
Abstract
Brain edema is a common and serious complication of ischemic stroke with limited effective treatment. We previously reported that methylene blue (MB) attenuated ischemic brain edema in rats, but the underlying mechanisms remained unknown. Aquaporin 4 (AQP4) in astrocytes plays a key role in brain edema. We also found that extracellular signal-regulated kinase 1/2 (ERK1/2) activation was involved in the regulation of AQP4 expression in astrocytes. In the present study, we investigated whether AQP4 and ERK1/2 were involved in the protective effect of MB against cerebral edema. Rats were subjected to transient middle cerebral artery occlusion (tMCAO), MB (3 mg/kg, for 30 min) was infused intravenously through the tail vein started immediately after reperfusion and again at 3 h after ischemia (1.5 mg/kg, for 15 min). Brain edema was determined by MRI at 0.5, 2.5, and 48 h after tMCAO. The decreases of apparent diffusion coefficient (ADC) values on diffusion-weighted MRI indicated cytotoxic brain edema, whereas the increase of T2 MRI values reflected vasogenic brain edema. We found that MB infusion significantly ameliorated cytotoxic brain edema at 2.5 and 48 h after tMCAO and decreased vasogenic brain edema at 48 h after tMCAO. In addition, MB infusion blocked the AQP4 increases and ERK1/2 activation in the cerebral cortex in ischemic penumbra at 48 h after tMCAO. In a cell swelling model established in cultured rat astrocyte exposed to glutamate (1 mM), we consistently found that MB (10 μM) attenuated cell swelling, AQP4 increases and ERK1/2 activation. Moreover, the ERK1/2 inhibitor U0126 (10 μM) had the similar effects as MB. These results demonstrate that MB improves brain edema and astrocyte swelling, which may be mediated by the inhibition of AQP4 expression via ERK1/2 pathway, suggesting that MB may be a potential choice for the treatment of brain edema.
Similar content being viewed by others
Introduction
Stroke is one of the leading causes of death and disability worldwide, and ischemic stroke accounts for 85% of all strokes [1, 2]. Ischemic stroke leads to neurological dysfunction, significant infarct volume, and serious neuronal damage [3, 4]. Brain edema is a very serious complication not only of ischemic stroke but also following treatment with thrombolysis and intracranial thrombectomy [5]. Ischemic brain edema mainly includes cytotoxic edema (oncotic cell swelling) and vasogenic edema. The former occurs immediately after cerebral ischemia and continues throughout the whole period of cerebral edema, and the latter occurs following damage to the brain–blood barrier (BBB) after brain ischemia [1]. Without treatment, brain edema leads to swelling of the cerebral parenchyma, reduces cerebral blood flow, and increases intracranial pressure that causes secondary brain damage, such as impaired vascular perfusion and life-threatening herniation of the brain. Therefore, improving the severity of brain edema has already become the main potential therapeutic strategy for the treatment of cerebral ischemia [6, 7]. Treatment of cerebral edema might relieve neuronal cell damage, reduce infarct volume, and improve neurological movement disorders. Thus far, the approved therapies for the treatment of cerebral edema include osmotherapy and decompressive craniectomy, which both aim to improve the downstream effects rather than address the molecular mechanisms underlying the development of brain edema. Therefore, the treatment effect is very limited [1]. It is of great importance to explore new therapeutic measures for brain edema after stroke.
Recently, the protective effect of methylene blue (MB) has been demonstrated in ischemic stroke in vitro and in vivo, potentially due to its capability to act as an alternative mitochondrial electron carrier [8]. MB is a blue dye synthesized by Heinrich Caro in 1876; it was initially employed in laboratory and clinical settings in the 1890s [9]. MB is currently used to treat carbon monoxide poisoning, methemoglobinemia and cyanide poisoning in the clinic [10, 11]. Due to its energy-enhancing and antioxidant properties, MB has been studied in ischemia–reperfusion damage [8], Alzheimer’s disease [12], Parkinson’s disease, and cerebral traumatic brain injury [13]. Wen et al. found that MB significantly reduced the ischemic lesion volume depicted by triphenyl tetrazolium chloride staining at 24 h after cerebral ischemia–reperfusion injury [8]. Using magnetic resonance imaging (MRI), Jiang et al. also confirmed that MB treatment reduced infarct volumes defined by apparent diffusion coefficient (ADC) or T2-weighted MRI (T2WI) in rat models of ischemic stroke [14]. Recently, we further found that MB not only reduced ADC and T2WI lesion volume in rats at 48 h after transient middle cerebral artery occlusion (tMCAO) but also remarkably decreased the corrected T2WI lesion volume, which referred to brain edema volume, suggesting that MB improved ischemic brain edema [15]. MRI is now widely used to guide acute stroke treatment in preclinical and clinical studies [16]. Diffusion-weighted MRI (DWI), in which image contrast is based on the water ADC, can examine ischemic injury within a few minutes after the onset of stroke and was used to evaluate cytotoxic brain edema [17]. T2WI is widely used to define final infarct volume and delineate vasogenic brain edema [18,19,20]. In the current study, to mimic clinical practice, we performed sequential MRI scans on rats after tMCAO at different stages and evaluated the effect of MB on cytotoxic and vasogenic brain edema by detecting the ADC and T2WI values.
Astrocytes are the most abundant cell type in the brain, and existing studies have confirmed that astrocyte swelling is the main type of brain edema and a lasting event after ischemic stroke [21]. Aquaporin 4 (AQP4), the major aquaporin in the central nervous system (CNS), is mainly expressed in astrocytic endfeet and plays a central role in brain edema [22]. Previous studies showed that genetic deletion or silencing of AQP4 improves brain edema following ischemia, while an accelerated progression of cytotoxic brain edema was observed in AQP4-overexpressing mice [23,24,25,26]. In an astrocyte swelling model induced by incubation with glutamate, which is one of the important factors leading to ischemic brain edema [27], we demonstrated that inhibition of AQP4 expression with AQP4 siRNA blocked astrocyte swelling [28]. Both in vivo and in vitro experimental results indicated that AQP4 is the target molecule for brain edema treatment. Accordingly, we speculated that the protective effect of MB against brain edema induced by ischemic stroke may be mediated by inhibition of AQP4 expression. Therefore, in the present study, we detected the effect of MB on AQP4 expression both in rat brain at 48 h after tMCAO and in cultured astrocytes incubated with glutamate.
