
Abstract
Oroxindin is a flavonoid isolated from the traditional Chinese medicine Huang-Qin, which has shown various pharmacological activities including anti-inflammatory, antitumor, antioxidant, etc. Thus far, the effect of oroxindin on colonic inflammation and the underlying mechanism remain unknown. In this study, we investigated the tissue distribution of oroxindin and its therapeutic effects on ulcerative colitis (UC) as well as the underlying mechanisms. UC model was established in mice by administrating dextran sulfate sodium (DSS) in drinking water for 7 d. We first showed that oroxindin was largely absorbed by the colon as an active ingredient after normal mice received Huang-Qin-Tang, a traditional Chinese medicine decoction. UC mice were then treated with oroxindin (12.5, 25, 50 mg ·kg−1 ·d−1, i.g.) for 10 d. We found that oroxindin treatment greatly suppressed massive macrophages infiltration and attenuated pathological changes in colonic tissue. Furthermore, oroxindin treatment significantly inhibited the generation of IL-1β and IL-18 in the colon via inhibiting the nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) inflammasome formation and activation. In cultured macrophages, LPS induced NLRP3 inflammasome formation and caspase-1 activation, which were suppressed by oroxindin (12.5–50 μM). In LPS-treated macrophages, oroxindin dose-dependently restored the expression of TXNIP protein, leading to suppressing TXNIP-dependent NF-κB activation. In conclusion, these results demonstrate that oroxindin could be absorbed by the colon and attenuate inflammatory responses via inhibiting NLRP3 inflammasome formation and activation, which is related to the inhibitory effect on TXNIP-dependent NF-κB-signaling pathway. Hence, oroxindin has the potential of becoming an effective drug for treating UC.
Similar content being viewed by others


Introduction
Ulcerative colitis (UC) is a chronic and relapsing inflammatory disease that occurs in the colon with abdominal pain, rectal bleeding, and diarrhea [1]. Moreover, patients with UC have an increased risk of developing colorectal carcinoma [2, 3]. The precise mechanisms involved in the initiation and development of UC have not yet been completely clarified. Numerous studies have indicated that genetic alterations and the impaired balance in the intestinal immune system may contribute to the development of UC [4, 5]. In particular, imbalance in the mucosal immune system causes the generation of pro-inflammatory cytokines, which in turn leads to chronic inflammation, ulceration, and lesions in the colonic mucosa [4,5,6,7]. The proinflammatory cytokines IL-1β and IL-18 are considered important pathogenic factors during the development of UC [8,9,10]. Meanwhile, the secretion of IL-1β and IL-18 is regulated by activated caspase-1, which is controlled by the nucleotide-binding oligomerization domain-like receptor (NLR) inflammasome. Intestinal inflammation has been studied in NLR-deficient mice [11], and evidence has confirmed the pathogenic role of the NLRP3 inflammasome in UC [12, 13], which makes the NLRP3 inflammasome a potential target for UC treatment.
Thioredoxin-interacting protein (TXNIP) possesses various functions including regulating cell metabolism, cell death, tumor formation, and inflammation [14, 15]. TXNIP was shown to compete with migration inhibitory factor (MIF) for NF-κB activation [16]. LPS is known to inhibit the expression of TXNIP mRNA and protein levels and activate the NF-κB-signaling pathway [17]. A previous study demonstrated that when treated with LPS or TNF-α, the expression of TXNIP was significantly decreased and the inhibition of TNF-α-induced NF-κB activation was mostly dependent on TXNIP in hepatocarcinogenesis mice [18], which suggested a protective role of TXNIP. Recently, some studies showed that TXNIP was down-regulated in patients with inflamed colonic mucosa and suggested that thioredoxin (TRX) might be a potential therapeutic agent for the treatment of inflammatory bowel disease (IBD) [19, 20]. In summary, these studies indicated that monomers that regulate TXNIP-dependent NF-κB activation and inhibit NLRP3 inflammasome activation have the potential to become new anti-inflammatory drugs for treating UC.
At present, UC has evolved into a global disease and is severely threatening people’s health in many Asian countries [21], while existing drugs for UC are usually associated with serious side effects [21,22,23]. As a traditional Chinese medicine decoction, Huang-Qin-Tang (HQT) is used for treating UC in China with a more than 2000-year history [23,24,25]. To determine the bioactive ingredients in HQT, in this study, we measured the accumulated constituents of HQT in colon tissue. Consequently, we chose oroxindin and explored its protective mechanism against UC. Oroxindin is a natural bioflavonoid isolated from Huang-Qin (one of the major ingredients in HQT). Some studies have demonstrated that oroxindin exhibits various bioactivities including anti-inflammatory, antitumor, antioxidant, etc. [26,27,28]. However, the protective effects of oroxindin on regulating the function of colonic macrophages during UC and the inhibitory mechanism of oroxindin on the NLRP3 inflammasome remain unclear. In the present study, we investigated the protective effects of oroxindin and the underlying mechanisms, and proved that oroxindin attenuates UC by inhibiting NLRP3 inflammasome activation via suppressing the TXNIP-dependent NF-κB-signaling pathway in colonic macrophages.
