodrug BUM5, but not bumetanide, potentiates the antiseizure effect of phenobarbital in adult epileptic mice. Epilepsia. 2016;57:698–705.
Huang H, Bhuiyan MIH, Jiang T, Song S, Shankar S, Taheri T, et al. A novel Na+-K+-Cl− cotransporter 1 inhibitor STS66∗ reduces brain damage in mice after ischemic stroke. Stroke. 2019;50:1021–5.
Lauf PK, Bauer J, Adragna NC, Fujise H, Zade-Oppen AM, Ryu KH, et al. Erythrocyte K-Cl cotransport: properties and regulation. Am J Physiol. 1992;263:C917–32.
Shen MR, Chou CY, Ellory JC. Volume-sensitive KCL transport associated with human cervical carcinogenesis. Pflügers Arch. 2000;440:751–60.
Ernest NJ, Weaver AK, Van Duyn LB, Sontheimer HW. Relative contribution of chloride channels and transporters to regulatory volume decrease in human glioma cells. Am J Physiol Cell Physiol. 2005;288:C1451–60.
Lauf PK, Di Fulvio M, Srivastava V, Sharma N, Adragna NC. KCC2a expression in a human fetal lens epithelial cell line. Cell Physiol Biochem. 2012;29:303–12.
Wei WC, AkermanCJ, Newey SE, Pan J, Clinch NWV, Jacob Y, et al. The potassium-chloride cotransporter 2 promotes cervical cancer cell migration and invasion by an ion transport-independent mechanism. J Physiol. 2011;589:5349–59.
Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, et al. Roles of the cation–chloride cotransporters in neurological disease. Nat Clin Pr Neurol. 2008;4:490–503.
Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, et al. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–37.
Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–5.
Arion D, Lewis DA. Altered expression of regulators of the cortical chloride transporters NKCC1 and KCC2 in schizophrenia. Arch Gen Psychiatry. 2011;69:21–31.
Di Cristo G, Awad PN, Hamidi S, Avoli M. KCC2, epileptiform synchronization, and epileptic disorders. Prog Neurobiol. 2018;162:1–16.
Coull JAM, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, et al. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–42.
Galeffi F, Sah R, Pond BB, George A, Schwartz-Bloom RD. Changes in intracellular chloride after oxygen-glucose deprivation of the adult hippocampal slice: effect of diazepam. J Neurosci. 2004;24:4478–88.
Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke. 2010;41:e151–9.
Bonislawski DP, Schwarzbach EP, Cohen AS. Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol Dis. 2007;25:163–9.
Wu H, Shao A, Zhao M, Chen S, Yu J, Zhou J, et al. Melatonin attenuates neuronal apoptosis through up-regulation of K+-Cl- cotransporter KCC2 expression following traumatic brain injury in rats. J Pineal Res. 2016;61:241–50.
Lee HHC, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAAreceptor-mediated currents. Nat Neurosci. 2011;14:736–43.
Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl Cotransporter KCC2 Mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–64.
Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol. 2002;159:747–52.
Aguado F. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–80.
Shulga A, Thomas-Crusells J, Sigl T, Blaesse A, Mestres P, Meyer M, et al. Posttraumatic GABAA-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J Neurosci. 2008;28:6996–7005.
Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–24.
Woo NS, Lu J, England R, McClellan R, Dufour S, Mount DB, et al. Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K-Cl cotransporter gene. Hippocampus. 2002;12:258–68.
Tornberg J, Voikar V, Savilahti H, Rauvala H, Airaksinen MS. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur J Neurosci. 2005;21:1327–37.
Pellegrino C, Gubkina O, Schaefer M, Becq H, Ludwig A, Mukhtarov M, et al. Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J Physiol. 2011;589:2475–96.
Sivakumaran S, Cardarelli RA, Maguire J, Kelley MR, Silayeva L, Morrow DH, et al. Selective inhibition of KCC2 leads to hyperexcitability and epileptiform discharges in hippocampal slices and in vivo. J Neurosci. 2015;35:8291–6.
