
Abstract
Cancer cells always require more nutrients, energy, and biosynthetic activity to sustain their rapid proliferation than normal cells. Previous studies have shown the impact of THZ1, a covalent inhibitor of cyclin-dependent kinase 7 (CDK7), on transcription regulation and cell-cycle arrest in numerous cancers, but its effects on cellular metabolism in cancer cells remain unknown. In this study we elucidated the anticancer mechanism of THZ1 in human non-small-cell lung cancer (NSCLC) cells. We showed that treatment with THZ1 (10−1000 nM) dose-dependently suppressed the proliferation of human NSCLC cell lines H1299, A549, H292, and H23, and markedly inhibited the migration of these NSCLC cells. Furthermore, treatment with THZ1 (50 nM) arrested cell cycle at G2/M phase and induced apoptosis in these NSCLC cell lines. More importantly, we revealed that treatment with THZ1 (50 nM) blocked the glycolysis pathway but had no effect on glutamine metabolism. We further demonstrated that THZ1 treatment altered the expression pattern of glutaminase 1 (GLS1) isoforms through promoting the ubiquitination and degradation of NUDT21. Combined treatment of THZ1 with a glutaminase inhibitor CB-839 (500 nM) exerted a more potent anti-proliferative effect in these NSCLC cell lines than treatment with THZ1 or CB-839 alone. Our results demonstrate that the inhibitory effect of THZ1 on the growth of human NSCLC cells is partially attributed to interfering with cancer metabolism. Thus, we provide a new potential therapeutic strategy for NSCLC treatment by combining THZ1 with the inhibitors of glutamine metabolism.
Similar content being viewed by others


Introduction
Non-small-cell lung cancer (NSCLC) accounts for ~80% of all lung cancers. It is the most commonly diagnosed malignant cancer leading to mortality worldwide, and its prevalence status in China is particularly serious [1]. In the face of this grim situation, more effective therapeutic options are urgently needed for patients suffering from NSCLC. THZ1 was first identified as a specific covalent inhibitor of cyclin-dependent kinase 7 (CDK7), which significantly suppresses various carcinomas [2]. Usually, CDK 7 together with cyclin H and MAT1 form a three-subunit cyclin-activating kinase (CAK), which is a critical component of the general transcription factor TFIIH [3]. In transcription initiation, elongation, and DNA repair processes, TFIIH plays a key role in phosphorylating the C-terminal domain (CTD) of RNA polymerase II (RNA Pol II) at Ser-2 [4], Ser-5 [5], and Ser-7 [6] residues with activated CDK7. Furthermore, CDK7 is also a unique CAK that modulates activation of most other CDKs through T-loop phosphorylation in metazoans [7]. The major difference between the new CDK7 inhibitor THZ1 and other inhibitors of CDKs is that THZ1 exerts its suppressive effect by targeting cysteine-312 (C312) of CDK7 rather than the highly conserved kinase domain [2, 8]. Recently, it has been reported that inhibition of CDK7 with a nanomolar THZ1 concentration not only affected the transcription of genes involved in apoptosis and cell cycle via CTD phosphorylation of RNA Pol II but also downregulated the expression of several transcription factors associated with superenhancers (SEs), such as RUNX1, MYC family members, and neuroendocrine lineage-specific factors, in T cell acute lymphoblastic leukemia [2], neuroblastoma [9], and small-cell lung cancer [10].
Cancer cells always require more nutrients, energy, and biosynthetic activity to sustain their rapid proliferation than normal cells [11]. Many studies have revealed that the cellular metabolism in cancer cells is reprogrammed to satisfy the altered metabolic demands for unrestrained cellular proliferation [12, 13], resulting in high rates of glucose utilization, followed by lactate production even under adequate oxygen conditions. Therefore, aerobic glycolysis has been recognized as a promising target for drug discovery [14, 15]. Recently, it was reported that the accumulation of lactate was involved in drug resistance in colon cancer [16] and cervical cancer [17]. Moreover, high levels of lactate also promoted tumor recurrence and metastatic potential [18].
Numerous studies have demonstrated that glutamine metabolism plays critical roles in many different cell types, especially cancer cells [19,20,21,22]. As the most abundant amino acid in human tissues, glutamine is one of the major nutrients that provides cancer cells with energy and intermediate metabolites for various biological processes [23]. Glutaminase is one of the key enzymes in glutamine metabolism. Glutaminase C (GAC) and kidney-type glutaminase (KGA) are the two isoforms of glutaminase 1 (GLS1), which catalyze the hydrolytic deamination of glutamine to glutamate and ammonia [24]. The expression of GAC is upregulated in many types of tumors, and the ratio of GAC/KGA is significantly increased in lung cancer [25]. A loss of GAC impacts cell proliferation more than a loss of KGA, suggesting that GAC plays a more important role in cancer cells [26].