Materials and methods
Reagents and antibodies
Oroxindin (>98% purity; Chengdu Pufei De Biotech Co., Ltd., China) was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 0.04 M and stored at −20 °C. The stock was then diluted with Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Suzhou, China) to the final concentration used in each experiment. Lipopolysaccharide (LPS) from Escherichia coli serotype O111:B4, 3-(4,5-dimethylthiazol-2-yl)−2,5-di-phenyltetrazolium bromide (MTT), 5-aminosalicylic acid, and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO, USA). 4′,6-Diamidino-2-phenylindole was purchased from Invitrogen (Carlsbad, CA, USA). Dextran sulfate sodium (DSS) and DMSO were purchased from MP Biomedicals (Solon, OH, USA). β-Actin and hematoxylin were obtained from Boster Biological Technology Co., Ltd. (Wuhan, China). Antibodies against caspase-1, mouse epidermal growth factor-like module-containing mucin-like hormone receptor-like 1 (also known as F4/80), and apoptosis-associated speck-like protein containing a caspase-associated recruitment domain (ASC) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). NLRP3, TXNIP, and p-P65 primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). An immunofluorescent NLRP3 antibody was purchased from Abcam (Cambridge, MA, USA). An Alexa Fluor 488 (Abcam) secondary antibody kit for immunohistochemistry was obtained from Cell Signaling Technology. Protein A/G PLUS-agarose beads were from Santa Cruz Biotechnology (sc-2003). An immunohistochemistry application solution kit was obtained from Cell Signaling Technology.
Liquid chromatography–mass spectrometry (LC–MS) analysis
The LC–MS system consisted of a Triple TOF™ 5600+ mass spectrometer (AB Sciex, Foster City, CA, USA) and a prominence ultra-high-performance LC system (Nexera LC-30A; Shimadzu, Kyoto, Japan). Data were analyzed using MultiQuant 3.0 software. After overnight fasting, mice were administered HQT decoctions at a dose of 20 g/kg, and tissue samples were collected at 0.5, 1, 3, and 12 h. Colon tissue samples were washed in normal saline and homogenized with methanol to a concentration of 500 mg/mL. The supernatant of the homogenate (100 μL) was deproteinized using a previously described method. Then, 25 μL of an internal standard solution (final concentration: 500 ng/mL) and 400 μL of methanol were added. Finally, 5 μL samples were injected into an Agilent SB-C18 column (2.1 mm × 100 mm, 1.8 μm) for analysis, using a mobile phase of (A) acetonitrile and (B) 0.5% formic acid in water at a flow rate of 0.25 mL/min. Paeoniflorin was detected by an electrospray ionization (ESI) anion, and the remaining constituents were measured by ESI-positive-ion detection and multiple reaction monitoring. To prepare standard samples, stock solutions of baicalin, oroxindin, glycyrrhizin, glycyrrhizinate, and paeoniflorin were diluted with methanol to nominal concentrations of 11.8652–48,600, 12.55–1200, 0.2563–1050, 1.8554–7600, and 2.05078–8400 ng/mL, respectively.
DSS-induced UC and drug treatment
Adult male C57BL/6 mice (6–8 weeks old, 18–22 g) were supplied by and maintained at the Laboratory Animal Services Center of Guangzhou University of Chinese Medicine (Guangzhou, China). The animal study followed the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the animal protocol was approved by the Animal Ethics Committee of Guangzhou University of Chinese Medicine. The animals were housed in cages under standard environmental conditions of a temperature of 20–25 °C under a 12-h light/dark cycle and were provided free access to standard food and water. They were randomly divided into six groups: control, DSS, oroxindin (12.5, 25, or 50 mg/kg) +DSS group, and sulfasalazine (50 mg/kg) +DSS-treatment groups. To establish the UC model, the model group received 3% (w/v) DSS in drinking water for 7 d, followed by regular water provided for 3 d, while the control group received regular drinking water. Oroxindin was dissolved in water to prepare an oral suspension, and oroxindin and sulfasalazine were administered via gastric intubation from d 1 to d 10 after DSS treatment until the termination of the experiment. The mice were sacrificed on d 11.
Histological analysis
The pieces of colonic tissues were fixed with 4% paraformaldehyde, embedded in paraffin and cut into 5-μm-thick sections. Hematoxylin–eosin (H&E) staining was performed. Colon histopathological changes were observed by a light microscope (Olympus, Tokyo, Japan). The extent of histopathological changes was scored according to previous studies [29] (Table 1).
Immunohistochemical analysis of colon tissues
Five-micrometer-thick paraffin sections were used for immunohistochemical experiments. For antigen retrieval, the samples were placed in citric acid solution (pH 9.0) and heated in a microwave oven for 10 min. Subsequently, the sections were incubated with an animal-free blocking solution for 1 h, prior to incubation with the F4/80 monoclonal antibody overnight at 4 °C. After that, the sections were labeled with secondary antibodies for 2 h. 3′-Diaminobenzidine was used to produce a brown signal, and hematoxylin was used as a counterstain. More detailed steps were performed according to the immunohistochemistry application solutions kit. The immunohistochemical images were visualized by a microscope (BX53; Olympus Corp., Tokyo, Japan) and analyzed by Image-Pro Plus software.
Cell culture and drug treatment
THP-1 cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C in DMEM supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 100 U/mL benzylpenicillin, and 100 mg/mL streptomycin. When cells reached ~80% confluence, they were plated in six-well plates at a density of ~1 × 106 cells/well and treated with various concentrations of oroxindin (12.5, 25, and 50 μM) for 2 h. Then, 1 μg/mL LPS was added, and the cells were incubated for an additional 24 h. Nitric oxide (NO) was measured according to the manufacturer’s protocols.
