
Abstract
Pancreatic adenocarcinoma is a highly malignant cancer that often involves a deregulation of c-Myc. It has been shown that c-Myc plays a pivotal role in the regulation of a variety of physiological processes and is involved in early neoplastic development, resulting in poor progression. Hence, suppression of c-Myc overexpression is a potential strategy for pancreatic cancer therapy. CUDC-907 is a novel dual-acting inhibitor of phosphoinositide 3-kinase (PI3K) and histone deacetylase (HDAC). It has shown potential efficiency in patients with lymphoma, multiple myeloma, or thyroid cancer, as well as in solid tumors with c-Myc alterations, but the evidence is lacking for how CUDC-907 regulates c-Myc. In this study, we investigated the effect of CUDC-907 on human pancreatic cancer cells in vitro and in vivo. Our results showed that CUDC-907 potently inhibited the proliferation of 9 pancreatic cancer cell lines in vitro with IC50 values ranging from 6.7 to 54.5 nM. Furthermore, we revealed the antitumor mechanism of CUDC-907 in Aspc-1, PANC-1, and Capan-1 pancreatic cancer cells: it suppressed the HDAC6 subunit, thus downregulating c-Myc protein levels, which was a mode of action distinct from the existing mechanisms. Consistently, the extraordinary antitumor activity of CUDC-907 accompanied by downregulation of c-Myc and Ki67 expression in tumor tissue was observed in a human pancreatic cancer Aspc-1 xenograft nude mouse model in vivo. Our results suggest that CUDC-907 can be a valuable therapeutic option for treating pancreatic adenocarcinoma.
Similar content being viewed by others


Introduction
Pancreatic adenocarcinoma is one of the most malignant and lethal common cancers [1, 2], and its diagnosis and treatment are challenging, resulting in a significant increases in the mortality rate in recent years, with an overall 5-year survival rate under 5% and a median survival of 6 months [3, 4]. Lately, emerging evidence has suggested that c-Myc, which is commonly amplified and/or overexpressed in tumor cells [5, 6], is often completely deregulated as a consequence of mutant KRAS [7]. c-Myc plays a pivotal role in the regulation of many physiological processes, including cell cycle control, apoptosis, protein synthesis, and cellular senescence [8,9,10,11]. In addition, c-Myc has been shown to regulate the expression of approximately 15% of all human genes. Thus c-Myc influences many growth-promoting signal transduction pathways, such as WNT, Hedgehog, Notch, and transforming growth factor-β, as well as many receptor tyrosine kinases [9]. Furthermore, c-Myc is involved in cell metabolism and microRNA regulation to further accelerate tumor progression [10, 12], and c-Myc is involved in early neoplastic development, resulting in poor progression. Although strategies have emerged to reduce c-Myc expression, no drug currently specifically and directly targets c-Myc protein [6, 10, 13, 14]. Hence, suppression of c-Myc overexpression is a potential strategy for pancreatic cancer therapy.
CUDC-907 was first reported by the Rudi Bao group in 2012, and it is a novel dual-acting inhibitor of phosphoinositide 3-kinase (PI3K) and histone deacetylase (HDAC) [15]. As a single small compound, CUDC-907 has significant potential against cancer growth and metastasis through the simultaneous disruption of multiple oncogenic signaling networks [15,16,17,18,19]. Apart from suppressing AKT activation and accumulation of histone acetylation (direct targets of PI3K and HDAC, respectively), changes in c-Myc level is one of the most vital changes after treatment. However, the mechanisms of how PI3K and HDAC decrease c-Myc expression remain unclear. At present, CUDC-907 is undergoing phase I and II clinical trials, showing potential efficiency in patients with lymphoma, multiple myeloma, or thyroid cancer as well as in solid tumors with c-Myc alterations [20]. However, evidence is lacking for the mechanism of CUDC-907 regulation of c-Myc, especially in refractory pancreatic adenocarcinoma models.
This study focused on c-Myc and aimed to investigate the therapeutic effect and underlying mechanism of CUDC-907 against pancreatic cancer in vitro and in vivo.
Materials and methods
Compounds
CUDC-907 was purchased from Med Chem Express (MCE) (NJ, USA), dissolved in dimethylsulfoxide at 10 mg/mL to form a stock solution and stored at −80 °C for in vitro studies. For in vivo studies, CUDC-907 was dissolved with 30% propylene glycol. GDC-0941 and vorinostat were purchased from Selleck (Houston, TX, USA).
Cell culture
Human pancreatic cancer cell lines, including Aspc-1, PANC-1, and Capan-1, were purchased from American Type Culture Collection (Manassas, VA, USA) and cultured according to the suppliers’ instructions. Cell lines were characterized by Genesky Biopharma Technology using short tandem repeat markers.
Proliferation assays
Pancreatic cancer cell lines were seeded into 96-well plates at an appropriate density of 3000–5000 cells per well with the recommended culture medium and cultured for 24 h before exposure to increasing doses of the test compounds for the indicated amounts of time. Dosages corresponding to the half maximal inhibition (IC50) were detected using an SRB assay and calculated with GraphPad Prism 5.0.