Delpire E, Baranczak A, Waterson AG, Kim K, Kett N, Morrison RD, et al. Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: development of a highly selective and more potent in vitro probe. Bioorg Med Chem Lett. 2012;22:4532–5.
Kimelberg HK. Volume activated anion channel and astrocytic cellular edema in traumatic brain injury and stroke. Adv Exp Med Biol. 2004;559:157–67.
Mongin AA. Volume-regulated anion channel—a frenemy within the brain. Pflug Arch. 2016;468:421–41.
Cahalan MD, Lewis RS. Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc Gen Physiol Ser. 1988;43:281–301.
Hazama A, Okada Y. Ca2+ sensitivity of volume-regulatory K+ and Cl− channels in cultured human epithelial cells. J Physiol. 1988;402:687–702.
Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 2014;157:447–58.
Voss FK, Ullrich F, Münch J, Lazarow K, Lutter D, Mah N, et al. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. 2014;344:634–8.
Deneka D, Sawicka M, Lam AKM, Paulino C, Dutzler R. Structure of a volume-regulated anion channel of the LRRC8 family. Nature. 2018;558:254–9.
Kern DM, Oh S, Hite RK, Brohawn SG. Cryo-EM structures of the DCPIB- inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. Elife. 2019;8:e42636.
Syeda R, Qiu Z, Dubin AE, Murthy SE, Florendo MN, Mason DE, et al. LRRC8 proteins form volume-regulated anion channels that sense ionic strength. Cell. 2016;164:499–511.
Zhang H, Cao HJ, Kimelberg HK, Zhou M. Volume regulated anion channel currents of rat hippocampal neurons and their contribution to oxygen-and-glucose deprivation induced neuronal death. PLoS ONE. 2011;6:e16803.
Yang J, Vitery MDC, Chen J, Osei-Owusu J, Chu J, Qiu Z. Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron. 2019;102:813–27.
Feustel PJ, Jin Y, Kimelberg HK. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke. 2004;35:1164–8.
Liu HT, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Gia. 2006;54:343–57.
Mongin AA, Kimelberg HK. ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am J Physiol Cell Physiol. 2002;283:C569–78.
Zhang Y, Zhang H, Feustel PJ, Kimelberg HK. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol. 2008;210:514–20.
Bowens NH, Dohare P, Kuo YH, Mongin AA. DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol Pharmacol. 2013;83:22–32.
Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim W-S, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–5.
Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–4.
Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–29.
Pifferi S, Cenedese V, Menini A. Anoctamin 2/TMEM16B: a calcium-activated chloride channel in olfactory transduction. Exp Physiol. 2012;97:193–9.
Ji Q, Guo S, Wang X, Pang C, Zhan Y, Chen Y, et al. Recent advances in TMEM16A: structure, function, and disease. J Cell Physiol. 2019;234:7856–73.
Brunner JD, Lim NK, Schenck S, Duerst A, Dutzler R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. 2014;516:207–12.
Paulino C, Neldner Y, Lam AK, Kalienkova V, Brunner JD, Schenck S, et al. Structural basis for anion conduction in the calcium-activated chloride channel TMEM16A. Elife. 2017;6:e26232.
Dang S, Feng S, Tien J, Peters CJ, Bulkley D, Lolicato M, et al. Cryo-EM structures of the TMEM16A calciumactivated chloride channel. Nature. 2017;552:426–9.
Xiao Q, Yu K, Perez-Cornejo P, Cui Y, Arreola J, Hartzell HC. Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc Natl Acad Sci USA. 2011;108:8891–6.
Wu MM, Lou J, Song BL, Gong YF, Li YC, Yu CJ, et al. Hypoxia augments the calcium-activated chloride current carried by anoctamin-1 in cardiac vascular endothelial cells of neonatal mice. Br J Pharmacol. 2014;171:3680–92.
Hoffmann EK, Sørensen BH, Sauter DPR, Lambert IH. Role of volume-regulated and calcium-activated anion channels in cell volume homeostasis, cancer and drug resistance. Channels. 2015;9:380–96.
Almaça J, Tian Y, Aldehni F, Ousingsawat J, Kongsuphol P, Rock JR, et al. TMEM16 proteins produce volume-regulated chloride currents that are reduced in mice lacking TMEM16A. J Biol Chem. 2009;284:28571–8.