Co-immunoprecipitation experiment
Co-immunoprecipitation experiments were performed according to the user’s protocol (Santa Cruz Biotechnology, Inc. sc-2003). Ice-cold immunoprecipitation lysis buffer was added to THP-1 cells before shaking for 15 min. After incubation on ice for 30 min, the cell lysate was centrifuged (12,000×g, 30 min, 4 °C) and the supernatant was incubated with 10 μL of PBS-suspended protein A/G PLUS-Agarose beads and incubated for 10 min at 4 °C. Subsequently, the supernatant was incubated with 2 μg of ASC antibody for 4 h at 4 °C. Immunoprecipitates were then collected by centrifugation at 1000×g for 5 min, and the pellet was resuspended in 2× sample buffer and boiled. The bound proteins of NLRP3 and Pro-Casp1 were detected by Western blot.
Western blot analysis
When cells reached ~80% confluence, they were cultured in six-well dishes and treated with various concentrations of oroxindin (12.5, 25, and 50 μM) for 2 h. Then, 1 μg/mL LPS was added, and the cells were incubated for additional 24 h to detect NLRP3, ASC, TXNIP, and Caspase-1 protein. To detect phospho-NF-κB p65 (p-P65) protein, LPS was incubated for an additional 1 h. Cellular proteins were extracted using a cytoplasmic and nuclear extraction kit (Beyotime Institute of Biotechnology, Shanghai, China). The Bradford method was used to determine the protein concentration. Subsequently, the proteins were separated in sodium dodecyl sulfate–polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA). The membranes were incubated at room temperature with 5% non-fat milk or 8% bovine serum albumin (BSA) in Tris-buffered saline with Tween 20 (TBST) to block non-specific binding for 2 h, followed by incubation with the specific antibodies targeting NLRP3 (1:1000), ASC (8:5000), TXNIP (1:1000), p-P65 (1:1000), Caspase-1 (3:1000), and iNOS (1:1000) at 4 °C overnight. On the second day, the membranes were washed with TBST three times, followed by incubation with an appropriate secondary antibody for 2 h at room temperature. Protein bands were visualized using enhanced chemiluminescence.
Measurement of IL-1β in cells
Cells were cultured at 1 × 106 cells/well in 96-well plates. Experiment groups and drug treatment have been stated above in the “Cell culture and drug treatment” section. The culture supernatant was collected to determine the concentration of IL-1β by an ELISA kits following the manufacture’s instructions.
Immunofluorescence microscopic analysis of colon tissues
Eight-micrometer-thick frozen colon tissue sections were used for immunofluorescence experiments. Briefly, the sections were placed in acetone for 10 min, permeabilized with 0.2% Triton X-100, and blocked with 10% BSA for 1 h at room temperature. For immunofluorescence, the sections were incubated with anti-F4/80 (1:100) plus anti-NLRP3 (1:100) or anti-caspase-1 (1:100) antibodies overnight. On the next day, the slides were washed and labeled with corresponding conjugated secondary antibodies (Alexa Fluor 488 and Dylight 594) for 2 h. Then, slides were visualized sequentially using a laser scanning confocal microscope (ZEN2.3, Zeiss) [30, 31]. Settings for image acquisition were identical for both control and experimental tissues.
Measurement of IL-1β and IL-18 in colon tissue
The colon tissues were homogenized in cold normal saline buffer and centrifuged at 3000×g for 5 min at 4 °C. The culture supernatant was collected to determine the concentration of IL-1β and IL-18 by ELISA kits following the manufacturer’s instructions. Subsequently, the protein concentration of the supernatant was detected using a BCA kit. The results are shown as the ratio of the amount of cytokines to the protein concentration.
Statistical analysis
Statistical analysis was performed using GraphPad Prism and SPSS 17.0 software. Data are expressed as the mean ± standard deviation (SD) or mean ± standard error of the mean (SEM) and are representative of at least three experiments. P < 0.05 was considered to indicate a statistically significant difference.
Results
Concentration of baicalin, oroxindin, glycyrrhizin, and paeoniflorin in the colon
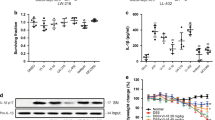
We analyzed the main ingredients of HQT and measured the concentration of four principal constituents (baicalin, oroxindin, glycyrrhizin, and paeoniflorin) in the colon tissue. After being fed with HQT (1 g/kg), normal mice were sacrificed at predetermined time points (0.5, 1, 3, and 12 h), and the results were shown in Fig. 1a. Fig. 1a shows that large amounts of baicalin and oroxindin accumulated in colon tissue. Subsequently, we calculated the ratio of monomer concentration in the colon tissue to the monomer concentration in HQT, and the ratio of oroxindin was higher than that of other compounds (Fig. 1b).

The normal mice were orally administered Huang-Qin-Tang (1 g/kg). a Concentrations of baicalin, oroxindin, glycyrrhizin, and paeoniflorin in the colon. b Relative concentrations of the compounds in the colon compared to their concentrations in Huang-Qing-Tang. Values are the mean ± SEM. Bai baicalin, Gly glycyrrhizin, Pae paeoniflorin, Oro oroxindin, Glyc glycyrrhizinate.