Western blotting
Cells and tumor tissues were lysed in buffer containing 2% sodium dodecyl sulfate (SDS) and quantified using the BCA protein assay (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts of protein were subjected to electrophoresis on SDS–polyacrylamide gel electrophoresis and were probed with the following primary antibodies: c-Myc (Y69, ab32072, Abcam); phospho-AKT (p-AKT) (ser473) (#4060, CST); AKT (#9272, CST); acetyl-histone H3 (Lys27) (H3K27ac) (#8173, CST); acetyl-histone H3 (Lys9) (H3K9ac) (#9649, CST); alpha actin (Proteintech); histone-H3 (17168-1-AP, Proteintech); and GAPDH (KangChen Bio-tech).
RNA interference
Cells were seeded in six-well plates, and the medium was replaced with Opti-MEM I Reduced Serum Media (Invivogen, USA) containing 50.0 nM small interfering RNA (siRNA; GenePharma, China) and Lipofectamine RNAiMAX transfection reagent (Invitrogen, USA) according to the manufacturer’s recommendations. The sense sequences were as follows: c-Myc siRNA-1# 5′-CUCAACGUUAGCUUCACCATT-3′; c-Myc siRNA-2# 5′-GAACACACAACGUCUUGGATT-3′; HDAC1 siRNA# 5′-CGGUCAUGUCCAAAGUAAUTT-3′; HDAC2 siRNA# 5′-CGGUGAUAUUGGAAAUUAUTT-3′; HDAC3siRNA# 5′-AAUCAGAACUCACGCCAGUTT-3′; HDAC4 siRNA# 5′-CGACAGGCCUCGUGUAUGATT-3′; HDAC6 siRNA-1# 5′-GCAAUGGAAGAAGACCUAATT-3′; HDAC6 siRNA-2# 5′-GAGGACAAUGUAGAGGAGATT-3′; FOXO1 siRNA-1# 5′-CCUACACAGCAAGUUCAUUTT-3′; and FOXO1 siRNA-2# 5′-CCAUGGACAACAACAGUAATT-3′.
Construction of stable MYC-overexpressing or HDAC6-overexpressing cell lines
Amphotropic Phoenix 293T packaging cells were transfected with the MSCV-MYC retroviral construct and empty vector (obtained from Addgene) using Lipofectamine 2000 (Invitrogen). The PLVX-HDAC6 lentiviral construct and MOCK vector were mixed with the psPAX2 and PMD2.G packaging plasmids, and the mixtures were transfected into 293T cells using Lipofectamine 2000. After 48 h, virus-containing medium was collected, filtered, and used to infect host cells in the presence of 6 mg/mL polybrene. Stable transfectants were obtained by selection with 2 mg/mL puromycin (Sigma) for 2 weeks. Cells were analyzed for c-Myc or HDAC6 protein expression using Western blotting with corresponding antibodies.
Quantitative real-time PCR
Total RNA was isolated from cells using TRIzol (Invitrogen) and transcribed into cDNA. Real-time PCR was performed using an ABI VIIA7 Fast Real-time PCR System and SYBR Green PCR Master Mix (Bio-Red). The primers used in this study were as follows:
ACTIN, 5′-AAGATCATTGCTCCTCCTGAGC-3′ (forward) and 5′-CATACTCCTGCTTGCTGATCCA-3′ (reverse); c-Myc-1, 5′-GGCTCCTGGCAAAAGGTCA-3′ (forward) and 5′-CTGCGTAGTTGTGCTGATGT-3′ (reverse); and c-Myc-2, 5′-TCCCTCCACTCGGAAGGAC-3′ (forward) and 5′-CTGGTGCATTTTCGGTTGTT-3′ (reverse).
Animal studies
Male Balb/c nude mice (4–5-week old) were purchased from Shanghai BK Laboratory Animal Limited Company. Aspc-1 cells at a density of 5 × 106 in 0.2 mL of phosphate-buffered saline were inoculated subcutaneously into the right flank of the nude mice. When the tumor volume reached approximately 100 mm3 at approximately 1 week, the mice were randomly assigned to the vehicle control and treated groups (n = 7), and they were treated with 30% propylene glycol or 300 mg/kg CUDC-907, respectively. The tumors and body weight of the mice were measured individually twice weekly. After treatment for 3 weeks, mice were sacrificed after the final therapy. Tumors were removed and lysed for Western blotting or fixed for immunohistochemistry.