Accumulating evidence has shown that AQP4 is dynamically regulated by different mechanisms. The extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway is a key signaling pathway involved in the mechanism of ischemic stroke [29]. p-ERK1/2 levels reflecting ERK1/2 activity have been shown to increase in tMCAO models [30, 31]. Our previous study found that the ERK1/2 signaling pathway was involved in the regulation of AQP4 expression. Therefore, we intended to explore whether the ERK1/2 pathway mediates the effect of MB on AQP4 expression to attenuate brain edema and astrocyte swelling. We examined the effects of MB on ERK1/2 activation in both the rat tMCAO model and astrocyte swelling model. Then, the ERK1/2 kinase inhibitor U0126 was used to investigate whether blocking ERK1/2 activation can imitate the effect of MB on reducing astrocyte swelling and AQP4 expression. Our results might provide a basis for the further application of MB in the treatment of cerebral edema caused by brain injuries, including ischemia, trauma, and epilepsy.
Materials and methods
Animals
Male neonatal (1 day) and adult (280–320 g) Sprague–Dawley rats were purchased from the Institute of Laboratory Animal Science, affiliated with the Chinese Academy of Military Medical Sciences (Certificate No, SCSK (Beijing) 2012–0004, Beijing, China). Adult rats were maintained under environmentally controlled conditions including a 12 h light–dark cycle, a standard diet and water.
All animal studies were carried out in accordance with the Guidelines for the Care and Use of Laboratory Animals. The animal experiments were approved by the Animal Care and Use Committee at Beijing Neurosurgical Institute (No. 201401007).
Transient middle cerebral artery occlusion (tMCAO) of rats and administration of MB
The tMCAO model was induced by inserting a nylon thread into the left middle cerebral artery in rats for 1 h followed by 47 h of reperfusion, as previously described [15]. Rats were randomly divided into three groups: (1) a sham surgery group (Sham group, n = 12), (2) a tMCAO with vehicle treatment group (tMCAO group, n = 16), and (3) an MB treatment group (tMCAO+MB group, n = 15). Two rats from the tMCAO group and two from the tMCAO+MB group prematurely died from subarachnoid hemorrhage and were excluded from further analysis. The mortality rate of rats in our experiment was ~10%.
For MB (Sigma Aldrich, St. Louis, MO, USA) treatment, MB (3 mg/kg, 4 mL·kg−1·h−1, ~30 min) was infused intravenously through the tail vein starting immediately after reperfusion and again at 3 h after ischemia (1.5 mg/kg, 4 mL·kg−1·h−1, ~15 min). An equivalent volume of 0.9% saline was infused intravenously for the tMCAO with vehicle treatment group. We performed MRI and histological and molecular biology examinations at the indicated time points according to the experimental design shown in Fig. 1a.

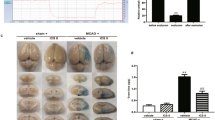
a Schematic drawing of the experimental design in rats. MCAO middle cerebral artery occlusion, MB methylene blue, Rep reperfusion, MRI magnetic resonance imaging, H&E hematoxylin–eosin staining, IHC immunohistochemistry, WB Western blotting, qRT-PCR quantitative reverse transcription polymerase chain reaction. b, c Apparent diffusion coefficient (ADC) and T2-weighted imaging (T2WI) images at 0.5, 2.5, and 48 h after tMCAO for tMCAO and tMCAO+MB rats; the left side is the ipsilateral side of the injury. The area of the dotted red or blue line is the range of brain edema after tMCAO in rats. n = 8 for tMCAO rats, n = 7 for tMCAO+MB rats. Values are the mean ± SD. *P < 0.05 vs. the tMCAO+MB group. #P < 0.05 vs. the 0.5 h group
Following ischemic stroke, there is usually an ischemic core and a surrounding area, defined as the ischemic penumbra, which has reduced cerebral blood flow but preserved energy metabolism [32]. The ischemic penumbra has been broadly defined as an area of severely hypoperfused but potentially rescuable tissue around the ischemic core and is regarded as a target for the current neuroprotective intervention [33]. In our study, brain tissues from the ischemic penumbra in tMCAO rats and the corresponding regions in sham rats were harvested for further experiments. As described previously [15, 34], a 2 mm section was cut 5 mm from the anterior tip of the frontal lobe. A longitudinal cut was made from top to bottom ~2 mm from the midline through the ischemic hemisphere of this section. Then, a transverse diagonal cut was made at approximately the “1 o′clock” position to separate the core from the penumbra in the adjacent cortex.
Magnetic resonance imaging (MRI)
In our experiment, MRI was performed in rats as previously described [15, 35]. The same rat was imaged at 0.5, 2.5, and 48 h after tMCAO in both the tMCAO group (n = 8) and tMCAO+MB group (n = 7) on a 7.0 T scanner (Bio Spec, Bruker, Bremerhaven, Germany) with a four-channel phase array rat head coil. DWI was obtained by a spin echo echo-planar-imaging sequence, and the parameters used were as follows: field of view (FOV) = 4 cm × 4 cm, slice thickness (THK) = 1 mm, matrix = 128 × 108, echo time (TE) = 35 ms, repetition time (TR) = 4500 ms, number of averages (NA) = 1, and b values = 0, 1000 s/mm2. Quantitative maps of the ADC were detected according to two different b values (0, 1000 s/mm2). OsiriX software was used to compute and calculate ADC maps. T2-weighted turbo-spin echo images (T2WI) were obtained using a fast spin echo sequence, and the detailed parameters were as follows: FOV = 4 cm × 4 cm, THK = 1 mm, matrix = 320 × 240, TE = 37 ms, TR = 3140 ms, and NA = 1.
To explore the effect of MB on brain edema at 0.5, 2.5, and 48 h after tMCAO, we calculated the relative signal intensity of ADC (ipsilateral/contralateral) (rADC) to represent cytotoxic brain edema and the relative signal intensity of T2WI (ipsilateral/contralateral) (rT2WI) to reflect vasogenic brain edema. First, the area of hypointense pixels in ADC or that of hyperintense pixels in T2WI was defined as the region of interest (ROI). Then, the ROIs on each of the lesion-containing slices in the ipsilateral hemisphere and the corresponding region in the contralateral hemisphere were delineated by two researchers who were blinded to the experimental groups. The signal intensities of ROIs were analyzed using imaging software (IMAGE, version 4.03; National Institutes of Health, Bethesda, MD, USA) on ADC maps and T2WI. According to previously described equations [36], the rADC was calculated as the ratio of ipsilateral ADC signal intensity to contralateral ADC signal intensity. Similarly, the rT2WI was the ratio of ipsilateral to contralateral T2WI signal intensity. Finally, the ratio of each rat was averaged from all slices calculated in one rat.