Oroxindin ameliorated DSS-induced UC
To determine the protective effect of oroxindin against UC, we established the acute colitis model by administering DSS [32]. Specifically, C57BL/6 mice were treated with 3% DSS in drinking water for 7 d and were then provided regular water for 3 d. Oroxindin (12.5, 25, and 50 mg/kg) and sulfasalazine (50 mg/kg) were orally administered from d 1 to d 10. Based on the data obtained, we found that oroxindin treatment attenuated body weight loss (percentage of that on d 1) compared with that of the model group (Fig. 2a). DSS caused colonic shortening, while the symptom was markedly improved by oroxindin treatment during colitis progression (Fig. 2b, c). As shown in Fig. 2d, we also measured the spleen index in each group, which was raised in the model group and reduced by sulfasalazine or oroxindin administration. Oroxindin also significantly decreased the white blood cell (WBC) count, an inflammation-related marker in peripheral blood (Fig. 2e). In addition, the ratios of monocytes in lymphocytes were significantly decreased in the oroxindin-treated groups (Fig. 2f). As shown in Fig. 3a, c, colonic inflammation and ulceration were evaluated by histopathological analysis using hematoxylin and eosin staining, and histological scores were determined by a standard method. Mucosal inflammation, missing epithelium, and submucosal infiltration were observed in colon tissues in the model group. Oroxindin remarkedly relieved these severe symptoms in the colon. In addition, oroxindin diminished DSS-induced massive colonic macrophage infiltration. To assess the extent of intestinal inflammation in the colons of the DSS-treated mice, colon tissue sections were stained with an anti-F4/80 antibody to detect macrophage infiltration (Fig. 3b, d). These data indicated that oroxindin is able to suppress pathological changes and massive macrophage infiltration in colonic tissues, which proves the protective effects of oroxindin against UC.

The mice were treated with 3% DSS in drinking water for 7 d to induce acute colitis and then sacrificed on d 11. a Body weight changes from the basal level in each group after induction of colitis with DSS. b Macroscopic appearances of the colon. c Colon length changes in each group. d The spleen index was measured in each group. e WBC counts in peripheral blood. f Mononuclear cells were detected in peripheral blood using an automatic blood analyzer. Values are the mean ± SD (n = 6). ##P < 0.01, ###P < 0.001 vs. the control group; *P < 0.05, **P < 0.01, ***P < 0.001 vs. the DSS-treated group.

a Serial sections of colon tissues were stained with hematoxylin and eosin. b Protein expression of F4/80 in colon tissues was detected by immunohistochemical analysis. Enlarged areas of interest are shown in merged images and shown in representative data. c Colitis histological scores. The results are expressed as the mean ± SD (n = 4). d The results of immunohistochemistry were analyzed using Image-Pro Plus software, and the automatic algorithm recognized the brown area as positive cells. The software operation followed the instructions. (n = 4). ###P < 0.001 vs. the control group; *P < 0.05, **P < 0.01 vs. the DSS-treated group.
Oroxindin inhibited NLRP3 inflammasome formation in macrophages
Macrophages are known to play critical roles in the pathogenesis of UC. In order to clarify the specific mechanism, we subsequently performed immunoprecipitation in cultured macrophages to determine whether oroxindin inhibits the formation of the NLRP3 inflammasome, one of the core protein complexes in inflammatory responses (the NLRP3–ASC–Pro–Casp1 assembly). As shown in Fig. 4a–c, after incubation with ASC primary antibody, the colocalization coefficient was increased in the model group based on the ratio of NLRP3 and pro-Casp1 to ASC, and this tendency could be significantly reversed by oroxindin, which indicated that inflammasome formation was suppressed by oroxindin.

a The images of co-immunoprecipitation depict the effect of LPS on the colocalization of “NLRP3–ASC–pro-casp1” assembly. b Summarized data show the colocalization efficiency of NLRP3. c Summarized data show the colocalization efficiency of Pro-casp1. Values are the mean ± SD (n = 3). #P < 0.05 vs. the control group; *P < 0.05 vs. the LPS-treated group.
Oroxindin inhibited LPS-induced NLRP3 expression and activation in macrophages
We further explored whether the protective effect of oroxindin was related to the inhibitory effect on NLRP3 inflammasome activation in vitro. As illustrated in Fig. 5a, b, we measured the protein level of NLRP3 in the cytoplasm of macrophages. Interestingly, the NLRP3 protein increased in the model group and was significantly suppressed by oroxindin treatment as expected. In addition, the expression level of ASC was not changed in any of the groups (Fig. 5c, d). Previous studies have indicated that the NLRP3 inflammasome activation could induce robust caspase-1 activation, as based on the equal amount of the cleaved and pro-enzyme form [33]. Importantly, the activation of caspase-1 was significantly inhibited by oroxindin (Fig. 5e, f). The concentration of IL-1β indicates the activation of the NLRP3 inflammasome. The results showed that the concentration of active IL-1β in the supernatant was increased in the model group and could be effectively reduced by oroxindin (Fig. 5g).

a Protein expression of NLRP3 was analyzed by Western blotting. b Densitometric analysis was used to determine the relative protein expression, normalized to that of β-actin. The results are representative of three independent experiments. c Protein expression of ASC was analyzed by Western blotting. d Densitometric analysis was used to determine the relative protein expression, normalized to that of β-actin. The results are representative of three independent experiments. e Protein expression of caspase-1 was analyzed by Western blotting. f Densitometric analysis was used to determine the ratios of cleaved caspase-1 (Cle-Casp1)/pro-caspase-1 (Pro-Casp1). g Summarized data show the concentration of IL-1β in the cell supernatant. The results are representative of three independent experiments and expressed as the mean ± SD. #P < 0.05 vs. the control group; *P < 0.05 vs. the LPS-treated group.
