
Abstract
Pulmonary arterial hypertension (PAH) is the most common form of pulmonary hypertension. Pulmonary arterial remodeling is closely related to the abnormal proliferation of pulmonary artery smooth muscle cells (PASMCs), which leads to the thickening of the medial layer of muscular arteries and then results in the narrowing or occlusion of the precapillary arterioles and PAH. However, the mechanisms underlying the abnormal proliferation of PASMCs remain unclear. In this study, we established rat primary PAH models using monocrotaline (MCT) injection or hypoxic exposure, then investigated the expression patterns of seven miRNAs associated with multiple pathogenic pathways central to pulmonary hypertension, and further explored the roles and the possible mechanisms of miR-135a during the development of PAH. In the rat primary PAH models, we observed that the expression of miR-135a-5p in lungs was drastically decreased at the initial stage of PAH development after MCT administration or hypoxic exposure, but it increased by 12-fold or 10-fold at the later stage. In vitro study in PASMCs showed a similar pattern of miR-135a-5p expression, with downregulation at 6 h but upregulation at 18, 24, and 48 h after hypoxic exposure. Early, but not late, administration of a miR-135a-5p mimic inhibited hypoxia-induced proliferation of PASMCs. The protective role of early miR-135a-5p agomir in the PAH rat model further supported the hypothesis that the early decrease in the expression of miR-135a-5p contributes to the proliferation of PASMCs and development of PAH, as early administration of miR-135a-5p agomir (10 nM, i.v.) reversed the elevated mean pulmonary arterial pressure and pulmonary vascular remodeling in MCT-treated rats. We revealed that miR-135a-5p directly bound to the 3′-UTR sequence of rat transient receptor potential channel 1 (TRPC1) mRNA and decreased TRPC1 protein expression, thus inhibiting PASMC proliferation. Collectively, our data suggest that dysregulation of miR-135a-5p in PASMCs contributes to the abnormal proliferation of PASMCs and the pathogenesis of PAH. Increasing miR-135a-5p expression at the early stage of PAH is a potential new avenue to prevent PAH development.
Similar content being viewed by others


Introduction
Pulmonary arterial hypertension (PAH) is the most common form of pulmonary hypertension, which is a substantial global health issue, particularly in countries with large aging populations. Patients with PAH share a similar pulmonary angioproliferative vasculopathy that predominantly affects the precapillary arterioles, resulting in increased right-ventricular afterload and finally leading to right heart failure [1]. Pulmonary arterial remodeling is closely related to the abnormal proliferation of pulmonary artery smooth muscle cells (PASMCs), which leads to the thickening of the medial layer of muscular arteries and then results in the narrowing or occlusion of the precapillary arterioles and PAH [1]. Although great advances have been made in the treatment of PAH over the past 20 years [2], the cellular mechanisms underlying the abnormal proliferation of PASMCs are still not entirely clear.
Recent studies have demonstrated that microRNAs (miRNAs) are implicated in the onset, progression or treatment responsiveness of PAH [3]. By using a network biology approach, Parikh predicted the miRNA profile, including miR-135a-5p, miR-211-5p, miR-204-5p, miR-224-5p, miR-93-5p, miR-17-5p, and miR-27b-5p, associated with multiple pathogenic pathways central to pulmonary hypertension [4]. However, the expression patterns of these miRNAs and their roles in the onset and progression of PAH remained largely unknown.
In this study, we determined the expression patterns of these seven miRNAs in the rat primary PAH model induced by monocrotaline (MCT, 60 mg/kg) [5] and hypoxic exposure [6]. We further explored the roles and the possible mechanisms of miR-135a-5p during the development of PAH.
Materials and methods
Experimental animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Third Military Medical University. Male Sprague-Dawley (SD) rats (200–250 g, from the Experimental Animal Center of the Third Military Medical University) were used for all animal studies. The rat model of PAH was established by a single subcutaneous injection of MCT (60 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) [5] or exposed to hypobaric hypoxia at a simulated altitude of 5 kilometers for 4 weeks (oxygen concentration of 10%) [6]. Animals were euthanized at the 1st, 2nd, 3rd, and 4th weeks after modeling, and their lungs were collected for further analysis of miRNA expression.
To explore the in vivo effects of miRNA-135a-5p on PAH development, 10 nM miRNA-135a-5p agomir was intravenously given to the PAH rat model every 3 days for 3 constitutive weeks after MCT administration. The miRNA-135a-5p agomir was modified by an O-methyl moiety at the 2′-ribose position and cholesteryl functionality at the 5′ end (RiboBio Co. Ltd., Guangzhou, China) and diluted in phosphate-buffered saline (PBS) buffer. Agomir negative control (NC), which was similar to agomir but with a scrambled seeding sequence, was used as a control. The rats were sacrificed for further experiments at the 4th week after MCT administration.