Wang H, Zou L, Ma K, Yu J, Wu H, Wei M, et al. Cell-specific mechanisms of TMEM16A Ca2+-activated chloride channel in cancer. Mol Cancer. 2017;16:152.
Liu PY, Zhang Z, Liu Y, Tang XL, Shu S, Bao XY, et al. TMEM16A inhibition preserves blood–brain barrier integrity after ischemic stroke. Front Cell Neurosci. 2019;13:360.
Bill A, Hall ML, Borawski J, Hodgson C, Jenkins J, Piechon P, et al. Small molecule-facilitated degradation of ANO1 protein: a new targeting approach for anticancer therapeutics. J Biol Chem. 2014;289:11029–41.
Bodedtkjer DMB, Kim S, Jensen AB, Matchkov VM, Andersson KE. New selective inhibitors of calcium-activated chloride channel- T16inh-A01, CaCCinh-A01 and MONNA- what do they inhibit? Br J Pharmacol. 2015;172:4158–72.
Ji B, Zhou F, Han L, Yang J, Fan H, Li S, et al. Sodium tanshinone IIA sulfonate enhances effectiveness Rt-PA treatment in acute ischemic stroke patients associated with ameliorating blood-brain barrier damage. Transl Stroke Res. 2017;8:334–40.
Liu F, Zhang Z, Csanády L, Gadsby DC, Chen J. Molecular structure of the human CFTR ion channel. Cell. 2017;169:85–92.
Živković SA, Jumaa M, Barišić N, McCurry K. Neurologic complications following lung transplantation. J Neurol Sci. 2009;280:90–3.
Goldstein AB, Goldstein LS, Perl MK, Haug MT, Arroliga AC, Stillwell PC. Cystic fibrosis patients with and without central nervous system complications following lung transplantation. Pediatr Pulmonol. 2000;30:203–6.
Zhang YP, Zhang Y, Xiao ZB, Zhang YB, Zhang J, Li ZQ, et al. CFTR prevents neuronal apoptosis following cerebral ischemia reperfusion via regulating mitochondrial oxidative stress. J Mol Med. 2018;96:611–20.
Solymosi EA, Kaestle-Gembardt SM, Vadász I, Wang L, Neye N, Chupin JCA, et al. Chloride transport-driven alveolar fluid secretion is a major contributor to cardiogenic lung edema. Proc Natl Acad Sci USA. 2013;110:E2308–16.
Ajonuma LC, He Q, Chan PKS, Ng EHY, Fok KL, Wong CHY, et al. Involvement of cystic fibrosis transmembrane conductance regulator in infection-induced edema. Cell Biol Int. 2008;32:801–6.
Lidington D, Fares JC, Uhl FE, Dinh DD, Kroetsch JT, Sauvé M, et al. CFTR therapeutics normalize cerebral perfusion deficits in mouse models of heart failure and subarachnoid hemorrhage. JACC Basic Transl Sci. 2019;4:940–58.
Zegarra-Moran O, Galietta LJV. CFTR pharmacology. Cell Mol Life Sci. 2017;74:117–28.
Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJV, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–37.
Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJV, et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–8.
Su X, Looney MR, Su HE, Lee JW, Song Y, Matthay MA. Role of CFTR expressed by neutrophils in modulating acute lung inflammation and injury in mice. Inflamm Res. 2011;60:619–32.
Thiagarajah JR, Broadbent T, Hsieh E, Verkman AS. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR Inhibitor. Gastroenterology. 2004;126:511–9.
Snyder DS, Tradtrantip L, Yao C, Kurth MJ, Verkman AS. Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J Med Chem. 2011;54:5468–77.
Friard J, Tauc M, Cougnon M, Compan V, Duranton C, Rubera I. Comparative effects of chloride channel inhibitors on LRRC8/VRAC-mediated chloride conductance. Front Pharmacol. 2017;8:328.