Hematoxylin–eosin staining and immunohistochemistry of rat brain
For morphological assays, rats that underwent 1 h of ischemia followed by 47 h of reperfusion were transcardially perfused with 4% paraformaldehyde. Brain tissue sections, ~4 mm thick, from the cerebral cortex in the ischemic penumbra were fixed in 4% paraformaldehyde for 1 day at 4 °C, embedded in paraffin and then sliced into 4 μm sections.
H&E staining was conducted to evaluate the histological injury. Immunohistochemistry staining for albumin was carried out to detect the permeability of the BBB [37]. Moreover, immunohistochemistry staining was used to examine the expression of glial fibrillary acidic protein (GFAP, DAKO, Glostrup, Denmark) and AQP4 (Abcam, Cambridge, MA, USA). The sections used for immunohistochemistry staining were prepared as previously described. The primary antibodies used were rabbit anti-rat albumin (1:400, Abcam), rabbit anti-rat GFAP (1:2000), and rabbit anti-rat AQP4 (1:2000). The antigen–antibody complexes were detected by an EnVisionTM Detection Kit (DAKO). Negative controls were made by omitting primary antibodies. Images were acquired with a microscope (Carl Zeiss, Oberkochen, Germany).
Astrocyte swelling and treatment with methylene blue or U0126
A cell swelling model induced by glutamate treatment of cultured astrocytes was used to evaluate the effect of MB on astrocyte swelling in vitro. According to our published method [38], primary cultured astrocytes were prepared from cerebral cortices of 1-day-old Sprague–Dawley rats and incubated in MEM (Gibco, Grand Island, NY, USA) supplemented with 10% FBS and maintained at 37 °C in a 5% CO2 incubator. When the astrocytes were 80%–90% confluent on the 10th day of primary culture, subculture was carried out to obtain more astrocytes. Secondary cultured astrocytes were used in this experiment. The purity of the cultured astrocytes was detected by GFAP immunofluorescence staining. Cell swelling of cultured astrocytes was induced by exposure to 1 mM glutamate for 48 h as previously described [28]. Then, the cell perimeter, AQP4 expression level and ERK1/2 activation were measured. In some experiments, MB at different concentrations (0, 0.1, 1, and 10 μM) was applied together with glutamate administration. Only the MEM was replaced in the control group.
To detect the effect of ERK1/2 activation on AQP4 levels in astrocytes, the ERK1/2 kinase inhibitor U0126 (Biomol, Hamburg, Germany) was used in the culture medium combined with glutamate and/or MB. U0126 was dissolved in dimethyl sulfoxide (DMSO), and a final concentration of 10 μM U0126 was used in the culture medium. The controls were treated with DMSO (0.2%). In the cell swelling model, in which cultured rat astrocytes were incubated with 1 mM glutamate for 48 h, reverse transcription quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot experiments were performed three times with three samples for each experimental condition in three independent cell culture experiments.
Immunocytochemistry of cultured astrocytes and measurements of cell perimeters
According to our previous description [28], cultured rat astrocytes were fixed in acetone for 10–15 min. Cells were blocked with 10% goat serum for 20 min and then incubated with rabbit polyclonal antibodies against GFAP (1:50) at 4 °C overnight. Subsequently, Alexa Fluor 488 IgG secondary antibody (1:200, Life Technologies, Carlsbad, CA, USA) was applied at room temperature (RT) for 1 h. Images were acquired under an inverted fluorescence microscope. The negative control was treated with PBS instead of the primary antibody.
The cell perimeter was measured to represent astrocyte volume according to our previous report [28]. After the cultured astrocytes were stained for GFAP, the cell perimeter was calculated by immunofluorescence using the Image-Pro Plus software package (Media Cybernetics, Rockville, MD, USA). Ten cells in each field under high magnification (×200) were randomly selected. Three fields in three wells were measured in every group, and the average values of nine fields were calculated as the cell perimeter of each group.
Reverse transcription quantitative real-time polymerase chain reaction (qRT-PCR)
The expression levels of AQP4 mRNA were measured by qRT-PCR. Total RNA from the brain tissue of the ischemic penumbra or cultured astrocytes were isolated using a Total RNA Extraction Kit (Promega, Madison, WI, USA). cDNA was generated from 1 μg of total RNA using a Reverse Transcription System (Promega). cDNA levels in all samples, including the housekeeping gene β-actin, were analyzed by PCR using a LightCycler 480 (Roche, Basel, Switzerland). The following primer sequences were used in this experiment: AQP4 forward: 5′-TGAATCCAGCTCGATCCTTTG-3′; reverse: 5′-TATCCAGTGGTTTTCCCAGTTTC-3′; β-actin (internal standard) forward: 5′-CGTTGACATCCGTAAAGACC-3′; reverse: 5′-CTAGGAGCCAGAGCAGTAATC-3′. The samples were treated at 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s, 58 °C for 30 s, and 72 °C for 30 s and a final extension at 72 °C for 10 min. The specificity of the measured signal was identified as a single peak in the melting curve. β-Actin was used as the internal control. The 2−ΔΔCt method was used for quantification. The results are shown as the AQP4/β-actin ratio.
Western blotting
AQP4 protein expression and ERK1/2 activation were detected by Western blot as previously described [38]. The ischemic penumbra of cortical regions in tMCAO rats and the corresponding cortical regions in sham rats were used. The brain tissue and cultured astrocytes were lysed in cell lysis buffer supplemented with phosphatase and protease inhibitor on ice using an ultrasonic instrument (Branson, Danbury, Conn, USA). The concentrations of extracted protein were estimated using a BCA protein assay kit. Samples containing approximately 30 μg of protein were separated on 10% SDS-PAGE gels and then transferred to 0.45 μm PVDF membranes. After blocking with 5% nonfat milk in Tris-saline buffer for 2 h at RT, the membranes were incubated with primary antibodies, including AQP4 (1:500), p-ERK1/2 and ERK1/2 (1:1000, Cell Signaling Technology, Danvers, MA, USA), at 4 °C overnight. Subsequently, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies at RT for 1 h. β-actin (1:5000, Sigma Aldrich) was used as a control. An enhanced chemiluminescence reagent kit (Applygen Technologies, Beijing, China) was used to detect the specific bands, which were quantified with a ChemiDocMP (Bio-Rad, Hercules, CA, USA). The levels of AQP4 protein expression were normalized to β-actin. The p-ERK1/2 levels were normalized to total ERK1/2.
Statistical analysis
Statistical analysis was carried out using SPSS 11.5 for Windows (SPSS Inc., Chicago, IL, USA). Data are presented as the mean ± SD. Student’s t test was used to compare the rADC or rT2WI between the tMCAO group and tMCAO+MB group at each time point in the MRI experiment. One-way repeated measures ANOVA followed by post hoc Tukey’s test was used to compare the rADC or rT2WI at 0.5, 2.5, and 48 h after tMCAO in the tMCAO group or tMCAO+MB group. One-way ANOVA followed by post hoc Tukey’s test was used to statistically analyze the cell perimeter, ERK1/2 activation, and AQP4 protein and mRNA expression data. A value of P < 0.05 was considered statistically significant.