Although several previous studies have demonstrated the impact of THZ1 on transcription regulation and cell-cycle arrest, its effects on cellular metabolism in cancer cells remain unknown. Here, we report that THZ1 effectively inhibited cell proliferation and migration, caused cell-cycle arrest at G2/M phase, and induced apoptosis of NSCLC cells. Furthermore, we showed that THZ1 suppressed glycolysis in cancer cells, and combined treatment with THZ1 and the glutaminase inhibitor CB-839 had a synergistic effect on inhibition of proliferation and cell-cycle progression in NSCLC cells.
Materials and methods
Cell culture
Human bronchial epithelial cells (HBE) and four NSCLC cell lines (H1299, H23, A549, H292) were purchased from American Type Culture Collection (ATCC) (Rockville, MD, USA) and cultured in roswell park memorial institute (RPMI) 1640 medium (Gibco, USA) supplemented with 10% (v/v) FBS (Gibco) at 37 °C with 5% CO2. THZ1, CB-839 (MedChemExpress, USA), and MG132 (Sigma-Aldrich, USA) were stored as 1 mM stock suspensions in DMSO and diluted into working solutions with RPMI 1640 medium. Chloroquine was dissolved in phosphate buffer saline (PBS) and diluted by RPMI 1640 medium to 200 µM.
Cell proliferation assay and colony-formation assay
For cell proliferation assay, cells (5 × 103 cells) were plated in 24-well plates in 0.5 mL RPMI 1640 medium supplemented with 10% FBS for 24 h. Then, cells were treated with THZ1 or DMSO. At the indicated time points, cells were fixed with 3.7% formaldehyde and stained with 0.1% crystal violet. The relative proliferation was determined by the absorbance of the 10% acetic acid extract at 595 nm. For the colony-formation assay, cells were seeded in 6-well plates at 500 cells per well and cultured for 14 days with 50 nM THZ1 or DMSO. After visible colonies were formed, they were fixed with 3.7% formaldehyde (Solarbio, China) and stained with 0.1% crystal violet (Sigma-Aldrich) solution.
Wound-healing assay
Cells were seeded in 6-well plate overnight in RPMI 1640 supplemented with 10% FBS to create a confluent monolayer. After drawing a straight line across the monolayer with a 200–μL pipet tip and a PBS wash, the medium was replaced with RPMI 1640 containing 1% FBS and THZ1 or DMSO. Then, the cells were photographed using a microscope (Olympus, IX71) at the indicated time points.
Phalloidin staining
The treated cells were fixed in 3.7% formaldehyde solution and permeated with 0.1% Triton X-100. Then, the cells were stained with the fluorescent phallotoxins (Thermo Fisher Scientific, USA), which were diluted 40-fold with PBS for 1 h at 4 °C in the dark. Photographs were taken with an inverted fluorescence microscope (Olympus, IX71).
RNA purification and quantitative RT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, USA), and a total of 1 μg RNA was used for reverse transcription using a PrimeScript RT reagent kit with a gDNA eraser (Takara, Japan) according to the manufacturer’s instructions. Quantitative RT-PCR was performed with SYBR Green (Applied Biosystems, USA) using ViiA7 Real-Time PCR System (Applied Biosystems). All quantitative RT-PCR reactions were performed in triplicate. The relative mRNA expression was calculated by the comparative Ct method using GAPDH as a control. All primer sequences are listed in Supplementary Table 1.
Western blot
Cells were lysed by RIPA buffer (Solarbio) with a protease and phosphatase inhibitor cocktail (Thermo Fisher). The protein concentration was measured by Pierce™ BCA Protein Assay Kit (Thermo Fisher). The protein lysates were separated on a 10% SDS-PAGE gel followed by immunoblotting with β-actin mouse monoclonal antibody (1:20000, Proteintech, USA), GAC rabbit polyclonal antibody (1:1000, Abcam, USA), Akt antibody (1:1000, Cell Signaling, USA), phospho-Akt antibody (Ser473, 1:2000, Cell Signaling), eIF4EPB1 antibody (1:1000, Cell Signaling), phospho-eIF4EBP1 antibody (Thr37, 1:1000, Cell Signaling), Cdc25c rabbit monoclonal antibody (1:1000, Cell Signaling), anti-caspase-3 mouse monoclonal antibody (1:1000, Cell Signaling), PCNA antibody (1:2000, Cell Signaling), cyclin B1 antibody (1:2000, Cell Signaling), CDK1 rabbit polyclonal antibody (1:500, OriGene, USA), NUDT21 rabbit polyclonal antibody (1:1000, Proteintech), and anti-ubiquitin rabbit polyclonal antibody (1:500, Proteintech). The signal was detected with an enhanced chemiluminescence western blot analysis system. All experiments were conducted in triplicate.
Immunoprecipitation
After cells were lysed with NP-40 buffer containing PMSF (Dingguo, China) and protease inhibitor cocktail (Thermo Fisher), the cell lysates were precleaned with protein G agarose at 4 ℃ for 1 h, then the supernatants were incubated with indicated antibodies and protein G agarose beads (Roche, USA) at 4 ℃ overnight. On the second day, immunocomplexes combined with beads were washed with lysis buffer and subjected to a western blot assay.