Immunohistochemistry assays
Tumor tissues were fixed in 4% paraformaldehyde, dehydrated, embedded in paraffin, deparaffinized, and incubated with Ki67 (1:50, Abcam, UK) and c-Myc (1:50, Y69, ab32072, Abcam) antibodies at 4 °C overnight, followed incubation with secondary antibody for 30 min at 37 °C. Slides were counterstained with hematoxylin and then cover slipped. Immunohistochemistry was independently assessed using the quick score method. Each specimen was assigned a score according to the intensity of the nucleic staining (no staining = 0; weak staining = 1; moderate staining = 2; and strong staining = 3), and the proportion of positive cells was estimated and given a percentage score on a scale from 1 to 6 (1%–4% = 1; 5%–19% = 2; 20%–39% = 3; 40%–59% = 4; 60%–79% = 5; and 80%–100% = 6). The final immune-reactive score was determined by multiplying the intensity score with the extent of score of stained cells, ranging from 0 (the minimum) to 18 (the maximum).
Statistical analyses
Data were presented as the mean ± SEM from at least three independent experiments, and differences were considered significant when P < 0.05 as determined by Student’s t test.
Results
CUDC-907 inhibits proliferation, causes G2/M arrest, and induces apoptosis in pancreatic cancer cells
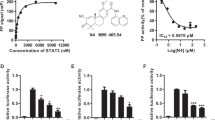
To identify compounds that inhibit the growth of pancreatic cancer cells in vitro, we screened a series of small molecule inhibitors, including Food and Drug Administration-approved drugs and agents undergoing clinical trials, to test the growth inhibition ratio against Aspc-1 pancreatic cancer cells. Four serial concentrations for each drug were tested, and the growth inhibition ratio was measured using a SRB assay after 6 days. Among the compounds tested, CUDC-907 showed the most promising effect (>95% under 0.37 μM) (Fig. 1a). To further evaluate the efficacy of this compound in inhibiting pancreatic cancer, nine pancreatic cancer cell lines were cultured for 3 days to assess the IC50 of CUDC-907. The mean IC50 values ranged from 6.7 to 54.5 nM (Fig. 1b). Accordingly, CUDC-907 revealed a dose-dependent inhibition of Aspc-1, PANC-1, and Capan-1 cell proliferation over time (Fig. 1c). Moreover, the colony assay validated the inhibitory effect of CUDC-907 on pancreatic cancer cell lines (Fig. 1d).


CUDC-907 displays potent antiproliferative activity against pancreatic cancer cell lines. a The growth inhibition ratio in Aspc-1 cells after 6 days of treatment with four serial concentrations of various inhibitors as indicated. Red to blue, cell growth inhibition ratio from 100% to 0%. b A panel of human pancreatic adenocarcinoma cells were treated with CUDC-907 at gradient concentrations for 3 days, and the half-maximal inhibitory concentration (IC50) was measured using the SRB assay. Error bars represent the mean ± SD from three independent experiments. c Dose-dependent proliferation curves of Aspc-1, PANC-1, and Capan-1 cells over time. Cells were treated with CUDC-907 at gradient concentrations for 0, 1, 2, 3, or 4 days, and the relative cell growth ratio (%) was calculated using SoftMax Pro. Error bars represent the mean ± SD from three independent experiments. d Colony-formation assays. Aspc-1, PANC-1, and Capan-1 cells were measured with crystal violet staining after treatment with CUDC-907 at 5 to 20 nM for 14 days. e–g CUDC-907 arrests pancreatic cancer cells in the G2/M phase and induces apoptosis. Aspc-1 and Capan-1 cells were incubated with various concentrations (as indicated) of CUDC-907 for 24 or 48 h, and the percentage of each cell cycle phase (e) and apoptosis (f) was assessed by flow cytometry; (g) Aspc-1 and Capan-1 cells were collected for Western blotting with the indicated antibodies after treatment with CUDC-907 for 48 h
Owing to the remarkable growth inhibition of pancreatic cancer cells caused by CUDC-907, flow cytometry was used to evaluate changes in the cell cycle and apoptosis of pancreatic cancer cells. Aspc-1 and Capan-1 cells were treated with 10, 25, 50, 100, and 500 nM CUDC-907 for 24 h for cell cycle assessment and 48 h for apoptosis assessment. CUDC-907 arrested Aspc-1 and Capan-1 cells at G2/M phase and strongly induced cell apoptosis in a concentration-dependent manner (Fig. 1e, f). Furthermore, key factors involved in cell apoptosis and cell cycle arrest were analyzed by Western blotting. CUDC-907 markedly upregulated p21 (CDKN1A) and cleaved-poly ADP-ribose polymerase, which was accompanied by decreases in the corresponding precursors, and CUDC-907 downregulated Bcl-2, Bax, and cyclin D1 (Fig. 1g). These results suggested that CUDC-907 may inhibit pancreatic cancer cell proliferation in part through cell cycle arrest and/or inducing cell apoptosis.