Results
MRI revealed that MB attenuated cytotoxicity and vasogenic brain edema in rats after tMCAO
The lower values of the rADC (the ipsilateral/contralateral ratio) indicated more diffusion of water in the intercellular space, namely, more serious cytotoxic brain edema [39].
In the current study, the rADC was decreased at 0.5 h (0.748 ± 0.009), 2.5 h (0.833 ± 0.040), and 48 h (0.772 ± 0.026) after tMCAO in rats, with a higher rADC at 2.5 h after tMCAO, indicating that tMCAO induced rapid cytotoxic brain edema in rats that lasted for 48 h, and the most serious cytotoxic brain edema occurred at 0.5 h after tMCAO.
After treatment with MB, the rADC in the MB group was 0.753 ± 0.013 at 0.5 h, 0.9138 ± 0.018 at 2.5 h, and 0.8161 ± 0.011 at 48 h after tMCAO. Compared with the tMCAO group, the MB-treated group showed no difference in rADC at 0.5 h, but a higher rADC was found at 2.5 and 48 h (P < 0.05, Fig. 1b), suggesting that MB attenuated the cytotoxic brain edema at 2.5 and 48 h after cerebral ischemia–reperfusion injury.
As local T2WI maps provide a quantitative evaluation of vasogenic brain edema severity [39], we utilized T2 imaging to determine the effect of MB on vasogenic brain edema in tMCAO rats.
The T2WI results showed that no difference was found between 0.5 and 2.5 h after cerebral ischemia–reperfusion injury in the tMCAO group (1.083 ± 0.037 and 1.122 ± 0.058, respectively), but the values of T2WI were significantly higher at 48 h after tMCAO (1.595 ± 0.028) than at 0.5 and 2.5 h after tMCAO, indicating that serious vasogenic brain edema occurred 48 h after tMCAO.
When MB was administered, T2WI did not reveal any differences between the tMCAO and MB groups at 0.5 h (1.083 ± 0.037, 1.100 ± 0.048, respectively) or 2.5 h (1.122 ± 0.058, 1.166 ± 0.044, respectively) after tMCAO. However, the value of T2WI at 48 h after tMCAO in the MB group (1.482 ± 0.045) was remarkably lower than that in the tMCAO (1.595 ± 0.028) group (P < 0.05, Fig. 1c), suggesting that MB attenuated vasogenic brain edema at 48 h after tMCAO.
MB blocked the increases in AQP4 expression in rats after tMCAO
In the rat model of tMCAO, H&E staining was used to visualize the changes in brain tissue structure after cerebral ischemia–reperfusion injury, which manifested as loose tissue structure, increased perivascular space, nuclear pyknosis and necrosis, and MB treatment attenuated these pathological changes (Fig. 2a). Immunohistochemical staining of albumin showed that the massive extravasation of albumin in the tMCAO group was decreased in the MB treatment group, indicating that MB alleviated the disruption of the BBB to improve vasogenic brain edema (Fig. 2a).

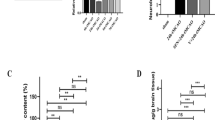
a H&E staining of the cerebral cortex in the ischemic penumbra and immunohistochemical staining of albumin, GFAP and AQP4. Scale bars: 50 μm. Arrows indicate AQP4 expression around blood vessels. b, c AQP4 mRNA and protein were detected by qRT-PCR and Western blot. The data are presented as the mean ± SD (n = 6 for each group), *P < 0.05 vs. ipsilateral injury in the sham group. #P < 0.05 vs. ipsilateral injury in the tMCAO group. &P < 0.05 vs. contralateral injury in the tMCAO group
Immunohistochemistry staining of GFAP, a marker of astrocytes, showed that the number of GFAP-positive astrocytes and the intensity of GFAP labeling were much greater in the tMCAO group than in the sham group but were decreased after MB treatment (Fig. 2a). Similarly, AQP4 expression, localized in astrocytes, increased in the ischemic penumbra at 48 h in tMCAO group compared with the sham group and was decreased in rats after treatment with MB; we also found that AQP4 is normally expressed in a highly polarized pattern, especially in endfeet around blood vessels, and the change in polarity distribution of AQP4 after cerebral ischemia–reperfusion injury and MB treatment attenuated the polarity distribution change (Fig. 2a).
qRT-PCR further revealed that the expression of AQP4 mRNA in the tMCAO group was significantly increased compared with that in the sham group, while MB blocked the increases in AQP4 after cerebral ischemia–reperfusion injury (Fig. 2b). Furthermore, Western blot results showed that the AQP4 protein in the tMCAO group was also remarkably elevated compared with that in the sham group and was decreased in the MB treatment group (Fig. 2c). Collectively, these results indicated that MB blocked the increases in AQP4 mRNA and protein levels in rats following tMCAO.
MB blocked the astrocyte swelling and AQP4 expression increase induced by glutamate in vitro
We subsequently examined the effect of MB on astrocyte swelling in a cell swelling model by using the cell perimeter to represent astrocyte volume. Cell perimeter measurements showed that, as in our previous report, cell swelling was induced in cultured rat astrocytes by exposure to 1 mM glutamate for 48 h; 0.1, 1, and 10 μM MB in media alone had no effect on the cell perimeter of astrocytes, but 10 μM MB significantly inhibited the astrocyte swelling induced by 1 mM glutamate (P < 0.05, Fig. 3a, b).

a Immunofluorescence staining of GFAP (shown in green); scale bar shows 200 μm. b Quantitative analysis of the imaging data of GFAP immunofluorescence staining (b). c, d AQP4 mRNA and protein were detected by qRT-PCR and Western blot. The data are presented as the mean ± SD (n = 9 for each group), *P < 0.05 vs. the control group (without glutamate and MB), #P < 0.05 vs. the glutamate group (1 mM glutamate and 0 mM MB)
The qRT-PCR results showed that 1 mM glutamate significantly increased AQP4 expression. Although MB at 0.1, 1, and 10 μM did not change AQP4 expression in cultured rat astrocytes, MB at 10 μM significantly blocked the increase in AQP4 expression induced by incubation with glutamate for 48 h (P < 0.05, Fig. 3c). Western blot analysis also confirmed that AQP4 expression was increased in cultured astrocytes incubated with 1 mM glutamate for 48 h (P < 0.05). Furthermore, 0.1, 1, and 10 μM MB had no effect on AQP4 expression under normal culture conditions, but MB at 10 μM had a remarkable antagonistic effect on the elevated AQP4 expression in the cell swelling model (P < 0.05, Fig. 3d). These results indicated that MB blocked astrocyte swelling and AQP4 expression increase in the cell swelling model.