Primary PASMC culture, transfection, infection, and luciferase reporter assay
Primary culture of PASMCs was performed as described in the reported literature [7]. Briefly, male SD rats were sacrificed, and their intrapulmonary arteries were isolated rapidly and placed in ice-cold PBS. After the endothelium and the adventitia were carefully cleaned, the vessels were washed, dissected into 1–3 mm3 pieces, placed in a 25 cm2 flask and cultured in Dulbecco's Modified Eagle's Medium (DMEM, high-glucose) supplemented with 10% Fetal Bovine Serum (FBS), 100 U/mL penicillin and 100 U/mL streptomycin in a humidified atmosphere (5% CO2) at 37 °C. After 72 h incubation, the aortic fragments were removed and the attached cells were allowed to grow for 5–7 days. The purity of the primary cultured PASMCs was verified by immunofluorescence using a specific antibody against alpha smooth muscle Actin(Abcam, MA, USA) and vWF (Abcam, USA). Cells between three and five passages were used for further experiments.
For transient transfection of miR-135a-5p mimic and inhibitor (Thermo Fisher Scientific, Waltham, MA, USA), primary PASMCs were transfected with miR-135a-5p mimic (30 nM, Thermo Fisher Scientific, USA) or inhibitor (30 nM, Thermo Fisher Scientific, USA) by using Lipofectamine RNAiMAX (Thermo Fisher Scientific, USA), according to the manufacturer’s protocol. Then, the transfected cells were put into a hypoxia chamber (3% O2, 5% CO2) for 48 h. To explore the role of late miR-135a-5p upregulation in the process of PAH, primary PASMCs were cultured under hypoxia for 12 h and then transfected with miR-135a-5p mimic (30 nmol/L, Thermo Fisher Scientific, USA) or inhibitor (30 nmol/L, Thermo Fisher Scientific, USA). PASMCs were cultured under hypoxia for another 48 h. Primary PASMCs cultured in normoxic conditions were used as control.
A recombinant adenovirus containing the rat transient receptor potential channel 1 (TRPC1) gene (Adv-rTRPC1) was prepared as described previously [8]. The coding sequence of the TRPC1 cDNA (lacking the 3′-UTR) and TRPC1-3′-UTR (including the 3′-UTR) were subcloned into the adenoviral recombination cassette vector (pACCMVpLpa plasmid) (Cqwestern, Chongqing, China). To study the role of miR-135a-5p in the TRPC1-based regulation of cell proliferation, PASMCs were infected with the purified Adv-rTRPC1 viral plaques with or without miR-135a-5p mimic (30 nmol/L, Thermo Fisher Scientific, USA). Then, the PASMCs were cultured in high-glucose DMEM supplemented with 10% FBS in normoxia (21% O2, 5% CO2) at 37 °C in a humidified atmosphere. A cell proliferation assay was conducted 48 h after the initial infection.
The 3′-UTR of the rat TRPC1 gene was amplified by PCR with C6 genomic DNA as template. The purified PCR products were inserted downstream of the firefly luciferase gene in the Firefly-Renilla dual reporter vector pmiR-RB-REPORT™ after digestion with XhoI and NotI, and the construct was designated wild-type 3′-UTR. The mutated 3′-UTR was amplified by PCR with wild-type 3′-UTR as the template using a mutation primer. The putative miR-135a-binding site 5′-AAAGCCA-3′ in the TRPC1-3′-UTR was mutated into 5′-TTCGGTA-3′. PASMC lysates were collected 24 h after transfection with miR-135a-5p (10 or 30 nmol/L, Thermo Fisher Scientific, USA) together with the pmiR-REPORT plasmid containing the wild-type or mutated 3′-UTR of TRPC1, and the luciferase activities were measured by a Dual-Luciferase Reporter System (Promega, Madison, WI, USA) using a TD 20/20 luminometer (Promega, USA) following the manufacturer’s protocol. The luminescence intensity of firefly luciferase was normalized to that of Renilla luciferase.