Oroxindin suppressed TXNIP-dependent NF-κB activities in macrophages
To further elucidate the molecular mechanisms that regulate NLRP3 inflammasome activation, we evaluated the levels of the TXNIP protein (Fig. 6a, b), which is involved in NF-κB activation. TXNIP was markedly reduced in macrophages cytoplasm in the model group and could be restored by oroxindin in a concentration-dependent manner. This result was especially confusing until we found the protective role of TXNIP in cells, because decreased TXNIP leads to TXNIP-dependent NF-κB activation. Therefore, we analyzed the levels of p-p65 in the cytoplasm. As shown in Fig. 6c, d, p-p65 expression was markedly up-regulated in the model group and was inhibited by oroxindin, suggesting that the protective effect of oroxindin is possibly related to the TXNIP-dependent NF-κB-signaling pathway.

a Protein expression of TXNIP was analyzed by Western blotting and shown as cumulative data from multiple experiments. b Densitometric analysis was used to determine the relative protein expression, normalized to that of β-actin. c Protein expression of p-p65 was analyzed by Western blotting. d Densitometric analysis was used to determine the relative protein expression, normalized to that of β-actin. The results are representative of three independent experiments. Values are the mean ± SD (n = 3). #P < 0.05 vs. the control group; *P < 0.05, **P < 0.01 vs. the LPS-treated group.
Oroxindin lessened UC by inhibiting the NLRP3 inflammasome
NLRP3 is initially activated during inflammatory injury, and its activation has been confirmed to play a crucial role in DSS-induced colitis [34]. To further confirm our hypothesis in vitro, we examined whether oroxindin could suppress NLRP3 inflammasome activation in UC mice. As shown in Fig. 7a, b, we examined the co-localization of F4/80 (red) and NLRP3 (green) or caspase-1 (green) in colon tissues. According to the results, the expression of NLRP3 and caspase-1 was increased in macrophages in the DSS-treated mice, while oroxindin exerted specific protective effects in the inflamed tissue. These findings indicated that oroxindin suppressed NLRP3 inflammasome formation in macrophages in colonic tissue. To further determine whether NLRP3 inflammasome activation was repressed in vivo, the concentrations of IL-1β and IL-18 (caspase-1 downstream effectors) were measured in colon tissue supernatants. The results showed that oroxindin could reduce the generation of IL-1β and IL-18, suggesting that oroxindin could quell the pathologic inflammation and ameliorate inflammatory symptoms in UC mice (Fig. 7c, d).

a Sections of colonic tissue were immunostained with F4/80 (red) and NLRP3 (green) antibodies and observed under a confocal laser scanning microscope and shown as representative data. b Sections of colonic tissue were immunostained with F4/80 (red) and caspase-1 (green) antibodies and observed under a confocal laser scanning microscope and are shown as representative data. Enlarged areas of interest are shown in merged images. c The concentration of IL-1β in colonic homogenates was determined by an enzyme-linked immunosorbent assay (ELISA) in triplicate. d The concentration of IL-18 in colonic homogenates was determined by ELISA in triplicate. Data are presented as the mean ± SD. ##P < 0.01 compared with the normal mice; *P < 0.05, **P < 0.01 compared with the DSS-induced colitis mice.
Discussion
Previous studies have identified NLRP3 inflammasome-mediated inflammatory responses that underlie the critical mechanism of intestinal inflammation in a DSS colitis model [34]. Thus, inhibition of NLRP3 inflammasome formation and activation may be an effective approach for UC treatment. In the present study, we confirmed that oroxindin, one of the important pharmacological substances in HQT, exerted protective effects against UC, and the underlying mechanism was associated with inhibition of the NLRP3 inflammasome.
According to the Introduction in Traditional Medicine, HQT could be used to prevent and treat UC. Our results showed that DSS administration to mice led to the loss of body weight (Fig. S1a) and induced mucosal inflammation, missing epithelium, and macrophage infiltration in colon tissues (Fig. S1b, c). Oral administration of HQT (5, 10, 20 g/kg) relieved mice weight lose and ameliorated the pathological changes and macrophage infiltration in colon tissues, which confirmed the protective effect of HQT against UC (Fig. S1a–c). However, the specific bioactive monomer in HQT that exerts protective effect on UC has not yet been reported. The tissue distribution of drugs measured by LC–MS is an important index in pharmacological effectiveness studies. Our data showed that the concentrations of baicalin and oroxindin were the highest monomers absorbed by the colon tissue (Fig. 1a). On the other hand, it is important to take the proportion of baicalin and oroxindin in HQT into consideration, because the concentration of baicalin is four times higher than that of oroxindin (data not shown). Therefore, we analyzed the relative content of oroxindin (the concentration in the colon relative to that in HQT), and the results suggested that the colon’s absorption of oroxindin was better than that of other constituents (Fig. 1b). This indicated that oroxindin could be one of the bioactive compounds in HQT with high colon absorption. Therefore, we were interested in whether oroxindin could prevent the development of UC.
As one of the innate immune cells, macrophages are vital in protecting the human body from luminal bacteria during UC [35]. In this study, we found that oroxindin could ameliorate colonic inflammatory symptoms caused by DSS and we focused on the numbers of macrophages in the blood (Fig. 2f) and colon. Intriguingly, oroxindin decreased the number of macrophages in the blood and reversed the infiltration of macrophages (F4/80+ cells) in colon tissues. Sun et al. have also demonstrated that oroxindin could ameliorate the infiltration of inflammatory cells in colon tissues, as well as the histopathological features of DSS-induced colitis [36, 37]. Thus, we aimed to determine the underlying mechanism of the regulating effect of oroxindin on macrophage function.