MB inhibited the activation of ERK1/2 in tMCAO rats and the cell swelling model
To identify the possibility of MB downregulating AQP4 through the ERK1/2 signaling pathway to reduce brain edema and astrocyte swelling, we first examined the effect of MB on ERK1/2 activation after brain edema caused by cerebral ischemia-reperfusion injury in vivo and in an astrocyte swelling model. Western blot results showed a significant increase in p-ERK1/2 expression in tMCAO rats, while MB inhibited activation of the ERK1/2 signaling pathway in rats in the tMCAO+MB group (P < 0.05, Fig. 4a).

a p-ERK1/2 levels were detected by Western blot at 48 h after tMCAO and MB treatment. n = 6. The data are presented as the mean ± SD, *P < 0.05 vs. ipsilateral injury in the sham group. #P < 0.05 vs. ipsilateral injury in the tMCAO group. b Activation of p-ERK1/2 was determined by Western blot after incubation with/without glutamate and MB in cultured astrocytes. The data are presented as the ratios of p-ERK/ERK (mean ± SD, n = 9 for each group), *P < 0.05 vs. the control group (without glutamate and MB), #P < 0.05 vs. the glutamate group (1 mM glutamate and 0 mM MB)
In cultured astrocytes, p-ERK1/2 expression was increased after incubation with glutamate (P < 0.05). No differences in p-ERK1/2 expression in astrocytes were found after pretreatment with 0.1, 1, or 10 μM MB. When coincubated with glutamate, 10 μM MB significantly blocked the ERK1/2 activation induced by glutamate (P < 0.05), while 0.1 and 1 μM MB had no effects (Fig. 4b). These results showed that the ERK1/2 signaling pathway was activated during brain edema and astrocyte swelling and that MB inhibited ERK1/2 activation by the same mechanism through which it exerts its effects on brain edema/astrocyte swelling and AQP4 expression.
The ERK1/2 inhibitor U0126 mimicked the effects of MB on astrocyte swelling and AQP4 expression in vitro
Next, the ERK1/2 inhibitor U0126 was used to evaluate the role of ERK1/2 activation in the protective effect of MB against astrocyte swelling. Western blot analysis showed that no difference in p-ERK1/2 expression levels was found between the control astrocytes and the astrocytes pretreated with U0126. Compared with that in the control group, p-ERK1/2 expression was increased after incubation with glutamate (P < 0.05), which was antagonized by either U0126 or MB treatment (P < 0.05), and U0126 had a stronger inhibitory effect than MB. Moreover, p-ERK1/2 expression was further decreased when combined treatment with MB and U0126 was applied (P < 0.05, Fig. 5a), indicating that U0126 and MB have a synergistic effect of reducing p-ERK1/2 activation induced by glutamate in cultured astrocytes.

a p-ERK1/2 expression was observed by Western blot. b Immunofluorescence staining of GFAP (shown in green). The scale bar represents 50 μm. c Quantitative analysis of the imaging data of GFAP immunofluorescence staining (b). d, e AQP4 mRNA and protein levels were detected by qRT-PCR and Western blot, respectively. β-actin was used as a loading control. The data are presented as the mean ± SD (n = 9 for each group), *P < 0.05 vs. the control (c) group, #P < 0.05 vs. the glutamate (Glu) group, &P < 0.05 vs. the Glu+MB group
Quantitative analysis of the cell perimeter showed that U0126 had no effect in the control group but blocked glutamate-induced astrocyte swelling. In addition, MB pretreatment alone blocked astrocyte swelling induced by glutamate (P < 0.05), but combination treatment with MB and U0126 did not have a stronger effect (Fig. 5b, c), indicating that U0126 had the same potency as MB in ameliorating astrocyte swelling in cultured rat astrocytes.
Furthermore, U0126 had no effect on AQP4 expression in cultured astrocytes, as determined by qRT-PCR and Western blot, but it blocked the AQP4 increases induced by glutamate (P < 0.05). Combined treatment with MB and U0126 had no synergistic effect on AQP4 expression (Fig. 5d, e), indicating that MB and U0126 may have a common pathway for the inhibition of astrocyte swelling and AQP4 expression increases.
Discussion
Brain edema is a serious complication of ischemic stroke, which is a complex and devastating neurological disease with limited treatment options. In the present study, we demonstrated that MB attenuated cytotoxic and vasogenic brain edema in rats after tMCAO, as evidenced by the increased rADC value and the decreased rT2WI value. In addition, the effect of MB against glutamate-induced cell swelling was observed in primary astrocyte cultures. Furthermore, we found that MB inhibited AQP4 expression and activation of ERK1/2 both in the rat tMCAO model and the astrocyte swelling model. Similarly, the ERK1/2 inhibitor U0126 reversed astrocyte swelling and AQP4 expression increases induced by glutamate. Taken together, our findings indicated that MB could ameliorate brain edema and astrocyte swelling, potentially through the inhibition of AQP4 expression via the ERK1/2 signaling pathway.
A previous study reported that MRI was a more sensitive measurement for detecting brain edema. Brian edema is one of the most serious complications of ischemic stroke and is classified into two major types based on its underlying mechanism and time course: cytotoxic and vasogenic. Cytotoxic brain edema usually takes place within minutes after ischemic stroke onset [25] and is mainly presented as astrocyte swelling [40]. The progression from cytotoxic to vasogenic edema occurred within 2–3 days because subsequent prolonged periods of ischemic stroke can prompt the breakdown of the BBB [41]. The cytotoxic and vasogenic forms of brain edema coexist in the late stage of ischemic stroke.
Loubinoux et al. reported that reduced ADC values reflect cytotoxic brain edema, whereas increased T2WI values correlate with the development of vasogenic edema [39]. Our current study found that transient cerebral ischemia decreased rADC at 0.5, 2.5, and 48 h and increased rT2WI at 48 h, consistent with the report of He et al. [42]. Importantly, we illustrated that MB alleviated the decrease in rADC at 2.5 and 48 h and attenuated the increase in rT2WI at 48 h after tMCAO. These results suggested that MB might play a crucial role in the protection against not only cytotoxic brain edema but also vasogenic brain edema. Interestingly, our previous study did not find an effect of MB on ADC-defined ischemic lesions at 2.5 h after tMCAO [15]. The MRI findings of the present study indicated that alterations of rADC are more sensitive than ADC-defined infarct volume during cerebral ischemia- reperfusion injury. The effect of MB against brain edema was further verified in an astrocyte swelling model in which MB inhibited the astrocyte swelling induced by glutamate in astrocyte cultures. These results were consistent with our previous transmission electron microscopy study showing that MB remarkably reduced astrocyte swelling after tMCAO in rats [15].