Cell-cycle analysis
Cells were incubated with 50 nM THZ1 or DMSO at indicated concentrations and harvested 48 h later. After fixing with 70% ethanol in phosphate-buffered saline (PBS) for 2 h at 4 °C, the cells were washed with PBS twice, and 2 × 105 cells were resuspended in 500 μL guava cell-cycle reagent (Millipore, Germany) and incubated for 15 min at 37 °C. Then, the stained cells were analyzed by the Millipore Guava easyCyte™ flow cytometer (Millipore).
Apoptosis assay
After treatment for 48 h with 50 nM THZ1 or DMSO, cells were treated with 0.25% trypsin (Gibco) without EDTA and washed twice with PBS. A total of 5 × 105 cells were resuspended in 500 μL binding buffer and incubated with 5 μL Annexin V-FITC and 5 μL propidium iodide (PI) solution (Annexin-V-FITC cell apoptosis assay kit, KeyGEN, China) at room temperature. After incubation for 15 min, the cells were analyzed by the Millipore Guava easyCyte™ flow cytometer (Millipore).
Measurements of metabolites
The ATP assay kit was purchased from Thermo Fisher. The glucose assay kit was purchased from Shanghai Rongsheng Biotech Co., Ltd. The lactic acid assay kit and the glutamine and glutamate assay kit were purchased from Nanjing Jiancheng Bioengineering Institute. All assays were performed following the protocols described in instruction manuals.
RNA interference
Knockdown of GAC was carried out with two distinct siRNAs targeting GLS1 (Thermo Fisher), and the indicated siRNAs or Stealth RNAiTM siRNA Negative Control were transiently delivered into NSCLC cell lines using Superfectin II In Vitro siRNA Transfection Reagent (Shanghai Pufei Biotech, China). The knockdown efficiency was determined by western blot with GAC antibodies.
Statistical analysis
All experiments were performed to assess statistical significance in triplicate, and the data are shown as the means ± SD. Data were analyzed with one-way ANOVA or unpaired t-test (GraphPad Prism, San Diego, CA). Differences in the results were considered significant at P < 0.05.
Results
THZ1 potently inhibits proliferation and migration of NSCLC cells
To investigate the effects of THZ1 on NSCLC cells, human bronchial epithelial cells (HBE) and four human non-small-cell lung carcinoma cell lines (H1299, H23, H292, A549) were treated with various concentrations of THZ1 or DMSO vehicle for 48 h, and cell proliferation was examined by crystal violet staining. The results showed that compared with HBE cells, THZ1 remarkably decreased proliferation of various NSCLC cells in a dose-dependent manner (Fig. 1a), and 50 nM of THZ1 significantly inhibited proliferation of NSCLC cells in a time-dependent manner (Fig. 1b–e). As shown in Fig. 1f THZ1 (50 nM) markedly suppressed colony formation in NSCLC cell lines, suggesting that THZ1 strongly inhibited proliferation of NSCLC cells. Notably, HBE cells showed less sensitivity to THZ1 treatment, and it seemed that THZ1 was much less toxic to normal cells than to cancer cells.

THZ1 significantly inhibits proliferation of NSCLC cells. a HBE, H1299, H23, H292, and A549 cells were seeded in 24-well plates at 5 × 103 cells per well and treated with THZ1 at different concentrations (0, 10, 20, 50, 100, 1000, 10000 nM) for 48 h. The cells were then fixed and stained with crystal violet, and the dye was extracted and detected by absorbance at 595 nm. b–e The NSCLC cell lines H1299 (b), A549 (c), H292 (d), and H23 (e) were treated with 50 nM THZ1 for 0, 1, 2, 3, 4 days, and then, the treated cells were fixed and stained with crystal violet at the indicated time points. The dye was extracted and detected by absorbance at 595 nm. f H1299, A549, H292, and H23 cells were plated at 500 cells per well in 6-well plates and treated with 50 nM THZ1 or DMSO for colony-formation assay. After 14 days, the colonies were fixed and stained with 0.1% crystal violet solution and then photographed. All results are from three independent experiments, and the data represent the mean ± SD (n = 3)
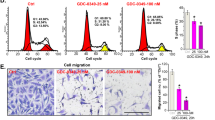
Next, the effects of THZ1 on cell migration were determined using wound-healing assays. As shown in Fig. 2a, b, the migration of H23 and H1299 cells was notably attenuated in the presence of 50 nM THZ1. Since the arrangement of the actin network is critical for cell motility [27], actin filaments in H1299 cells were stained with fluorescent phallotoxins. As shown by fluorescence microscopy, cellular projections of THZ1-treated H1299 cells were significantly reduced when compared with the untreated group, suggesting that THZ1 efficiently inhibited the migration of NSCLC cells (Fig. 2c).