CUDC-907 downregulates c-Myc protein levels to inhibit proliferation
According to previous research on CUDC-907, the downregulation of c-Myc protein is an early event induced by CUDC-907 treatment, resulting in the growth restriction of c-Myc-driven cancers [16]. Pancreatic cancer is a type of refractory cancer with predominant c-Myc overexpression, and the proliferation of pancreatic cancer can be inhibited with a low concentration of CUDC-907 as suggested by the aforementioned results. To explore the mechanisms underlying this inhibition, we evaluated c-Myc expression in pancreatic cancer cell lines and the normal pancreatic H6C7 cell line. The pancreatic cancer cell lines exhibited higher c-Myc expression than that of the normal H6C7 cell line (Fig. 2a), which confirmed that c-Myc expression is generally higher in pancreatic cancer, suggesting an underlying reliance on c-Myc. Next, we investigated the ability of CUDC-907 to inhibit HDACs, PI3K activity, and c-Myc protein, which are targets of CUDC-907. CUDC-907 reduced AKT phosphorylation and caused H3K27ac and H3K9ac accumulation in Aspc-1, PANC-1, and Capan-1 cells in a dose-dependent manner after 24 h (Fig. 2b). CUDC-907 treatment also inhibited c-Myc protein expression at a similar dose used to reduce AKT phosphorylation and accumulate histone acetylation (Fig. 2b). The real-time PCR results for c-Myc expression were consistent with these findings (Fig. 2c), indicating that c-Myc is a crucial protein regulated by CUDC-907 treatment in cells.


CUDC-907 downregulates c-Myc protein to inhibit cell proliferation. a c-Myc expression in pancreatic cancer cell lines compared with the normal cell line H6C7 evaluated by Western blotting. b c-Myc protein level alteration. Protein levels in Aspc-1, PANC-1, and Capan-1 cells treated with CUDC-907 (0, 25, 50, 100, and 500 nM) for 24 h were measured. The cell lysates were subjected to Western blotting analysis using the indicated antibodies. c c-Myc mRNA level alteration. Cells pretreated as described in b followed by qRT-PCR analysis of c-Myc level. Error bars represent the mean ± SD from three independent experiments. d Cell growth assay. Pancreatic cancer cells were transfected with c-Myc-specific siRNAs for 24 h and then transferred to a 96-well plate to be cultured for 3 or 6 days or to a 6-well plate for 14 days to form colonies. Relative cell proliferation rates were calculated by OD values, and colonies were scanned for crystal violet staining. Interference efficiency was verified by Western blotting. e Aspc-1-MYC and PANC-1-MYC cells, which were stably transfected with a mock or MYC-overexpression plasmid, were transfected with siRNAs against c-Myc or scramble for 24 h and then were transferred to 96-well plates to be cultured for 6 days, and the relative cell proliferation rate was calculated. Overexpression efficiency was verified by Western blotting. Error bars represent the mean ± SD from triplicates or three independent experiments. ***P < 0.001
We next investigated the role of c-Myc in pancreatic cancer cell proliferation using two independent siRNAs to knockdown c-Myc in Aspc-1, PANC-1, and Capan-1 cells. After transfection of siRNAs for 24 h, cells were transferred to a 96-well plate for culture for 3 or 6 days or to a 6-well plate to form colonies. Relative cell proliferation was calculated based on optical density (OD) values. Knockdown of c-Myc in cells suppressed proliferation by approximately 50% (Fig. 2d, i, ii), and a similar result was observed using colony-formation assays (Fig. 2d, iii). The interference efficiency was verified by Western blotting (Fig. 2d, iv). Consistently, c-Myc overexpression increased the proliferation of Aspc-1 and PANC-1 cells by approximately 40% and 20%, respectively, whereas c-Myc suppression in Aspc-1-MYC and PANC-1-MYC cells partially, but not entirely, attenuated the increased cell proliferation (Fig. 2e).
Taken together, these data revealed that c-Myc overexpression positively correlates with pancreatic cancer cell proliferation. Thus inhibition of c-Myc by CUDC-907 offers a potential therapeutic effect.
CUDC-907 inhibits the PI3K pathway to suppress c-Myc function
Previous studies have demonstrated that HDAC inhibitors act synergistically with PI3K inhibitors to inhibit tumor growth in a MYC-dependent manner in different types of cancer [18, 21,22,23]. Because CUDC-907 is a dual inhibitor of PI3K and HDACs, we evaluated the effect of individually inhibiting the PI3K pathway and/or HDACs on c-Myc protein regulation in pancreatic cancer cells. Aspc-1, PANC-1, and Capan-1 pancreatic cancer cells were treated individually with the HDAC inhibitor, vorinostat (SAHA), and the PI3K inhibitor, GDC-0941, for 24 h. Western blotting analysis revealed that both PI3K and HDAC inhibitors regulated c-Myc protein levels at various concentrations (Fig. 3a, i, ii). Moreover, the IC50 values of vorinostat and GDC-0941 alone or in combination against Aspc-1, PANC-1, and Capan-1 cells were measured. The combination index was calculated to be 0.74, which indicated a synergistic effect between PI3K and HDAC inhibitors (Fig. 3b).