AQP4 is the principal water channel protein mainly expressed in astrocytes throughout the CNS. Many studies have demonstrated that AQP4 participates in the formation and resolution of cytotoxic brain edema and vasogenic brain edema [43, 44]. Studies have found that the ADC value was negatively correlated with AQP4 expression [45, 46], indicating that AQP4 is involved in brain edema. Existing research shows that the water permeability of AQP4 is increased within a short time of cell swelling [47, 48] and that the change in AQP4 protein and mRNA expression mainly occurs several hours after the onset of cell swelling. In the present study, increased AQP4 was detected both in the tMCAO rat model and astrocyte swelling model, providing direct evidence that AQP4 participated in ischemic brain edema and astrocyte swelling. Accumulating evidence has indicated that increased AQP4 in astrocytes mediates not only cytotoxic brain edema but also vasogenic brain edema [49]. Research evidence has indicated that increased AQP4 not only is associated with brain edema but also reflected the elevated permeability of the BBB [50]. In the current study, we found that cerebral ischemia–reperfusion causes the leakage of albumin concurrent with increases in AQP4 expression and that the blockage of increased AQP4 by MB not only reduced astrocyte swelling but also attenuated BBB damage. In accordance with our results, Miclescu et al. showed that MB played an important role in the neuroprotective effect against brain damage related to cerebral ischemia–reperfusion injury and ameliorated BBB disruption [51]. We found that MB blocked AQP4 increases and alleviated the decrease in rADC and increase in rT2WI in the tMCAO rat model, suggesting a protective effect of MB against cytotoxic and vasogenic brain edema induced by ischemic stroke and that MB improved brain edema by inhibiting AQP4 expression. Therefore, we proposed that MB may provide a new choice for the treatment of brain ischemic edema.
We further explored the signaling pathway that mediated the effect of MB on the alleviation of brain edema and inhibition of AQP4 expression. Several studies have suggested that ERK1/2 activation is involved in the regulation of AQP4 expression in astrocytes under pathophysiological conditions. Qi et al. reported that activation of the ERK1/2 and p38 MAPK pathways upregulated AQP4, which led to oxygen–glucose deprivation/reperfusion-induced injury [52]. Nito et al. showed that ERK1/2 mediated AQP4 expression in MCAO rats and rat cortical astrocytes exposed to OGD [53]. In the present study, we also found the activation of ERK1/2 both in the tMCAO rat model and cell swelling model. We further found that MB treatment blocked ERK1/2 activation and inhibited the increase in AQP4 expression. Moreover, in astrocyte cultures, we found that U0126, an ERK1/2 pathway inhibitor, blocked ERK1/2 activation as well as astrocyte swelling and AQP4 increases induced by glutamate. No synergistic effects of U0126 and MB on astrocyte swelling and AQP4 expression were observed, indicating that the effect of MB on astrocyte swelling and AQP4 expression was likely mediated by ERK1/2 signaling. Prior studies have shown that the inhibition of ERK1/2 activation improved the neurological outcome following focal cerebral ischemia [54]. Activation of ERK1/2 after cerebral ischemia is involved in dysregulation of the intracellular Ca2+ concentration, inflammation, oxidative stress, and cell death [55,56,57]. Our results suggested that the ERK1/2 signaling pathway may also participate in brain edema/astrocyte swelling and AQP4 regulation after brain ischemia and that MB may attenuate ischemic brain edema and astrocyte swelling by inhibiting ERK1/2 activation.
MB has already been used in some clinical treatments. Recent research has also shown that MB can pass through the BBB and reach the brain, where the concentration of MB is 10 times higher than that in normal circulation in rats, indicating its potential use in CNS diseases [58]. Furthermore, there are no side effects with long-term use of an equivalent dose of MB in humans, pointing to the possibility of future clinical treatment. In the current study, we found that MB attenuated brain edema at 2.5 and 48 h after cerebral ischemia–reperfusion injury in rats. Given that such a rapid application of MB to patients with ischemic stroke in the clinic is unlikely, further research on the therapeutic time window of MB for cerebral edema is required. In addition, future studies are also needed to establish the optimal drug delivery method and the MB administration treatment duration, frequency and/or dose. The therapeutic efficacy of MB for brain edema should also be evaluated in other animal models. Existing studies have shown that MB, as an alternative to the mitochondrial electron transport chain, may participate in neuroprotection by improving energy metabolism. Dysfunction of energy metabolism is an important mechanism of brain edema [59]. Much research suggests that MB increases ATP content in stroke models [11, 60]. Whether MB can reduce brain edema induced by ischemic stroke by improving energy metabolism is worth further study. Potential interactions between AQP4 and Na/K-ATPase have been indicated. Ouabain, a Na/K-ATPase inhibitor, was reported to be involved in ammonia-induced astrocyte swelling by activation of the ERK1/2 signaling pathway [61]. Further studies are needed to explore the role of Na/K-ATPase in the MB-mediated effect on AQP4 against brain edema.
In summary, the current study demonstrated that MB could reduce cytotoxic brain edema and vasogenic cerebral edema after tMCAO and ameliorate astrocyte swelling induced by glutamate in cultured astrocytes. The mechanism of the effect of MB on cerebral edema might be mediated by suppression of AQP4 overexpression through inhibition of the ERK1/2 signaling pathway. These results suggested that MB might provide a new choice for the treatment of cerebral edema caused by brain ischemia.
References
Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36:513–38.
Wang W, Jiang B, Sun H, Ru X, Sun D, Wang L, et al. Prevalence, incidence, and mortality of stroke in China: results from a nationwide population-based survey of 480 687 adults. Circulation. 2017;135:759–71.
Posada-Duque RA, Barreto GE, Cardona-Gomez GP. Protection after stroke: cellular effectors of neurovascular unit integrity. Front Cell Neurosci. 2014;8:231.
Seyedsaadat SM, Kallmes DF. Memantine for the treatment of ischemic stroke: experimental benefits and clinical lack of studies. Rev Neurosci. 2019;30:203–20.
Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 2007;6:258–68.
Walberer M, Ritschel N, Nedelmann M, Volk K, Mueller C, Tschernatsch M, et al. Aggravation of infarct formation by brain swelling in a large territorial stroke: a target for neuroprotection? J Neurosurg. 2008;109:287–93.
Wang WW, Xie CL, Zhou LL, Wang GS. The function of aquaporin4 in ischemic brain edema. Clin Neurol Neurosurg. 2014;127:5–9.
Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 2011;286:16504–15.
Wiklund L, Sharma A, Sharma HS. Neuroprotection by methylene blue in cerebral global ischemic injury induced blood-brain barrier disruption and brain pathology: a review. CNS Neurol Disord Drug Targets. 2016;15:1181–7.
Tucker D, Lu Y, Zhang Q. From mitochondrial function to neuroprotection-an emerging role for methylene blue. Mol Neurobiol. 2018;55:5137–53.
Yang SH, Li W, Sumien N, Forster M, Simpkins JW, Liu R. Alternative mitochondrial electron transfer for the treatment of neurodegenerative diseases and cancers: methylene blue connects the dots. Prog Neurobiol. 2017;157:273–91.
Delport A, Harvey BH, Petzer A, Petzer JP, Azure B. and a synthetic structural analogue of methylene blue, ethylthioninium chloride, present with antidepressant-like properties. Life Sci. 2014;117:56–66.
Talley Watts L, Long JA, Chemello J, Van Koughnet S, Fernandez A, Huang S, et al. Methylene blue is neuroprotective against mild traumatic brain injury. J Neurotrauma. 2014;31:1063–71.
Jiang Z, Watts LT, Huang S, Shen Q, Rodriguez P, Chen C, et al. The effects of methylene blue on autophagy and apoptosis in MRI-defined normal tissue, ischemic penumbra and ischemic core. PLoS One. 2015;10:e0131929.
Fang Q, Yan X, Li S, Sun Y, Xu L, Shi Z, et al. Methylene blue ameliorates ischemia/reperfusion-induced cerebral edema: an MRI and transmission electron microscope study. Acta Neurochir Suppl. 2016;121:227–36.
Schlaug G, Benfield A, Baird AE, Siewert B, Lovblad KO, Parker RA, et al. The ischemic penumbra: operationally defined by diffusion and perfusion MRI. Neurology. 1999;53:1528–37.
Moseley ME, Kucharczyk J, Mintorovitch J, Cohen Y, Kurhanewicz J, Derugin N, et al. Diffusion-weighted MR imaging of acute stroke: correlation with T2-weighted and magnetic susceptibility-enhanced MR imaging in cats. AJNR Am J Neuroradiol. 1990;11:423–9.
Shen Q, Meng X, Fisher M, Sotak CH, Duong TQ. Pixel-by-pixel spatiotemporal progression of focal ischemia derived using quantitative perfusion and diffusion imaging. J Cereb Blood Flow Metab. 2003;23:1479–88.
Shen Q, Fisher M, Sotak CH, Duong TQ. Effects of reperfusion on ADC and CBF pixel-by-pixel dynamics in stroke: characterizing tissue fates using quantitative diffusion and perfusion imaging. J Cereb Blood Flow Metab. 2004;24:280–90.
Shen Q, Ren H, Cheng H, Fisher M, Duong TQ. Functional, perfusion and diffusion MRI of acute focal ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:1265–79.
Schitine C, Nogaroli L, Costa MR, Hedin-Pereira C. Astrocyte heterogeneity in the brain: from development to disease. Front Cell Neurosci. 2015;9:76.
Vella J, Zammit C, Di Giovanni G, Muscat R, Valentino M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front Cell Neurosci. 2015;9:108.
Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–63.
Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–3.
Yao X, Derugin N, Manley GT, Verkman AS. Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci Lett. 2015;584:368–72.
He ZP, Lu H. Aquaporin-4 gene silencing protects injured neurons after early cerebral infarction. Neural Regen Res. 2015;10:1082–7.
Yuan F, Wang T. Glutamate-induced swelling of cultured astrocytes is mediated by metabotropic glutamate receptor. Sci China C Life Sci. 1996;39:517–22.
Shi Z, Zhang W, Lu Y, Lu Y, Xu L, Fang Q, et al. Aquaporin 4-mediated glutamate-induced astrocyte swelling is partially mediated through metabotropic glutamate receptor 5 activation. Front Cell Neurosci. 2017;11:116.
Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–88.
Hu Y, Duan M, Liang S, Wang Y, Feng Y. Senkyunolide I protects rat brain against focal cerebral ischemia-reperfusion injury by up-regulating p-Erk1/2, Nrf2/HO-1 and inhibiting caspase 3. Brain Res. 2015;1605:39–48.
Lopez-Morales MA, Castello-Ruiz M, Burguete MC, Jover-Mengual T, Aliena-Valero A, Centeno JM, et al. Molecular mechanisms underlying the neuroprotective role of atrial natriuretic peptide in experimental acute ischemic stroke. Mol Cell Endocrinol. 2018;472:1–9.
Liu R, Yuan H, Yuan F, Yang SH. Neuroprotection targeting ischemic penumbra and beyond for the treatment of ischemic stroke. Neurological Res. 2012;34:331–7.
Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–98.
Ashwal S, Tone B, Tian HR, Cole DJ, Pearce WJ. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke. 1998;29:1037–46.
Huang YC, Tzeng WS, Wang CC, Cheng BC, Chang YK, Chen HH, et al. Neuroprotective effect of agmatine in rats with transient cerebral ischemia using MR imaging and histopathologic evaluation. Magn Reson Imaging. 2013;31:1174–81.
Qian Q, Huang HT, Xu L, Jin P, Lin M. Prediction of infarct lesion volumes by processing magnetic resonance apparent diffusion coefficient maps in patients with acute ischemic stroke. J Stroke Cerebrovasc Dis. 2016;25:2821–7.
Krueger M, Mages B, Hobusch C, Michalski D. Endothelial edema precedes blood-brain barrier breakdown in early time points after experimental focal cerebral ischemia. Acta Neuropathol Commun. 2019;7:17.
Shi ZF, Zhao WJ, Xu LX, Dong LP, Yang SH, Yuan F. Downregulation of aquaporin 4 expression through extracellular signal-regulated kinases1/2 activation in cultured astrocytes following scratch-injury. Biomed Environ Sci. 2015;28:199–205.
Loubinoux I, Volk A, Borredon J, Guirimand S, Tiffon B, Seylaz J, et al. Spreading of vasogenic edema and cytotoxic edema assessed by quantitative diffusion and T2 magnetic resonance imaging. Stroke. 1997;28:419–26.
Stokum JA, Kurland DB, Gerzanich V, Simard JM. Mechanisms of astrocyte-mediated cerebral edema. Neurochem Res. 2015;40:317–28.
Previch LE, Ma L, Wright JC, Singh S, Geng X, Ding Y. Progress in AQP research and new developments in therapeutic approaches to ischemic and hemorrhagic stroke. Int J Mol Sci. 2016;17:1146.