THZ1 markedly suppresses the migration of NSCLC cells. H23 cells (a) and H1299 cells (b) were treated with 50 nM THZ1 or DMSO for 18 h. Cell migration was quantified by measuring the difference in area between the leading edge at the beginning of the experiment and after 18 h. The wound area was assessed by the ImageJ software (Olympus, ×100). Data represent the average of three independent experiments (mean ± SD) (n = 3; **P < 0.01; ***P < 0.001 vs. DMSO). c H1299 cells were treated with 50 nM THZ1 for 18 h and then stained with the fluorescent phallotoxin. Photographs were taken with an inverted fluorescence microscope (Olympus, Phalloidin stain, ×200)
THZ1 causes significant arrest of NSCLC cells at G2/M phase of cell cycle
CDK7 is a crucial regulator of RNA Pol II phosphorylation and phosphorylation of CDK1 and CDK2 by CAK during the cell cycle [28]. The effects of THZ1 on cell-cycle progression were examined. The results showed that the numbers of cells at G2/M phase were increased to various degrees in NSCLC cell lines treated with THZ1 (Fig. 3a). Cyclins and cyclin-dependent kinases (CDKs) are regulators of cell-cycle progression. With the results of the initial assay, we analyzed the mRNA expression levels of the genes closely involved in cell-cycle progression of H1299 cells by real-time RT-PCR. As shown in Fig. 3b, THZ1 dramatically suppressed mRNA expression of cell-cycle-regulatory genes associated with G2/M phase, such as CDK1, CCNB2, and CDC25B, in a dose-dependent manner. In addition, the mRNA expression of genes associated with G1 phase, such as CCND1, CCND2, CCNE1, CCNE2, CDK2, CDK4, and CDK6, and the E2F family (E2F1, E2F2) were also progressively reduced with the increase in THZ1 concentrations. To further confirm the effects of THZ1 on G2/M phase in NSCLC cells, we examined the expression of proteins associated with G2/M phase in H1299 cells. As expected, the expression levels of cyclin B1 and Cdc25c, both activators of CDK1, were significantly decreased with THZ1 treatment (Fig. 3c). At the same time, the expression of CDK1 was diminished in a dose-dependent manner. These data demonstrated that THZ1 resulted in cell-cycle arrest at G2/M phase in NSCLC cells.

THZ1 induces cell-cycle arrest at G2/M phase. a H1299, A549, H23, and H292 cells were treated with 50 nM THZ1 or DMSO for 48 h, and the cells were collected, stained by propidium iodide and analyzed by flow cytometry (pink: G0/G1 phase, green: S phase, and blue: G2/M phase). The quantification of cell numbers in each phase of the cell cycle in the right figures were derived from the left figures and marked with different colors (red: G0/G1 phase, yellow: S phase, and blue: G2/M phase). b The mRNA expression levels of the cell-cycle-related genes in H1299 cells were measured by quantitative RT-PCR after treatment with various concentrations of THZ1 for 48 h. All data are represented as the mean ± SD (n = 3; **P < 0.01, ***P < 0.001 vs. DMSO). c The expression levels of the genes involved in G2/M phase in H1299 cells treated with 0, 50, and 100 nM THZ1 for 48 h were detected by immunoblotting using the corresponding antibodies
THZ1 induces apoptosis of NSCLC cells
The effects of THZ1 on apoptosis of NSCLC cells were analyzed by flow cytometry with Annexin V-FITC and PI staining. As shown in Fig. 4a, the numbers of the cells undergoing apoptosis were significantly increased when treated with THZ1. In addition, the mRNA expression levels of anti-apoptotic genes (BCL-XL, BCL2, BIRC2, BIRC3, and XIAP), which are critical regulators in the mitochondrial apoptosis pathway, were significantly decreased (Fig. 4b). Moreover, as shown in Fig. 4c, the expression of cleaved-caspase-3 was progressively induced, whereas the expression of caspase-3 precursor displayed a downregulation trend, accompanied by increasing concentrations of THZ1 in H1299, H23, and H292 cells. Accordingly, these results suggested that THZ1 significantly induced apoptosis in NSCLC cells.

THZ1 induces marked apoptosis in NSCLC cells. a H1299, H23, and H292 were treated with 50 nM THZ1 for 48 h and analyzed by flow cytometry with Annexin-V-FITC and propidium iodide (PI) staining to detect apoptosis. b The mRNA expression levels of anti-apoptotic genes in H1299 cells treated with various concentrations of THZ1 for 48 h were measured by quantitative RT-PCR. All data are represented as the mean ± SD (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001 vs. DMSO). c The expression levels of pro-caspase 3 and cleaved-caspase-3 in H1299, H23, and H292 cells treated with 0, 50, and 100 nM THZ1 for 48 h were detected by immunoblotting
THZ1 modulates glycolysis in NSCLC cells
Christensen et al. demonstrated that the genes affected by THZ1 were closely involved in the metabolism of SCLC cells [10]. However, to date no study has been published to investigate whether THZ1 inhibits the growth of cancer cells by interfering with glycolysis. Thus, we measured the intra- and extracellular glucose and lactate concentrations, and the relative ATP levels in THZ1-treated H1299, H23, and HBE cells. We found that compared with the control group, THZ1 significantly decreased the intracellular glucose concentration and increased the extracellular glucose concentration in H1299 cells (Fig. 5a). Moreover, both intra- and extracellular lactate concentrations and the relative ATP levels were decreased in H1299 cells treated with THZ1 (Fig. 5b, c). Similar results were also observed in H23 cells (Supplementary Figure 1a-1c), unlike those in HBE cells (Supplementary Figure 2a-2c). These results demonstrated that THZ1 suppressed the glucose uptake, lactate secretion, and relative ATP production, which had an inhibitory effect on cancer cell metabolism. Furthermore, along with an increasing concentration of THZ1, the mRNA expression of HK2, PFKL, PFKM, PFKP, PGK1, PKM2, LDHA, and GLUT1 was inhibited, and the protein expression level of HK2, the rate-limiting enzyme that phosphorylates glucose to glucose 6-phosphate during the first step of glucose metabolism, was greatly reduced (Fig. 5d). These findings showed that THZ1 inhibited the key glycolysis steps leading to the reduction of glucose uptake and lactate secretion and preventing an adequate supply of energy for NSCLC cells proliferation.