CUDC-907 inhibits both the PI3K pathway and HDACs to suppress c-Myc function. a Individual PI3K or HDACs inhibitors suppress c-Myc. Aspc-1, PANC-1, and Capan-1 cells were treated with the indicated concentrations of the PI3K inhibitor GDC-0941 or the HDAC inhibitor vorinostat for 24 h, and the expression of c-Myc, phosphorylation of AKT and the acetylation of histone proteins were detected by Western blotting. b Synergistic effect of PI3K and HDAC inhibitors. IC50 values of vorinostat and GDC-0941 alone or in combination against Aspc-1, PANC-1, and Capan-1 cells were tested, and the combination index (CI) was calculated using the CompuSyn software. CI < 1, synergic effect; CI = 1, additive effect; CI > 1, antagonistic effect. Three independent experiments were exhibited. Error bars represent the mean ± SD from three independent experiments
To identify the mechanism of CUDC-907 suppression of c-Myc through the PI3K pathway, Aspc-1 or PANC-1 cells were individually treated with CUDC-907 or the PI3K inhibitor, GDC-0941, for 2 h, and the c-Myc and PI3K signaling pathways were evaluated by Western blotting analysis. CUDC-907 and GDC-0941 upregulated c-Myc Thr58 phosphorylation protein levels but downregulated c-Myc protein levels. The relative accumulation of phospho-c-Myc (Thr58) to total c-Myc ratios are shown in Fig. 4a. The potential involvement of proteasomes in the early accumulation of phospho-c-Myc (Thr58) induced by CUDC-907 was investigated. The proteasome inhibitor, MG-132, blocked the reduction of c-Myc protein levels caused by CUDC-907, as shown in Fig. 4b. These results suggested that the PI3K pathway is inactivated after CUDC-907 treatment, resulting in accumulation of c-Myc Thr58 phosphorylation and leading to the proteasome-mediated reduction of c-Myc protein.

CUDC-907 inhibits the PI3K pathway and leads to degradation of c-Myc. a PI3K pathway inhibition upregulates phosphor-c-Myc (Thr58) protein level. Aspc-1 and PANC-1 cells were treated with CUDC-907 or the PI3K inhibitor GDC-0941 for 2 h, and phosphor-c-Myc (Thr58) and total c-Myc protein levels were detected by Western blotting analysis using the indicated antibodies. Relative accumulation of phosphor-c-Myc (Thr58) to total c-Myc (normalized by the same Actin using the Image J software) was shown. b Aspc-1 and PANC-1 cells were treated with CUDC-907 (0, 25, 50, 100, and 500 nM) with (right) or without (left) proteasome inhibitor MG-132 for 2 h and then the cell lysates were subjected to Western blotting analysis to detect the c-Myc protein level
CUDC-907 regulates c-Myc expression through HDAC6 inhibition
The above results indicated that CUDC-907 inhibits cancer cell proliferation through decreasing the expression of c-Myc protein and that HDACs play a vital role in this process. The HDAC family contains 18 HDACs, which have been shown to regulate several biological pathways through the deacetylation of lysine residues on histone and non-histone proteins, indicating that HDACs have different functions [24,25,26,27]. As a pan-HDAC inhibitor, CUDC-907 can inhibit multiple subunits of the HDAC family. To further confirm the mechanism underlying the effect of CUDC-907 on HDACs, five common HDACs (HDAC1, HDAC2, HDAC3, HDAC4, and HDAC6) were investigated by measuring histone acetylation changes and c-Myc expression following transfection with individual siRNAs for 72 h in Aspc-1 and Capan-1 cells. HDAC6 exhibited a distinct role in regulating histone acetylation and c-Myc protein levels compared with other HDACs. Transfection of HDAC6 siRNAs reduced c-Myc levels with no influence on histone acetylation, whereas knockdown of other HDAC subunits increased c-Myc levels and slightly increased H3K27ac and H3K9ac levels (Fig. 5a). Subsequently, Aspc-1, PANC-1, and Capan-1 cells were transfected with individual HDAC siRNAs for 3 days to evaluate cell proliferation. A significant reduction in cell proliferation was detected following the transfection of HDAC6 siRNAs but not the other siRNAs (Fig. 5b). Moreover, Aspc-1 cells stably overexpressing HDAC6 were generated (Aspc-1-HDAC6), and cell proliferation was evaluated. Overexpression HDAC6 partially reversed the antiproliferation effect induced by CUDC-907 (Fig. 5c). Next, the relation between HDAC6 and c-Myc was further examined. FOXO transcription factors, especially FOXO1, have profound roles in the remodeling and regulation of many genes [28]. As a tumor suppressor, FOXO1 inactivation has been documented in many kinds of human cancer [29], and inactivation of FOXO1 has an antagonistic effect on c-Myc [30]. The present study showed that siHDAC6 causes accumulation of FOXO1 and represses c-Myc expression (Fig. 5d, i), which was consistent with previous research on FOXO1 and c-Myc [22]. Conversely, overexpression of HDAC6 or siFOXO1 in Aspc-1 cells upregulated the c-Myc protein levels (Fig. 5d, ii, iii). Thus these results indicated that HDAC6 regulates c-Myc through the HDAC6–FOXO1–c-Myc axis. Consistently, CUDC-907 treatment, resulting in the accumulation of FOXO1 and inhibition of c-Myc, further supported this mechanism (Fig. 5e). These results implied that CUDC-907 treatment inhibits HDAC enzyme activity although the protein subunit levels were not affected. Thus HDAC6 may be an upstream regulator of c-Myc expression and may influence cancer cell proliferation.