He Z, Wang X, Wu Y, Jia J, Hu Y, Yang X, et al. Treadmill pre-training ameliorates brain edema in ischemic stroke via down-regulation of aquaporin-4: an MRI study in rats. PLoS One. 2014;9:e84602.
Verkman AS, Smith AJ, Phuan PW, Tradtrantip L, Anderson MO. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin Ther Targets. 2017;21:1161–70.
Hubbard JA, Szu JI, Binder DK. The role of aquaporin-4 in synaptic plasticity, memory and disease. Brain Res Bull. 2018;136:118–29.
Tan Y, Zhang H, Wang XC, Qin JB, Wang L. The value of multi ultra high-b-value DWI in grading cerebral astrocytomas and its association with aquaporin-4. Br J Radiol. 2018;91:20170696.
Lu H, Hu H, He ZP. Reperfusion of the rat brain tissues following acute ischemia: the correlation among diffusion-weighted imaging, histopathology, and aquaporin-4 expression. Chin Med J. 2011;124:3148–53.
Gunnarson E, Zelenina M, Axehult G, Song Y, Bondar A, Krieger P, et al. Identification of a molecular target for glutamate regulation of astrocyte water permeability. Glia. 2008;56:587–96.
Farr GW, Hall CH, Farr SM, Wade R, Detzel JM, Adams AG, et al. Functionalized phenylbenzamides inhibit aquaporin-4 reducing cerebral edema and improving outcome in two models of CNS injury. Neuroscience. 2019;404:484–98.
Chu H, Huang C, Ding H, Dong J, Gao Z, Yang X, et al. Aquaporin-4 and cerebrovascular diseases. Int J Mol Sci. 2016;17:1249.
Ceccariglia S, D’Altocolle A, Del Fa A, Silvestrini A, Barba M, Pizzolante F, et al. Increased expression of Aquaporin 4 in the rat hippocampus and cortex during trimethyltin-induced neurodegeneration. Neuroscience. 2014;274:273–88.
Miclescu A, Sharma HS, Martijn C, Wiklund L. Methylene blue protects the cortical blood-brain barrier against ischemia/reperfusion-induced disruptions. Crit Care Med. 2010;38:2199–206.
Qi LL, Fang SH, Shi WZ, Huang XQ, Zhang XY, Lu YB, et al. CysLT2 receptor-mediated AQP4 up-regulation is involved in ischemic-like injury through activation of ERK and p38 MAPK in rat astrocytes. Life Sci. 2011;88:50–6.
Nito C, Kamada H, Endo H, Narasimhan P, Lee YS, Chan PH. Involvement of mitogen-activated protein kinase pathways in expression of the water channel protein aquaporin-4 after ischemia in rat cortical astrocytes. J Neurotrauma. 2012;29:2404–12.
Ahnstedt H, Mostajeran M, Blixt FW, Warfvinge K, Ansar S, Krause DN, et al. U0126 attenuates cerebral vasoconstriction and improves long-term neurologic outcome after stroke in female rats. J Cereb Blood Flow Metab. 2015;35:454–60.
Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12:698–714.
Shenoda B. The role of Na+/Ca2+ exchanger subtypes in neuronal ischemic injury. Transl Stroke Res. 2015;6:181–90.
Siniscalchi A, Gallelli L, Malferrari G, Pirritano D, Serra R, Santangelo E, et al. Cerebral stroke injury: the role of cytokines and brain inflammation. J Basic Clin Physiol Pharmacol. 2014;25:131–7.
Huang L, Lu J, Cerqueira B, Liu Y, Jiang Z, Duong TQ. Chronic oral methylene blue treatment in a rat model of focal cerebral ischemia/reperfusion. Brain Res. 2018;1678:322–9.
Ryou MG, Choudhury GR, Li W, Winters A, Yuan F, Liu R, et al. Methylene blue-induced neuronal protective mechanism against hypoxia-reoxygenation stress. Neuroscience. 2015;301:193–203.
Roy Choudhury G, Winters A, Rich RM, Ryou MG, Gryczynski Z, Yuan F, et al. Methylene blue protects astrocytes against glucose oxygen deprivation by improving cellular respiration. PLoS One. 2015;10:e0123096.
Dai H, Song D, Xu J, Li B, Hertz L, Peng L. Ammonia-induced Na, K-ATPase/ouabain-mediated EGF receptor transactivation, MAPK/ERK and PI3K/AKT signaling and ROS formation cause astrocyte swelling. Neurochem Int. 2013;63:610–25.
Acknowledgements
We sincerely appreciate the help of Prof. Shao-hua Yang from the Department of Pharmacology and Neuroscience, University of North Texas Health Science Center, for critical comments on the paper. We also thank Prof. Shao-wu Li from the Neurological Imaging Center, Beijing Neurosurgical Institute, for help with MRI data acquisition and Dr Ying Jiang from the Department of Functional Neurosurgery, Beijing Neurosurgical Institute, for guidance related to the MRI data analysis. This project is supported by the National Natural Science Foundation of China (81801159, 8127128, and 51707012).
Author information
Authors and Affiliations
Contributions
ZFS carried out the experiments and contributed to the acquisition, analysis, and interpretation of data and drafting of the paper; QF, YC, LXX, MW, MJ, YL, XXW, YJW, XY, and LPD conducted the experiments, acquired the data, and analyzed the data; FY conceived and designed the experiments for the work and contributed to acquisition, analysis, and interpretation of data and drafting of the paper. All authors read and approved the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Shi, Zf., Fang, Q., Chen, Y. et al. Methylene blue ameliorates brain edema in rats with experimental ischemic stroke via inhibiting aquaporin 4 expression. Acta Pharmacol Sin 42, 382–392 (2021). https://doi.org/10.1038/s41401-020-0468-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0468-5
Keywords
This article is cited by
-
Non-coding RNAs and Aquaporin 4: Their Role in the Pathogenesis of Neurological Disorders
Neurochemical Research (2024)
-
Crosstalk Among Glial Cells in the Blood–Brain Barrier Injury After Ischemic Stroke
Molecular Neurobiology (2024)
-
Macrophage migration inhibitory factor exacerbates asthmatic airway remodeling via dynamin-related protein 1-mediated autophagy activation
Respiratory Research (2023)
-
Brain-Targeting Emodin Mitigates Ischemic Stroke via Inhibiting AQP4-Mediated Swelling and Neuroinflammation
Translational Stroke Research (2023)
-
Tanshinone IIA reduces AQP4 expression and astrocyte swelling after OGD/R by inhibiting the HMGB1/RAGE/NF-κB/IL-6 pro-inflammatory axis
Scientific Reports (2022)