THZ1 modulates glucose metabolism in NSCLC cells. a–c H1299 cells were seeded in 6-well plates and treated with 50 nM THZ1 for 48 h, and the glucose (a) and lactate (b) concentrations in the supernatant (left) or the collected cells (right), as well as the intracellular ATP level (c) were measured. d The mRNA expression levels of the genes involved in glycolysis were measured by quantitative RT-PCR in H1299 cells treated with various concentrations of THZ1 for 48 h. All data are represented as the mean ± SD (n = 3; *P < 0.05, ***P < 0.001 vs. DMSO). e The expression levels of HK2, Akt, phospho-Akt (Ser473), eIF4EBP1, and phospho-eIF4EBP1 (Thr37), were detected by immunoblotting using the corresponding antibodies in H1299 cells treated with 0, 50, and 100 nM THZ1 for 48 h
Previous studies have revealed that activated Akt promoted tumor cell-autonomous increases in aerobic glycolysis [29], and active mammalian target of rapamycin complex 1 (mTORC1) inhibited its downstream effector eIF4E binding protein (eIF4EBP1), which restrained the protein translation initiation process by binding and inactivating eukaryotic translation initiation factor 4E (eIF4E), leading to the induction of GLUT1 and hexokinase 2 and thereby increasing glucose uptake and glycolysis [30]. To better understand the underlying mechanism of THZ1-induced inhibition of glycolysis, we measured the expression of proteins related to the Akt-mTOR pathway. As shown in Fig. 5e, THZ1 significantly inhibited the expression of phospho-Akt (Ser473) and phospho-eIF4EBP1 (Thr37), the downstream protein of the activated mTORC1, in H1299 cells in a dose-dependent manner, suggesting that THZ1 interfered with the glycolysis-associated signaling pathway in NSCLC cells.
THZ1 alters the expression pattern of two GLS1 isoforms and exerts a synergistic inhibitory effect with glutaminase inhibitor CB-839
Since glutamine is an essential metabolite in cancer metabolism [31], we measured the concentrations of intra- and extracellular glutamate and extracellular glutamine. As shown in Fig. 6a, b, THZ1 had no effect on glutamine metabolism in H1299. We next detected the mRNA expression of GLS1, the key enzyme in glutamine metabolism. As seen in Fig. 6c, no significant changes were observed in the mRNA expression of GLS1 in H1299 cells treated with THZ1. As we know, GLS1 expresses two splicing variants, GAC and KGA, and GAC has a higher protein expression and catalytic activity than KGA in transformed cells and cancers [32]. Interestingly, we observed that the ratio of GAC/KGA mRNA expression was significantly reduced by THZ1 treatment (Fig. 6d). Moreover, the protein expression of the KGA was significantly increased, whereas GAC was gradually decreased when the cells were treated with increasing THZ1 concentrations (Fig. 6e). Considering that NUDT21 could bind to the intron 14 of GLS1 gene inducing the preferential expression of GAC [33], we assessed the NUDT21 protein and found a significant reduction in NUDT21 protein expression, as expected. To further confirm the degradation pathway of NUDT21 protein in NSCLC cells treated with THZ1, we inhibited two major protein degradation pathways in eukaryotic cells, the ubiquitin-proteasome system and autophagy, with MG132 (carbobenzoxyl-leucinyl-leucinyl-leucine-H, a selective reversible inhibitor of the 26S proteasome) and chloroquine (an autophagy inhibitor), respectively. By comparison, MG132, rather than chloroquine, blocked the decrease in protein expression of NUDT21, which was induced by THZ1, indicating that degradation of NUDT21 induced by THZ1 treatment was mediated by the ubiquitin-proteasome system. We further detected the ubiquitination of NUDT21 treated with THZ1. Figure 6g shows that the ubiquitination level of NUDT21 gradually increased upon treatment with increasing THZ1 concentrations. These results suggest that THZ1 treatment altered the expression pattern of the two GLS1 isoforms by increasing the ubiquitination and degradation of NUDT21 in NSCLC cells.