CUDC-907 regulates c-Myc expression and cell proliferation through HDAC6 inhibition. a HDAC6 interference inhibits c-Myc protein levels. Aspc-1 and Capan-1 cells were transfected with individual siRNA against HDAC1, 2, 3, 4, and 6. HDAC6 interference inhibits c-Myc protein levels, as detected by Western blotting analysis using the indicated antibodies. b HDAC6 interference suppresses cell proliferation. Interference was performed as shown in Fig. 4a, and cells were transferred to a 96-well plate to be cultured for 3 days. The relative cell proliferation rate was calculated by OD values. Error bars represent the mean ± SD from three independent experiments. c HDAC6 reverses antiproliferation effect induced by CUDC-907 partially. Aspc-1-MOCK and Aspc-1-HDAC6 cells, which were stably transfected with a MOCK or HDAC6-overexpressing plasmid, were treated with CUDC-907 (0, 2, 5 nM) for 3 days, and the relative cell proliferation rate was calculated using SoftMax Pro. Error bars represent the mean ± SD from three independent experiments. d, e HDAC6 regulates c-Myc through the HDAC6–FOXO1–c-Myc axis. Aspc-1 cells were transfected with individual siRNA against HDAC6, FOXO1, or HDAC6-overexpressing plasmid, and the protein levels were detected by Western blotting analysis using the indicated antibodies (d). Also, a series of concentration of CUDC-907 were treated in Aspc-1, PANC-1, and Capan-1 cells for 24 h, and the FOXO1 and c-Myc protein levels were evaluated by Western blotting (e)
CUDC-907 inhibits the growth of pancreatic xenografts in vivo
Based on the encouraging CUDC-907 in vitro results, the antitumor effect of CUDC-907 in vivo was evaluated next. A human pancreatic xenograft model was established by subcutaneously injecting human pancreatic cancer Aspc-1 cells into nude mice. After 19 days of intragastric administration, Aspc-1 xenograft-bearing mice treated with 300 mg/kg CUDC-907 once daily exhibited significantly reduced tumor growth compared to the vehicle group with a T/C (%) of 43.9% (P < 0.01) (Fig. 6a, i). Importantly, there was no reduction in body weight (Fig. 6a, ii). After the final treatment on day 20, all the nude mice were sacrificed, and tumors were resected, imaged, measured, and weighed as shown in Fig. 6b. Additionally, the expression levels of key intratumoral downstream signaling molecules were examined. The CUDC-907-treated group exhibited a profound inhibition of c-Myc, phospho-AKT, and phospho-Erk levels as well as a slight increase of H3K27ac (Fig. 6c). Immunohistochemistry analysis of the CUDC-907-treated group showed that Ki67 and c-Myc levels were downregulated compared to those in the untreated group (Fig. 6d). All together, these findings suggested that CUDC-907 had a potential therapeutic effect on pancreatic xenografts.

CUDC-907 inhibits pancreatic cancer growth in vivo. a Tumor growth inhibition upon CUDC-907 treatment in Aspc-1 pancreatic xenografts. The curve of relative tumor volume after 20 days of treatment with CUDC-907 or vehicle and the corresponding curve of body weight are shown. Error bars represent the mean ± SD from seven individuals in each group. **P < 0.01. b Tumor morphology and weight. After 20 days, all nude mice were sacrificed, and the tumors were resected to be imaged and weighed. c Protein levels' alteration. Equal amounts of proteins from tumor tissues were evaluated for the phosphorylation of AKT and Erk and the acetylation of histone. d Immunohistochemistry staining. (i) Representative images of Ki67 and c-Myc staining of untreated xenografts and xenografts treated with CUDC-907. Scale bar, 100 μm. (ii) Quantification of immunohistochemistry staining was calculated by quick score (QS) method. Ten random areas from three individually stained tumors were captured. Error bars represent the mean ± SD from three independent experiments. **P < 0.01
Discussion
Owing to the extremely high frequency of KRAS gene mutations (>90%) detected in pancreatic cancer patients [31, 32], attempts to target KRAS directly have been investigated in recent years but have failed in the clinic. Thus KRAS is widely assumed to be untreatable by drugs [33, 34]. As previous studies have identified, c-Myc overexpression is also present at a high rate in pancreatic adenocarcinomas, and the inhibition of MYC family proteins may eradicate KRAS-driven cancer [5, 35, 36]. c-Myc is a member of the MYC family, and its overexpression is positively correlated with the degree of tumor malignancy; thus inhibiting c-Myc protein may benefit PDAC treatment. CUDC-907, a dual-acting inhibitor of PI3K and HDAC, exhibits predominant anticancer effects in c-Myc-driven tumors, such as diffuse large B cell lymphoma [16, 18, 19], indicating that this drug may be an effective therapy across a wide range of hematologic and solid cancers known to be dependent on c-Myc. This study demonstrated CUDC-907-induced growth inhibition of a panel of human pancreatic cancer cell lines, indicating the potent antiproliferative activity of CUDC-907 in vitro.