Melis N, Tauc M, Cougnon M, Bendahhou S, Giuliano S, Rubera I, et al. Revisiting CFTR inhibition: a comparative study of CFTRinh-172 and GlyH-101 inhibitors. Br J Pharmacol. 2014;171:3716–27.
Poroca DR, Pelis RM, Chappe VM. ClC channels and transporters: structure, physiological functions, and implications in human chloride channelopathies. Front Pharmacol. 2017;8:151.
D’Anglemont De Tassigny A, Souktani R, Ghaleh B, Henry P, Berdeaux A. Structure and pharmacology of swelling-sensitive chloride channels, ICl,swell. Fundam Clin Pharmacol. 2003;17:539–53.
Roman RM, Smith RL, Feranchak AP, Clayton GH, Doctor RB, Fitz JG. ClC-2 chloride channels contribute to HTC cell volume homeostasis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G334–53.
Blanz J, Schweizer M, Auberson M, Maier H, Muenscher A, Hübner CA, et al. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci. 2007;27:6581–9.
Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 2013;12:659–68.
Duan D, Winter C, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–21.
Zhang YP, Zhang H, Duan DD. Chloride channels in stroke. Acta Pharmacol Sin. 2013;34:17–23.
Wang L, Chen L, Jacob TJC. The role of ClC-3 in volume-activated chloride currents and volume regulation in bovine epithelial cells demonstrated by antisense inhibition. J Physiol. 2000;524:63–75.
Qian Y, Du YH, Tang YB, Lv XF, Liu J, Zhou JG, et al. ClC-3 chloride channel prevents apoptosis induced by hydrogen peroxide in basilar artery smooth muscle cells through mitochondria dependent pathway. Apoptosis. 2011;16:468–77.
Reyes RC, Verkhratsky A, Parpura V. Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neuro. 2012;4:e00075.
Giladi M, Tal I, Khananshvili D. Structural features of ion transport and allosteric regulation in sodium-calcium exchanger (NCX) proteins. Front Physiol. 2016;7:30.
Hilge M, Aelen J, Vuister GW. Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol Cell. 2006;22:15–25.
Reeves JP, Hale CC. The stoichiometry of the cardiac sodium-calcium exchange system. J Biol Chem. 1984;259:7733–9.
Hilgemann DW, Collins A, Matsuoka S. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Secondary modulation by cytoplasmic calcium and ATP. J Gen Physiol. 1992;100:933–61.
Kang TM, Hilgemann DW. Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature. 2004;427:544–8.
Pignataro G, Tortiglione A, Scorziello A, Giaccio L, Secondo A, Severino B, et al. Evidence for a protective role played by the Na+/Ca2+ exchanger in cerebral ischemia induced by middle cerebral artery occlusion in male rats. Neuropharmacology. 2004;46:439–48.
Shenoda B. The role of Na+/Ca2+ exchanger subtypes in neuronal ischemic injury. Transl Stroke Res. 2015;6:181–90.
Floyd CL, Gorin FA, Lyeth BG. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia. 2005;51:35–46.
Pignataro G, Gala R, Cuomo O, Tortiglione A, Giaccio L, Castaldo P, et al. Two sodium/calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke. 2004;35:2566–70.
Jeon D, Chu K, Jung KH, Kim M, Yoon BW, Lee CJ, et al. Na+/Ca2+ exchanger 2 is neuroprotective by exporting Ca2+ during a transient focal cerebral ischemia in the mouse. Cell Calcium. 2008;43:482–91.
Molinaro P, Cuomo O, Pignataro G, Boscia F, Sirabella R, Pannaccione A, et al. Targeted disruption of Na+/Ca2+ exchanger 3 (NCX3) gene leads to a worsening of ischemic brain damage. J Neurosci. 2008;28:1179–84.
Cross JL, Meloni BP, Bakker AJ, Sokolow S, Herchuelz A, Schurmans S, et al. Neuronal injury in NCX3 knockout mice following permanent focal cerebral ischemia and in NCX3 knockout cortical neuronal cultures following oxygen-glucose deprivation and glutamate exposure. J Exp Stroke Transl Med. 2009;2:3–9.