Primary PASMC proliferation assay
The Cell Counting Kit-8 (CCK-8) assay kit (Dojindo, Rockville, MD, USA) was used to determine the cell viability of the PASMCs. The primary PASMCs were seeded in 96-well plates at a density of 3 × 105/mL and transfected with rno-miR-135a-5p mimic or inhibitor at a final concentration of 30 nmol/L. Then, the cells were cultured under hypoxia (3% O2) or normoxia (21% O2) for another 48 h. After the cells were incubated with 10 μL CCK-8 solution for 1 h, the optical density (OD) values were measured at 450 nm using a microplate reader (Bio-Rad 680, Hercules, California, USA).
[3H]-leucine incorporation
The primary PASMCs were seeded in 96-well plates at a density of 3 × 105/mL and transfected with rno-miR-135a-5p mimic or inhibitor at a final concentration of 30 nmol/L in a hypoxic environment (3% O2). Then, [3H]-leucine incorporation solution (PerkinElmer, Waltham, MA, USA) was added to each well to a final concentration of 1 μCi/mL. After 48 h coculture with [3H]-leucine, disintegrations per minute (DPM) values were determined by using a liquid scintillation counter (LKB-BasK-Bata 1217, LKB, Bromma, Sweden).
Quantitative reverse transcription-PCR (qRT-PCR)
MiRNAs were extracted from lung tissue samples using the miRcute miRNA Isolation Kit (Tiangen, Beijing, China). MiRNA was quantified and assessed using a NanoDrop 2000 (Thermo Fisher Scientific, USA), and the reverse transcription of miRNA was performed using miRcute miRNA First-Strand cDNA synthesis kit according to the manual (Tiangen, China). The primers for rno-miR-135a-5p, miR-211-5p, miR-204-5p, miR-224-5p, miR-93-5p, miR-17-5p, miR-27b-5p, and 5S RNA were designed by Primer Premier 5.0, and the reverse primer for miR-135a-5p was obtained from Tiangen (Table 1). qRT-PCR was performed using the miRcute miRNA qPCR detection kit (Tiangen, China) on a MasterCycler RealPlex (Eppendorf, Hamburg, Germany) under the following conditions: 94 °C for 2 min followed by 40 cycles at 94 °C for 20 s and 60 °C for 34 s. The relative expression levels, in fold change, of each miRNA were determined by the ΔΔCt method and normalized to the internal control, 5S rRNA. After the PASMCs were incubated under normoxia (21% O2, 5% CO2) or hypoxia (3% O2, 5% CO2) for 6 h, 12 h, 18 h, 24 h or 48 h, the level of miR-135a-5p expression was measured as above.
Total RNA was extracted from the primary PASMCs using the RNAsimple Total RNA Kit (Tiangen, China). The reverse transcription was performed using the PrimeScript™ RT reagent Kit (TaKaRa, Dalian, China) according to the manual. The primers of PCNA and β-actin were designed by Primer Premier 5.0 (Table 2). qRT-PCR was performed using SYBR Green I mix (TaKaRa, China) under the following conditions: 95 °C for 30 s followed by 40 cycles at 95 °C for 5 s and 60 °C for 30 s. The relative expression levels, in fold change, of the PCNA mRNA was determined by the ΔΔCt method and normalized to the internal control, β-actin, as described previously [9, 10].
Western blot
Cells were lysed using RIPA Lysis Buffer (Beyotime, Shanghai, China), and the protein concentration was detected by the BCA Protein Assay Kit (Beyotime, China), as previously described [11]. Equal amounts of cellular protein extracts were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore, MA, USA). Each membrane was incubated overnight at 4 °C with a primary antibody against transient receptor potential channel 1 (TRPC1, 1:3000) (Abcam, USA) or β-actin (1:20000) (Abcam, USA) and subsequently incubated with appropriate secondary antibody (1:1000) (ZSGB-BIO, Beijing, China) at room temperature for 1 h. The membranes were exposed to Enhanced Chemiluminescence Plus reagents (Thermo Fisher Scientific, USA). Protein quantification was performed with a BioSpectrum 510 imaging system (UVP, Upland, CA, USA).
Hemodynamic measurements
At the 4th week after MCT treatment, rats were anesthetized with ethyl carbamate (1 g/kg, intraperitoneally). To measure right-ventricular systolic pressure (RVSP), a polyvinyl catheter (internal diameter, 0.28 mm) was inserted into the right ventricle (RV) via the right external jugular vein. Then, the catheter was further guided to the pulmonary artery to measure the mean pulmonary arterial pressure (mPAP) [12]. The signals were continuously recorded by using PowerLab 4/35 (AD Instruments, New South Wales, Australia) with a physiological pressure transducer (SP 844, MEMSCAP Inc., Crolles Cedex, France).