To better address the underlying mechanisms, the pleiotropic effects of CUDC-907, which can inhibit both HDACs and PI3K signaling, were investigated. These two distinct targets unequally contribute to cell growth inhibition. For example, the PI3K pathway was inactivated after CUDC-907 treatment, resulting in a reduction in AKT phosphorylation, which was thought to lead to the accumulation of c-Myc Thr58 phosphorylation and the caspase-independent reduction of c-Myc protein. This process plays a pivotal role in regulating the biologic effects of CUDC-907 [16]. By comparing to the PI3K pathway influence on c-Myc, we found that HDAC inhibition altered c-Myc levels in other distinct ways. This finding was consistent with the existing knowledge that HDAC inhibitors reduce c-Myc protein levels by suppressing transcription [18, 37]. To further determine which HDAC family members are vital for the regulation of c-Myc protein levels, we tested a panel of siRNAs that sufficiently knockdown HDAC1, 2, 3, 4, and 6. HDAC6 knockdown strongly reduced c-Myc protein and suppressed proliferation but resulted in almost no change in histone acetylation in pancreatic cancer cells. In contrast, siRNAs targeting HDAC1, 2, 3, and 4 exhibited the opposite effects on c-Myc and histone acetylation levels and showed minimal activity against pancreatic cancer cell growth. These findings were supported by a previous study demonstrating that enhanced HDAC6 expression induces c-Myc expression, resulting in the promotion of proliferation and motility in estrogen receptor-negative breast cancer cells [38]. Moreover, we also demonstrated that HDAC6 influenced c-Myc protein through the HDAC6–FOXO1–c-Myc axis. Thus HDAC6 is likely to be the predominant upstream protein regulating c-Myc expression and likely serves as a mediator of tumor cell survival in the present model. Based on these in vitro findings, we also evaluated the antitumor activities of CUDC-907 in human pancreatic cancer models. Importantly, CUDC-907 potently inhibited xenograft growth.
In summary, these data suggested that CUDC-907 is an effective drug in pancreatic cancer treatment that warrants further clinical development. In addition, it would be interesting to identify additional specific HDAC6 inhibitors that may possess better anticancer properties and improve survival in advanced pancreatic cancer.
References
Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–17.
Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–49.
Lin QJ, Yang F, Jin C, Fu DL. Current status and progress of pancreatic cancer in China. World J Gastroenterol. 2015;21:7988–8003.
Michl P, Gress TM. Current concepts and novel targets in advanced pancreatic cancer. Gut. 2013;62:317–26.
Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol. 2002;15:462–9.
McKeown MR, Bradner JE. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med. 2014;4:a014266
Tsang AT, Dudgeon C, Yi L, Yu X, Goraczniak R, Donohue K, et al. U1 adaptors suppress the KRAS-MYC oncogenic axis in human pancreatic cancer xenografts. Mol Cancer Ther. 2017;16:1445–55.
Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4:a014241.
Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
Fletcher S, Prochownik EV. Small-molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta. 2015;1849:525–43.
Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11.
Wang L, Zhang X, Jia LT, Hu SJ, Zhao J, Yang JD, et al. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology. 2014;59:1850–63.
Horiuchi D, Anderton B, Goga A. Taking on challenging targets: making MYC druggable. Am Soc Clin Oncol Educ Book. 2014:e497–e502.
Posternak V, Cole MD. Strategically targeting MYC in cancer. F1000Res. 2016. https://doi.org/10.12688/f1000research.7879.1.
Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu GX, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012;18:4104–13.
Sun K, Atoyan R, Borek MA, Dellarocca S, Samson ME, Ma AW, et al. Dual HDAC and PI3K inhibitor CUDC-907 downregulates MYC and suppresses growth of MYC-dependent cancers. Mol Cancer Ther. 2017;16:285–99.
Kotian S, Zhang L, Boufraqech M, Gaskins K, Gara SK, Quezado M, et al. Dual inhibition of HDAC and tyrosine kinase signaling pathways with CUDC-907 inhibits thyroid cancer growth and metastases. Clin Cancer Res. 2017;23:5044–54.
Mondello P, Derenzini E, Asgari Z, Philip J, Brea EJ, Seshan V, et al. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget. 2017;8:14017–28.
Ferreira AC, de-Freitas-Junior JC, Morgado-Diaz JA, Ridley AJ, Klumb CE. Dual inhibition of histone deacetylases and phosphoinositide 3-kinases: effects on Burkitt lymphoma cell growth and migration. J Leukoc Biol. 2016;99:569–78.