O’Donnell JC, Jackson JG, Robinson MB. Transient oxygen/glucose deprivation causes a delayed loss of mitochondria and increases spontaneous calcium signaling in astrocytic processes. J Neurosci. 2016;36:7109–27.
Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, et al. SEA0400, a novel and selective inhibitor of the Na+/Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–56.
Arakawa N, Sakaue M, Yokoyama I, Hashimoto H, Koyama Y, Baba A, et al. KB-R7943 inhibits store-operated Ca2+ entry in cultured neurons and astrocytes. Biochem Biophys Res Commun. 2000;279:354–7.
Iwamoto T, Kita S, Uehara A, Imanaga I, Matsuda T, Baba A, et al. Molecular determinants of Na+/Ca2+ Exchange (NCX1) inhibition by SEA0400. J Biol Chem. 2004;279:7544–53.
Koyama Y, Matsui S, Itoh S, Osakada M, Baba A, Matsuda T. The selective Na+-Ca2+ exchange inhibitor attenuates brain edema after radiofrequency lesion in rats. Eur J Pharmacol. 2004;489:193–6.
Molinaro P, Sirabella R, Pignataro G, Petrozziello T, Secondo A, Boscia F, et al. Neuronal NCX1 overexpression induces stroke resistance while knockout induces vulnerability via Akt. J Cereb Blood Flow Metab. 2016;36:1790–803.
Molinaro P, Cantile M, Cuomo O, Secondo A, Pannaccione A, Ambrosino P, et al. Neurounina-1, a novel compound that increases Na+/Ca2+ exchanger activity, effectively protects against stroke damage. Mol Pharmacol. 2013;83:142–56.
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–23.
Díaz-García CM, Yellen G. Neurons rely on glucose rather than astrocytic lactate during stimulation. J Neurosci Res. 2019;97:883–9.
Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia. 1997;21:2–21.
Deng D, Yan N. GLUT, SGLT, and SWEET: structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016;25:546–58.
Farrell CL, Pardridge WM. Blood-brain barrier glucose transporter is asymmetrically distributed on brain capillary endothelial lumenal and ablumenal membranes: an electron microscopic immunogold study. Proc Natl Acad Sci USA. 1991;88:5779–83.
Yu AS, Hirayama BA, Timbol G, Liu J, Basarah E, Kepe V, et al. Functional expression of SGLTs in rat brain. Am J Physiol Cell Physiol. 2010;299:C1277–84.
Vemula S, Roder KE, Yang T, Bhat GJ, Thekkumkara TJ, Abbruscato TJ. A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J Pharmacol Exp Ther. 2009;328:487–95.
Yu AS, Hirayama BA, Timbol G, Liu J, Diez-Sampedro A, Kepe V, et al. Regional distribution of SGLT activity in rat brain in vivo. Am J Physio Cell Physiol. 2013;304:C240–7.
Sebastiani A, Greve F, Gölz C, Förster CY, Koepsell H, Thal SC. RS1 (Rsc1A1) deficiency limits cerebral SGLT1 expression and delays brain damage after experimental traumatic brain injury. J Neurochem. 2018;147:190–203.
Yuan H, Frank JE, Hong Y, An H, Eldeniz C, Nie J, et al. Spatiotemporal uptake characteristics of [18]F-2-fluoro-2-deoxy- d-glucose in a rat middle cerebral artery occlusion model. Stroke. 2013;44:2292–9.
Arnberg F, Grafström J, Lundberg J, Nikkhou-Aski S, Little P, Damberg P, et al. Imaging of a clinically relevant stroke model glucose hypermetabolism revisited. Stroke. 2015;46:835–42.
Thorén M, Azevedo E, Dawson J, Egido JA, Falcou A, Ford GA, et al. Predictors for cerebral edema in acute ischemic stroke treated with intravenous thrombolysis. Stroke. 2017;48:2464–71.
Broocks G, Kemmling A, Aberle J, Kniep H, Bechstein M, Flottmann F, et al. Elevated blood glucose is associated with aggravated brain edema in acute stroke. J Neurol. 2020;267:440–8.