THZ1 alters the expression of two GLS isoforms by promoting the ubiquitin-mediated degradation of NUDT21. H1299 cells were seeded in 6-well plates and treated with 50 nM THZ1 for 48 h, and the glutamate concentrations in the supernatant ((a) left) and collected cells ((a) right) and the glutamine concentrations in the supernatant (b) were assayed. c The mRNA expression levels of GLS1 in H1299 cells treated with various concentrations of THZ1 for 48 h were measured by quantitative RT-PCR. d The mRNA expression ratio of GAC/KGA was determined in H1299 cells treated with 50 nM THZ1 or DMSO for 48 h. e The expression levels of GAC, KGA were detected by immunoblotting using the antibody recognizing both isoforms, and the NUDT21 expression using indicated antibodies in H1299 cells treated with 0, 50, and 100 nM THZ1 for 48 h. The KGA and GAC intensities were calculated by normalizing against β-actin. f H1299 cells were treated with 50 nM THZ1 or DMSO. 42 h later, 20 μM MG132 or 20 μM chloroquine was added to the cells and treated for 6 h. Then, the western blot was performed to detect the NUDT21 expression using indicated antibodies. g H1299 cells were treated with 0, 50, and 100 nM THZ1 for 48 h. Then, the cells were lysed, and immunoprecipitation was performed using anti-NUDT21 antibody. The protein expression levels were examined by western blotting using indicated antibodies. All data are represented as the mean ± SD (n = 3; **P < 0.01 vs. DMSO)
Studies have shown that glutaminase might be an important therapeutic target for cancer treatment due to its crucial role in cancer metabolism [34]. As THZ1 did not affect the glutamine metabolism in NSCLC, we decided to test whether the inhibition of glutaminase has a synergy with THZ1 anti-tumor effects. As shown in Fig. 7a, a low dose of THZ1 applied alone had only minor inhibitory effects on cell growth, however, upon a combined treatment with CB-839 [35] (a glutaminase inhibitor), cell growth was remarkably inhibited, even at 10 nM THZ1 treatment in H1299 and H292 cells. The protein expression levels of several cell-cycle-related genes and GAC were more significantly decreased in H1299 cells treated with both 20 nM THZ1 and CB-839 than in cells treated with 20 nM THZ1 or CB-839 alone (Fig. 7b). To determine if the observed effects resulted from glutaminase inhibition, we knocked down the GLS1 gene in H1299 cells with siRNAs (Fig. 7c) followed by treatment with 20 nM THZ1. As shown in Fig. 7d, a potent inhibitory effect of 20 nM THZ1 was observed in the cells with GLS1 knockdown than in control cells. These results suggested that THZ1 treatment in combination with glutamine metabolism inhibition might serve as a promising therapeutic strategy for NSCLC cells.

The combination of THZ1 and CB-839 treatments has a synergistic inhibitory effect on the viability of NSCLC cells. The proliferation of H1299 (a, left) and H292 cells (a, right) was assessed upon treatment with 500 nM CB-839 alone or in combination with THZ1 at different low concentrations (0, 10, 20, and 50 nM). b The protein expression levels of the genes related to cell cycle and metabolism were detected by immunoblotting using the corresponding antibodies in H1299 cells treated with 20 nM THZ1 alone or in combination with 500 nM CB-839 for 48 h. c GLS1 siRNAs were transfected into H1299 cells. 48 h later, the cells were lysed, and the protein expression levels of GLS1 were detected by western blot. d H1299 cells with GLS1 knockdown were treated with 20 nM THZ1 for the indicated time, and cell growth assay was performed. All data are represented as the mean ± SD (n = 3; ***P < 0.001 vs. 20 nM THZ1 or 500 nM CB-839 treatment alone)
Discussion
Uncontrolled proliferation is one of the hallmarks of cancer cells. A variety of small molecule inhibitors that target genes related to cancer cell growth and survival have been investigated as candidate therapeutic agents for NSCLC [36]. Transcription factors play a key role in the development of diverse cancers; however, therapeutically targeting of transcription factors remains a significant challenge. By screening a diverse panel of 1151 cancer cell lines, Nicholas et al. have found that THZ1, a newly developed covalent inhibitor of CDK7, potently suppresses most of the cancer cell lines, especially in T-ALL cell lines [2]. In this study, we assessed the therapeutic potential of THZ1 as a promising drug candidate for the treatment of NSCLC. We found that THZ1 significantly inhibited proliferation of NSCLC cells and arrested the cell cycle in G2/M phase along with downregulation of cyclins and CDKs. Furthermore, THZ1 also induced apoptosis in NSCLC cells accompanied by reduced expression of anti-apoptosis genes. These results are consistent with the results of previous studies that CDK7 activated by cyclin H and MAT1 is closely associated with the cell cycle and transcription [37]. Importantly, we discovered that HBE cells were not sensitive to THZ1. In previous studies, the nontransformed BJ fibroblast and retinal pigment epithelial (RPE-1) cells displayed cell-cycle arrest rather than initiation of apoptosis or cell death when treated with relatively high doses of THZ1. There was also no loss of body weight, behavioral changes, or overt toxicity observed in the subsequent studies on animals, implying that normal cells might tolerate transcriptional disruption, and the broad therapeutic window of THZ1 treatment displayed its unique superiority in distinguishing normal cells and cancer cells compared with chemotherapy [2, 10]. However, the minor toxicity in normal cells could not be well explained by targeting RNA Pol II and CDK7 because these two essential enzymes are ubiquitous in all cell types and are physiologically important. Thus, we speculated that the effect of THZ1 on NSCLC cells might not be necessarily related to CDK7 and RNA Pol II targeting. Other new molecular mechanisms need to be further clarified.