When exposed to significant interference, the antigenic receptors on the surface of macrophages interact with the antigenic receptors in the cytoplasm. In particular, a previous study has suggested the pivotal role of the NLRP3 inflammasome in the pathogenesis of UC [38], and it may be a potential target for developing novel therapeutics for UC [39]. The gastrointestinal microbiota is known as one of the pathogenic factors of UC. In particular, Gram-negative bacteria, with an LPS-containing outer membrane, account for a large part of the gut microbiota. To further clarify the results of our in vivo study, we evaluated the anti-inflammatory effects of oroxindin in LPS-stimulated THP-1 cells in vitro. First, we performed a cell proliferation assay to assess the effects of oroxindin in macrophages. Compared to the control group, the proliferation of LPS-stimulated cells was significantly decreased, whereas oroxindin (12.5, 25, 50 μM) could alter this effect (Fig. S2). Therefore, we chose these three doses in our in vitro experiments. It was shown that oroxindin reduced NLRP3 inflammasome protein expression in LPS-stimulated THP-1 cells (Fig. 5a). NLRP3 is a critical component of the NLRP3 inflammasome. It can form a single oligomeric structure, recognizing pathogen-associated or damage-associated molecular patterns. Once the inflammasome is formed, ASC, a significant adaptor protein for pyrin domain (PYD)–PYD interaction, is recruited, and intracellular redistribution of ASC is essential for NLRP3 inflammasome activation [40]. In our present study, the ASC proteins were found in the cytoplasm at a consistent level. ASC subsequently bridges to pro-caspase-1 through caspase-associated recruitment domain (CARD)–CARD interaction and, thereby, to the assembly of a cytosolic protein complex called the “inflammasome” [37, 41], which induces autocleavage of caspase-1 into a cleaved form [42]. Additionally, our results showed that the amount of the multiprotein complex (NLRP3–ASC–caspase-1 assembly), as well as the activation ratio of caspase-1 (cleaved form to pro-enzyme form), was significantly diminished by oroxindin (Figs. 4a and 5e). NLRP3 inflammasome activation leads to the generation of the proinflammatory cytokine IL-1β. ELISA analysis revealed that oroxindin suppresses the concentration of IL-1β in macrophages (Fig. 5g). Thus, the in vitro study indicated that oroxindin could inhibit NLRP3 inflammasome formation and activation.
In this study, the expression of the NLRP3 inflammasome proteins was repressed by oroxindin in vitro, and we attempted to clarify the upstream mechanism. Previous studies have demonstrated that TXNIP expression is related to NF-κB activity. LPS stimulation decreased the protein level of TXNIP, which could regulate TXNIP-dependent NF-κB activation and prime NLRP3, pro-IL-1β, and pro-IL-18 expression. Therefore, we detected the expression of TXNIP and NF-κB in macrophages [43,44,45]. The results showed that the TXNIP protein level was significantly reduced in the LPS-stimulated group and that the level was restored by oroxindin (Fig. 6a, b). Correspondingly, the activation of NF-κB was repressed by oroxindin, suggesting the involvement of the TXNIP-dependent NF-κB-signaling pathway. As shown in Fig. 8, all results indicated that oroxindin suppressed the NLRP3 inflammasome in a TXNIP-dependent manner, suggesting that oroxindin could be a powerful tool for inhibiting TXNIP-dependent NF-κB activation and repressing NLRP3 inflammasome activation. In addition, it has been reported that the expression of TXNIP is closely related to the inducible nitric oxide synthase (iNOS)–NO-signaling pathway [17, 46]. Forrester et al. [47] proved that TXNIP expression in macrophages is suppressed by endogenously synthesized NO, which is produced by iNOS. Therefore, to further confirm the role of TXNIP, we investigated the effect of oroxindin on the iNOS-NO signaling pathway. It has been proven that TXNIP expression in macrophages is suppressed by iNOS-induced NO production [23]. Therefore, we measured the concentration of NO in the cell supernatant, and the results showed that LPS induced robust NO production, while this effect could be suppressed by oroxindin (Fig. S3a). Subsequently, we detected the expression of iNOS, and the results showed that the increase in iNOS induced by LPS could be down-regulated by oroxindin (Fig. S3b, c). To further identify the participance of the iNOS-NO signaling pathway, nicaraven, a well-known NO scavenger, was used to abate NO production. The results showed that Nica could obviously decrease the expression of iNOS (Fig. S3d, e), while the expression of TXNIP was correspondingly up-regulated (Fig. S3f, g). Oroxindin has a similar effect as Nica (Fig. S3d–g), suggesting that oroxindin has an inhibitory effect on the iNOS-NO signaling pathway, followed by the restoration of TXNIP expression and suppression of NF-κB activities.

DSS-induced intestinal epithelium bacterial translocation results in large amounts of LPS entering the cytoplasm, leading to a dramatic decrease in TXNIP, followed by TXNIP-dependent NF-κB activation. NF-κB activation, which is also regarded as the first signal that activates the NLRP3 inflammasome, is responsible for the increased transcription levels of pro-NLRP3, pro-IL-1β, and pro-IL-18. The activation of the NLRP3 inflammasome complex induces auto-cleavage of caspase-1 from pro-caspase-1 into cleaved caspase-1 and clips cleavage of pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into their active forms, which results in intestinal inflammation and tissue injury. Therefore, the protective effect of oroxindin on restoring TXNIP expression and abating the downstream NF-κB-signaling pathway helps to reduce inflammatory cytokine release and ameliorate pathological changes during colitis.