Younes A, Berdeja JG, Patel MR, Flinn I, Gerecitano JF, Neelapu SS, et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016;17:622–31.
Simmons JK, Michalowski AM, Gamache BJ, DuBois W, Patel J, Zhang K, et al. Cooperative targets of combined mTOR/HDAC inhibition promote MYC degradation. Mol Cancer Ther. 2017;16:2008–21.
Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K antagonists cooperate to inhibit growth of MYC-driven medulloblastoma. Cancer Cell. 2016;29:311–23.
Grinshtein N, Rioseco CC, Marcellus R, Uehling D, Aman A, Lun X, et al. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget. 2016;7:59360–76.
Nebbioso A, Carafa V, Conte M, Tambaro FP, Abbondanza C, Martens J, et al. c-Myc modulation and acetylation is a key hdac inhibitor target in cancer. Clin Cancer Res. 2017;23:2542–55.
Aramsangtienchai P, Spiegelman NA, He B, Miller SP, Dai L, Zhao Y, et al. HDAC8 catalyzes the hydrolysis of long chain fatty acyl lysine. ACS Chem Biol. 2016;11:2685–92.
Damaskos C, Garmpis N, Karatzas T, Nikolidakis L, Kostakis ID, Garmpi A, et al. Histone deacetylase (HDAC) inhibitors: current evidence for therapeutic activities in pancreatic cancer. Anticancer Res. 2015;35:3129–35.
Mishra VK, Wegwitz F, Kosinsky RL, Sen M, Baumgartner R, Wulff T, et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017;45:6334–49.
Tan P, Guan H, Xie L, Mi B, Fang Z, Li J, et al. FOXO1 inhibits osteoclastogenesis partially by antagonizing MYC. Sci Rep. 2015;5:16835.
Wang Y, Zhou Y, Graves DT. FOXO transcription factors: their clinical significance and regulation. Biomed Res Int. 2014;2014:925350.
Peck B, Ferber EC, Schulze A. Antagonism between FOXO and MYC regulates cellular powerhouse. Front Oncol. 2013;3:96.
Gharibi A, Adamian Y, Kelber JA. Cellular and molecular aspects of pancreatic cancer. Acta Histochem. 2016;118:305–16.
Mangge H, Niedrist T, Renner W, Lyer S, Alexiou C, Haybaeck J. New diagnostic and therapeutic aspects of pancreatic ductal adenocarcinoma. Curr Med Chem. 2017;24:3012–24.
Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817–22.
Karandish F, Mallik S. Biomarkers and targeted therapy in pancreatic cancer. Biomark Cancer. 2016;8:27–35.
Liu H, Ai J, Shen A, Chen Y, Wang X, Peng X, et al. c-Myc alteration determines the therapeutic response to FGFR inhibitors. Clin Cancer Res. 2017;23:974–84.
Hahm ER, Singh KB, Singh SV. c-Myc is a novel target of cell cycle arrest by honokiol in prostate cancer cells. Cell Cycle. 2016;15:2309–20.
Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101:1241–6.
Hsieh TH, Tsai CF, Hsu CY, Kuo PL, Lee JN, Chai CY, et al. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J. 2012;26:778–87.
Acknowledgements
This work was supported by the Shanghai Talent Development Funds (No. 201663 to XH), the Open Project of State Key Laboratory of Drug Research (No. SIMM1705KF-10 to HH), and the Youth Innovation Promotion Association CAS (to XH).
Author contributions
XH and MYG designed research; XHF performed research; XHF, XZ, HY, XWX, ZLH, JY, XLZ, RRW, ZQZ. and SRT analyzed the data; XH, XHF, and XZ drafted the manuscript; and XH and MYG supervised the work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Fu, Xh., Zhang, X., Yang, H. et al. CUDC-907 displays potent antitumor activity against human pancreatic adenocarcinoma in vitro and in vivo through inhibition of HDAC6 to downregulate c-Myc expression. Acta Pharmacol Sin 40, 677–688 (2019). https://doi.org/10.1038/s41401-018-0108-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0108-5
Keywords
This article is cited by
-
CUDC-907 exhibits potent antitumor effects against ovarian cancer through multiple in vivo and in vitro mechanisms
Cancer Chemotherapy and Pharmacology (2024)
-
Synergistic effect of adavosertib and fimepinostat on acute myeloid leukemia cells by enhancing the induction of DNA damage
Investigational New Drugs (2024)
-
Antitumor activity and mechanism of resistance of the novel HDAC and PI3K dual inhibitor CUDC-907 in pancreatic cancer
Cancer Chemotherapy and Pharmacology (2021)
-
A novel derivative of valepotriate inhibits the PI3K/AKT pathway and causes Noxa-dependent apoptosis in human pancreatic cancer cells
Acta Pharmacologica Sinica (2020)
-
Mission Possible: Advances in MYC Therapeutic Targeting in Cancer
BioDrugs (2019)