Cancer cells undergo metabolic reprogramming to support rapid cell proliferation. The metabolism in cancer cells is characterized by increased rates of glucose consumption, lactate and ATP production and biosynthesis of lipids and other macromolecules [38]. Recently, by performing the gene ontology analysis in SCLC cell lines, Christensen et al. observed that THZ1 significantly downregulated the expression of the genes associated with metabolism, besides the alterations in the expression of genes involved in transcription and cell cycle [10]. However, they mainly focused on the effect of THZ1 on the transcription by regulating the phosphorylation of RNA Pol II CTD or transcription factors at superenhancers [37, 39, 40], while little attention was paid to the effect of THZ1 on cancer cell metabolism. In our study, we observed remarkable decreases in glucose utilization, lactate secretion and ATP production in NSCLC cells treated with THZ1. In addition, we also observed that THZ1 significantly downregulated the expression of genes involved in glycolysis. It is well-known that the Akt/mTOR signaling pathway is responsible for regulation of glycolysis [29]. Previous studies showed that HK2 mRNA translation was increased by the deletion of PTEN through the activation of the AKT-mTORC1-4EBP1 axis in prostate cancer cells [41], and Akt also regulated glucose metabolism through phosphorylation and inactivation of GSK3β (glycogen synthase kinase 3β) [42]. Our study demonstrated that the expression levels of the activated form of AKT and its downstream targets were sharply decreased when NSCLC cells were treated with THZ1. Thus, we speculated that THZ1 inhibited glycolysis in NSCLC cells through the Akt/mTOR pathway, and the deficiency in glucose metabolism resulted in a shortage of energy and intermediates for biosynthesis. The consequences of these changes are the cell-cycle arrest and apoptosis.
Glutamine metabolism is an alternative pathway to gain anaplerosis for cancer cell growth, and evidence has shown that abundant glutamine is a main source of anaplerotic precursor in proliferating glioma cells, both in human and rats [20, 43]. Numerous studies have demonstrated that blocking glutamine metabolism disturbs proliferation of cancer cells. In this study, although we did not observe alterations in the metabolic products of glutamine metabolism, such as glutamine and glutamate, in H1299 cells treated with THZ1, the expression patterns of GAC and KGA, two isoforms of GLS1, were significantly changed by THZ1 treatment. We further demonstrated that THZ1 altered the expression patterns of the two isoforms of GLS1 by ubiquitination and degradation of NUDT21. As GAC is more abundant and catalytically active than KGA in many cancers, it plays a very important role in accelerating the metastases and cell proliferation. Reducing the expression of GAC by THZ1 may block the progression of cancer to a certain extent. Our results suggested that THZ1-mediated inhibition of cancer growth might be associated with regulation of glutaminase instead of a direct interference with glutamine metabolism.
The identification of vulnerability in cancer metabolism has been used to develop novel strategies for cancer treatment. Momcilovic et al. reported that a combined inhibition of glycolysis and glutamine metabolism led to rapid tumor regression in a mouse xenograft model of lung cancer [44]. Although there was no significant inhibitory effect of THZ1 on glutamine metabolism, our results revealed that THZ1 could block glucose metabolism and affect the expression pattern of GLS1. Thus, a combination treatment with low doses of THZ1 and GLS1 inhibitor CB-839 in NSCLC cells was designed to explore whether this combination could enhance the efficiency of NSCLC treatment. As expected, the combination showed a much better anti-proliferative effect in NSCLC cells than in those treated with a single agent. In addition, the expression levels of the genes related to cell cycle and metabolism were remarkably decreased upon treatment with both agents, indicating a more severe disturbance of cell-cycle progression and metabolism. Combination therapy with THZ1 and CB-839 showed a better anti-tumor effect, and the use of low-dose THZ1 might be tolerated better and lead to fewer negative effects in normal cells.
Conclusion
In conclusion, our study explored a new molecular mechanism of THZ1-mediated inhibition of proliferation and migration of NSCLC cells and provide a potential therapeutic strategy for NSCLC treatment by combining THZ1 and inhibitors of glutamine metabolism.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–20.
Yee A, Nichols MA, Wu L, Hall FL, Kobayashi R, Xiong Y. Molecular cloning of CDK7-associated human MAT1, a cyclin-dependent kinase-activating kinase (CAK) assembly factor. Cancer Res. 1995;55:6058–62.
Ni Z, Schwartz BE, Werner J, Suarez JR, Lis JT. Coordination of transcription, RNA processing, and surveillance by P-TEFb kinase on heat shock genes. Mol Cell. 2004;13:55–65.