To further confirm our hypothesis in vitro, we examined whether oroxindin could suppress NLRP3 inflammasome activation in UC mice by immunofluorescence co-localization. We found that the NLRP3 inflammasome components, NLRP3 and the downstream protein, caspase-1, were upregulated in macrophages in colonic tissue in the UC model and were dramatically suppressed by oroxindin (Fig. 7a, b). The levels of IL-1β and IL-18, central mediators of the inflammatory responses, were also decreased, which confirmed the anti-inflammatory effects of oroxindin (Fig. 7c, d). Taken together, these data supported our in vivo study and confirmed our hypothesis that oroxindin inhibits colonic inflammation by inhibiting NLRP3 inflammasome formation and activation.
In conclusion, this study demonstrated that oroxindin was absorbed into the colon at a high level and ameliorated acute intestinal inflammation in a mouse UC model by suppressing NLRP3 inflammasome activation. Oroxindin could also suppress NLRP3 inflammasome activation by restoring TXNIP expression and inhibiting the TXNIP-dependent NF-κB-signaling pathway induced by LPS in macrophages. Considering the effective protective effect of oroxindin on inflammatory responses, this compound could be regarded as a potential therapeutic drug for UC. However, the relationship between TXNIP and the UC model remains to be further investigated.
References
Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology 1998;115:182–205.
Choi CR, Bakir IA, Hart AL, Graham TA. Clonal evolution of colorectal cancer in IBD. Nat Rev Gastroenterol Hepatol. 2017;14:218–29.
Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol 2016;17:230–40.
Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–66.
Abraham C, Cho JH. Inflammatory bowel disease. N. Engl J Med. 2009;361:2066–78.
Piechota-Polanczyk A, Fichna J. Review article: the role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn-Schmiedeberg’s Arch Pharmacol. 2014;387:605–20.
Dzutsev A, Trinchieri G. Proteus mirabilis: the enemy within. Immunity 2015;42:602–4.
Shouval DS, Biswas A, Kang YH, Griffith AE, Konnikova L, Mascanfroni ID, et al. Interleukin 1β mediates intestinal inflammation in mice and patients with interleukin 10 receptor deficiency. Gastroenterology 2016;151:1100–4.
Zherebiatiev A, Kamyshnyi A. Expression levels of proinflammatory cytokines and NLRP3 inflammasome in an experimental model ofoxazolone-induced colitis. Iran J Allergy Asthma Immunol. 2016;15:39–45.
Fukata M, Arditi M. The role of pattern recognition receptors in intestinal inflammation. Mucosal Immunol 2013;6:451–63.
Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell 2016;165:792–800.
Hirota SA, Ng J, Lueng A, Khajah M, Parhar K, Li Y, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17:1359–72.
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti T. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010;32:379–91.
Kim SY, Suh HW, Chung JW, Yoon SR, Choi I. Diverse functions of VDUP1 in cell proliferation, differentiation, and diseases. Cell Mol Immunol. 2007;4:345–51.
Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol 2014;4:514.
Kim MJ, Kim WS, Kim DO, Byun J, Huy H, Lee SY, et al. Macrophage migration inhibitory factor interacts with thioredoxin-interacting protein and induces NF-κB activity. Cell Signal 2017;34:110–20.
Kanari Y, Sato Y, Aoyama S, Muta T. Thioredoxin-interacting protein gene expression via MondoA is rapidly and transiently suppressed during inflammatory responses. PLoS One 2013;8:e59026.
Kwon HJ, Won YS, Suh HW, Jeon JH, Shao Y, Yoon SR, et al. Vitamin D3 upregulated protein 1 suppresses TNF-α-induced NF-κB activation in hepatocarcinogenesis. J Immunol. 2010;185:3980–9.
Tamaki H, Nakamura H, Nishio A, Nakase H, Ueno S, Uza N, et al. Human thioredoxin-1 ameliorates experimental murine colitis in association with suppressed macrophage inhibitory factor production. Gastroenterology 2006;131:1110–21.
Takahashi Y, Masuda H, Ishii Y, Nishida Y, Kobayashi M, Asai S. Decreased expression of thioredoxin interacting protein mRNA in inflamed colonic mucosa in patients with ulcerative colitis. Oncol Rep. 2007;18:531–5.
Vegh Z, Kurti Z, Lakatos PL. Epidemiology of inflammatory bowel diseases from west to east. J Dig Dis. 2017;18:92–8.
Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet 2007;369:1641–57.
Ju L, Ke F, Yadav P. Herbal medicine in the treatment of ulcerative colitis. Saudi J Gastroenterol. 2012;18:3.
Kamdar A, Alsafi A, Halse O. Cerebral sinus thrombosis occurring in a patient with ulcerative colitis treated with the Chinese herbal medicine YanNan BaiYao. QJM 2015;108:239–40.
Wang C, Tang X, Zhang L. Huangqin-Tang and ingredients in modulating the pathogenesis of ulcerative colitis. Evid Based Complement Alternat Med. 2017;2017:1–7.
Li H, Hui H, Xu J, Yang H, Zhang X, Liu X, et al. Wogonoside induces growth inhibition and cell cycle arrest via promoting the expression and binding activity of GATA-1 in chronic myelogenous leukemia cells. Arch Toxicol 2016;90:1507–22.