Oerter S, Förster C, Bohnert M. Validation of sodium/glucose cotransporter proteins in human brain as a potential marker for temporal narrowing of the trauma formation. Int J Leg Med. 2019;133:1107–14.
Sherwood TW, Lee KG, Gormley MG, Askwith CC. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J Neurosci. 2011;31:9723–34.
Sherwood TW, Frey EN, Askwith CC. Structure and activity of the acid-sensing ion channels. Am J Physiol Cell Physiol. 2012;303:C699–710.
Deval E, Gasull X, Noël J, Salinas M, Baron A, Diochot S, et al. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther. 2010;128:549–58.
Yin T, Lindley TE, Albert GW, Ahmed R, Schmeiser PB, Grady MS, et al. Loss of acid sensing ion channel-1a and bicarbonate administration attenuate the severity of traumatic brain injury. PLoS ONE. 2013;8:e72379.
Leng T, Shi Y, Xiong ZG, Sun D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: new therapeutic targets for stroke? Prog Neurobiol. 2014;115:189–209.
Immke DC, McCleskey EW. Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat Neurosci. 2001;4:869–70.
Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA. 2004;101:6752–7.
Duan B, Wang YZ, Yang T, Chu XP, Yu Y, Huang Y, et al. Extracellular spermine exacerbates ischemic neuronal injury through sensitization of ASIC1a channels to extracellular acidosis. J Neurosci. 2011;31:2101–12.
McCarthy CA, Rash LD, Chassagnon IR, King GF, Widdop RE. PcTx1 affords neuroprotection in a conscious model of stroke in hypertensive rats via selective inhibition of ASIC1a. Neuropharmacology. 2015;99:650–7.
Pignataro G, Simon RP, Xiong ZG. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–8.
Zhao X, Gorin FA, Berman RF, Lyeth BG. Differential hippocampal protection when blocking intracellular sodium and calcium entry during traumatic brain injury in rats. J Neurotrauma. 2008;25:1195–205.
Kintner DB, Su G, Lenart B, Ballard AJ, Meyer JW, Ng LL, et al. Increased tolerance to oxygen and glucose deprivation in astrocytes from Na+/H+ exchanger isoform 1 null mice. Am J Physiol Cell Physiol. 2004;287:12–21.
Luo J, Chen H, Kintner DB, Shull GE, Sun D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J Neurosci. 2005;25:11256–68.
Begum G, Song S, Wang S, Zhao H, Bhuiyan MIH, Li E, et al. Selective knockout of astrocytic Na+/H+ exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia. 2018;66:126–44.
O’Donnell ME, Chen YJ, Lam TI, Taylor KC, Walton JH, Anderson SE. Intravenous HOE-642 reduces brain edema and Na uptake in the rat permanent middle cerebral artery occlusion model of stroke: evidence for participation of the blood-brain barrier Na+/H+ exchanger. J Cereb Blood Flow Metab. 2013;33:225–34.
Acknowledgements
This work was supported by grants to ZPF from Canadian Institutes of Health Research (CIHR PJT-153155), HSS from Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2016-04574), and BXY from National Natural Science Foundation of China grant 81620108029.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Luo, Zw., Ovcjak, A., Wong, R. et al. Drug development in targeting ion channels for brain edema. Acta Pharmacol Sin 41, 1272–1288 (2020). https://doi.org/10.1038/s41401-020-00503-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00503-5
Keywords
This article is cited by
-
MRI of cerebral oedema in ischaemic stroke and its current use in routine clinical practice
Neuroradiology (2024)
-
Protein nanoparticle-induced osmotic pressure gradients modify pulmonary edema through hyperpermeability in acute respiratory distress syndrome
Journal of Nanobiotechnology (2022)
-
Dual-modal in vivo assessment for electrophysical and hemodynamic characteristics of cerebral edema induced by lipopolysaccharide
BioMedical Engineering OnLine (2022)
-
Post-stroke Impairment of the Blood–Brain Barrier and Perifocal Vasogenic Edema Is Alleviated by Endovascular Mesenchymal Stem Cell Administration: Modulation of the PKCδ/MMP9/AQP4-Mediated Pathway
Molecular Neurobiology (2022)