Ho CK, Shuman S. Distinct roles for CTD Ser-2 and Ser-5 phosphorylation in the recruitment and allosteric activation of mammalian mRNA capping enzyme. Mol Cell. 1999;3:405–11.
Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, et al. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–2.
Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–80.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–46.
Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–39.
Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26:909–22.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20.
Warburg O. Iron, the Oxygen-Carrier of Respiration-Ferment. Science. 1925;61:575–82.
Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–77.
Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, et al. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011;117:2926–38.
Fischer CP, Bode BP, Souba WW. Adaptive alterations in cellular metabolism with malignant transformation. Ann Surg. 1998;227:627–34; discussion 34-6.
Kaplan O. Correspondence re: M. Fanciulli et al. Energy metabolism of human LoVo colon carcinoma cells: correlation to drug resistance and influence fo lonidamine. Clin. Cancer Res. 2000;6:1590–7, 4166–7.
Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283:28106–14.
Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, et al. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60:916–21.
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178:93–105.
Portais JC, Voisin P, Merle M, Canioni P. Glucose and glutamine metabolism in C6 glioma cells studied by carbon 13 NMR. Biochimie. 1996;78:155–64.
Forbes NS, Meadows AL, Clark DS, Blanch HW. Estradiol stimulates the biosynthetic pathways of breast cancer cells: detection by metabolic flux analysis. Metab Eng. 2006;8:639–52.
Brand K. Glutamine and glucose metabolism during thymocyte proliferation. Pathw glutamine glutamate Metab Biochem J. 1985;228:353–61.
Eagle H, Oyama VI, Levy M, Horton CL, Fleischman R. The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J Biol Chem. 1956;218:607–16.
Elgadi KM, Meguid RA, Qian M, Souba WW, Abcouwer SF. Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genom. 1999;1:51–62.
Szeliga M, Matyja E, Obara M, Grajkowska W, Czernicki T, Albrecht J. Relative expression of mRNAS coding for glutaminase isoforms in CNS tissues and CNS tumors. Neurochem Res. 2008;33:808–13.
van den Heuvel AP, Jing J, Wooster RF, Bachman KE. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol Ther. 2012;13:1185–94.
Cunningham CC, Stossel TP, Kwiatkowski DJ. Enhanced motility in NIH 3T3 fibroblasts that overexpress gelsolin. Science. 1991;251:1233–6.
Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25:839–50.
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101.
Szeliga M, Albrecht J. Opposing roles of glutaminase isoforms in determining glioblastoma cell phenotype. Neurochem Int. 2015;88:6–9.
Wilson KF, Erickson JW, Antonyak MA, Cerione RA. Rho GTPases and their roles in cancer metabolism. Trends Mol Med. 2013;19:74–82.
Redis RS, Vela LE, Lu W, Ferreira de Oliveira J, Ivan C, Rodriguez-Aguayo C, et al. Allele-specific reprogramming of cancer metabolism by the long non-coding RNA CCAT2. Mol Cell. 2016;61:520–34.
Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–19.
Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13:890–901.
Minguet J, Smith KH, Bramlage P. Targeted therapies for treatment of non-small cell lung cancer--Recent advances and future perspectives. Int J Cancer. 2016;138:2549–61.
Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163:174–86.
Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–34.
Jiang YY, Lin DC, Mayakonda A, Hazawa M, Ding LW, Chien WW. et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2017;66:1358–68.
Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene. 2017;36:2286–96.
Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014;8:1461–74.
Bondy CA, Cheng CM. Signaling by insulin-like growth factor 1 in brain. Eur J Pharmacol. 2004;490:25–31.
Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281:37372–80.
Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Magyar C, Braas D, et al. Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Rep. 2017;18:601–10.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (91639106 to H-bX, 81660041 to L-bD), the National Basic Research Program of China (2013CB531103 to H-bX and L-bD), and the Jiangxi Province Science Foundation for Youths (20122BAB215020 to H-wC).
Author contribution
H-bX and H-dS designed the experiments and interpreted data. Z-jC, D-lM and Q-yS performed the experiments. X-lT, X-lW, L-bD and Z-jC analyzed data. Z-jC and H-bX wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no financial, professional, or other conflicts related to this manuscript.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Cheng, ZJ., Miao, DL., Su, QY. et al. THZ1 suppresses human non-small-cell lung cancer cells in vitro through interference with cancer metabolism. Acta Pharmacol Sin 40, 814–822 (2019). https://doi.org/10.1038/s41401-018-0187-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0187-3
Keywords
This article is cited by
-
CDK7 inhibitor THZ1 enhances antiPD-1 therapy efficacy via the p38α/MYC/PD-L1 signaling in non-small cell lung cancer
Journal of Hematology & Oncology (2020)
-
Glutaminases regulate glutathione and oxidative stress in cancer
Archives of Toxicology (2020)
-
The P2X7 purinergic receptor: a potential therapeutic target for lung cancer
Journal of Cancer Research and Clinical Oncology (2020)

{kind=link}
{kind=link}