Li-Weber M. New therapeutic aspects of flavones: the anticancer properties of scutellaria and its main active constituents wogonin, baicalein and baicalin. Cancer Treat Rev. 2009;35:57–68.
Yang Y, Tang Y, Liu Y. Wogonoside displays anti-inflammatory effects through modulating inflammatory mediator expression using RAW264.7 cells. J Ethnopharmacol 2013;148:271–6.
Liu X, Zhou W, Zhang X, Lu P, Du Q, Tao L, et al. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem Pharmacol 2016;112:37–49.
Zhou X, Wu Y, Ye L, Wang Y, Zhang K, Wang L, et al. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS-induced vascular injury. Acta Pharm Sin B. 2019;9:711–23.
Lian D, Yuan H, Yin X, Wu Y, He R, Huang Y, et al. Puerarin inhibits hyperglycemia-induced inter-endothelial junction through suppressing endothelial Nlrp3 inflammasome activation via ROS-dependent oxidative pathway. Phytomedicine 2019;55:310–9.
Lorenz RG, McCracken VJ, Elson CO. Animal models of intestinal inflammation: ineffective communication between coalition members. Springer Semin Immunopathol. 2005;27:233–47.
Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR. Potentiation of caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-kappaB-driven protein synthesis. J Immunol 2005;175:7611–22.
Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010;59:1192–9.
Kühl AA, Erben U, Kredel LI, Siegmund B. Diversity of intestinal macrophages in inflammatory bowel diseases. Front Immunol 2015;6:613.
Sun Y, Zhao Y, Yao J, Zhao L, Wu Z, Wang Y, et al. Wogonoside protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting NF-κB and NLRP3 inflammasome activation. Biochem Pharmacol 2015;94:142–54.
Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol 2003;171:6154–63.
Zhang H, Wang Z, Lu X, Wang Y, Zhong J, Liu J. NLRP3 gene is associated with ulcerative colitis (UC), but not Crohn’s disease (CD), in Chinese Han population. Inflamm Res 2014;63:979–85.
Zmora N, Levy M, Pevsner-Fischer M, Elinav E. Inflammasomes and intestinal inflammation. Mucosal Immunol. 2017;10:865–83.
de Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J Biol Chem. 2009;284:32932–41.
Taniguchi SI, Sagara J. Regulatory molecules involved in inflammasome formation with special reference to a key mediator protein, ASC. Semin Immunopathol 2007;29:231–8.
Yamin TT, Ayala JM, Miller DK. Activation of the native 45-kDa precursor form of interleukin-1-converting enzyme. J Biol Chem. 1996;271:13273–82.
Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol 2017;8:36.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015;21:677–87.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–91.
Park YJ, Yoon SJ, Suh HW, Kim DO, Park JR, Jung H, et al. TXNIP deficiency exacerbates endotoxic shock via the induction of excessive nitric oxide synthesis. PLoS Pathog 2013;9:e1003646.
Forrester MT, Seth D, Hausladen A, Eyler CE, Foster MW, Matsumoto A, et al. Thioredoxin-interacting protein (Txnip) is a feedback regulator of S-nitrosylation. J Biol Chem. 2009;284:36160–6.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81603587, 81603668); the Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2018B030306027); the Key projects of basic research in Guangdong Province (No. A1-AFD018191A100); the Science & Technology Award for Young-aged Talents of China Association of Traditional Chinese Medicine (No. CACM-2017-QNRC2-C12); the Project of Guangzhou University of Chinese Medicine (No. A3-040219415009, A1-AFD018191A41); and by the Key Research and Development Program of Guangdong Province (2018KZDXM023).
Author information
Authors and Affiliations
Contributions
QL and RZ contributed to the animal experiments, cell experiments, and data analysis. KW, QL and FFN contributed to the LC–MS experiments. YJF and XL contributed to the study analysis and data analysis. QL, ZFP, and XLD, SWH, YZ, and XXZ contributed to the revision of the manuscript. YC and LZ contributed to the conception and design of the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Liu, Q., Zuo, R., Wang, K. et al. Oroxindin inhibits macrophage NLRP3 inflammasome activation in DSS-induced ulcerative colitis in mice via suppressing TXNIP-dependent NF-κB pathway. Acta Pharmacol Sin 41, 771–781 (2020). https://doi.org/10.1038/s41401-019-0335-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0335-4
Keywords
This article is cited by
-
Progress on Regulation of NLRP3 Inflammasome by Chinese Medicine in Treatment of Ulcerative Colitis
Chinese Journal of Integrative Medicine (2023)
-
Melatonin, an endogenous hormone, modulates Th17 cells via the reactive-oxygen species/TXNIP/HIF-1α axis to alleviate autoimmune uveitis
Journal of Neuroinflammation (2022)
-
Ainsliadimer C, a disesquiterpenoid isolated from Ainsliaea macrocephala, ameliorates inflammatory responses in adipose tissue via Sirtuin 1-NLRP3 inflammasome axis
Acta Pharmacologica Sinica (2022)
-
Modelling and targeting mitochondrial protein tyrosine phosphatase 1: a computational approach
In Silico Pharmacology (2022)
-
Identification of anti-inflammatory compounds from Zhongjing formulae by knowledge mining and high-content screening in a zebrafish model of inflammatory bowel diseases
Chinese Medicine (2021)

{kind=link}
{kind=link}
{kind=link}