Histomorphometric analysis
The histological structures of the left lung were determined by hematoxylin-eosin (HE) staining, as described previously [13], at the 4th week after MCT treatment [14]. Briefly, the left lung of each rat was fixed in 10% formalin, embedded in paraffin and then sliced (5 μm). After standard HE staining, morphological images of the pulmonary arterioles were captured under a Leica confocal laser-scanning microscope (Leica, Wetzlar, Germany). Pulmonary vascular structure remodeling was evaluated by determining the percent medial wall area (WA%) of vessels with diameter between 50 to 200 µm via using ImageJ software. WA% was calculated using the following formula: WA (%) = (Total vessel area - Lumen area)/Total vessel area × 100.
Statistical analysis
All data were shown as the means ± SEM or means ± SD and analyzed for statistical significance by a two-tailed Student’s t-test for two independent groups or ANOVA for three or more independent groups using SPSS 13.0. P-values less than 0.05 were considered statistically significant.
Results
The expression profile of miR-135a-5p in PAH models in vivo
To confirm the expression levels of the seven miRNAs at the crucial times of hypoxia-induced PAH in rat lung [4], real-time quantitative PCR (qRT-PCR) was performed at the end of the 4th week after MCT treatment or hypoxic exposure. The data showed that the expression of miR-135a-5p, miR-211-5p, miR-204-5p, miR-224-5p, and miR-93-5p was significantly increased after MCT treatment (Fig. 1a), and the expression of miR-135a-5p, miR-211-5p, miR-204-5p, miR-224-5p, and miR-17-5p was significantly upregulated after hypoxic exposure (Fig. 1b). Moreover, miR-135a-5p was the highest in both of these PAH models (Fig. 1a, b). To further explore the dynamic changes in miR-135a-5p expression, qRT-PCR was also performed at the end of the first, second, third and fourth weeks after MCT treatment or hypoxic exposure. The expression of miR-135a-5p was remarkably downregulated at the end of the first week but increased gradually at the end of the second week. At the end of the fourth week, the miR-135a-5p expression level was increased by 12-fold in the MCT model (Fig. 1c) and tenfold in the hypoxia model (Fig. 1d) compared with that in the control animals. Thus, these findings suggested that miR-135a-5p expression is decreased at the beginning of PAH but soon increases with the development of the disease, indicating an important function of miR-135a-5p in the development of PAH.

Dynamic changes in miR-135a-5p expression in the lung tissue of the PAH rat model and in vitro hypoxic exposure. a, b The levels of miRNAs, including miR-135a-5p, miR-211-5p, miR-204-5p, miR-224-5p, miR-93-5p, miR-17-5p, and miR-27b-5p in rat lung tissue at the 4th week after MCT treatment or hypoxic exposure via real-time PCR. c, d The expression level of miR-135a-5p in the lung tissue of rats at the indicated time points after MCT treatment or hypoxic exposure via real-time PCR. e The level of miR-135a-5p expression in pulmonary artery smooth muscle cells (PASMCs) exposed to 3% O2 (hypoxia) at the indicated time points. The levels of miR-135a-5p were normalized to that of 5S RNA. Data are presented as the means ± s.e.m. in a, b and c, d and the means ± s.d. in e. n = 4–6 rats for a, b and c, d; n = 3 independent experiments for e. *P < 0.05, **P < 0.01 vs. control in a, b, vs. 0 w in c, d, and vs. 0 h in e
The expression profile of miR-135a-5p in vitro
To further explore the expression changes of miR-135a-5p in vitro, PASMCs were treated with hypoxia, and then, the level of miR-135a-5p was assessed by qRT-PCR. We found that the expression of miR-135a-5p was decreased at 6 h, while it was upregulated significantly at 18 h, 24 h and 48 h after hypoxic exposure (Fig. 1e). These results revealed that the level of miR-135a-5p was related to the duration of hypoxia in PASMCs.
Early, but not late, miR-135a-5p mimic administration inhibited the proliferation of PASMCs exposed to hypoxia in vitro
To explore the effect of miR-135a-5p expression reversal on the proliferation of PASMCs after exposure to hypoxia, PASMCs were transfected with miR-135a-5p mimic and inhibitor at final concentrations of 30 nmol/L before or after 12 h hypoxic (3% O2) treatment. At the end of the treatment, the proliferation of PASMCs was determined by CCK-8 and [3H]-leucine incorporation, and the gene expression of a well-known cell proliferation marker, PCNA, was also determined by qRT-PCR. The CCK-8 assay data showed that hypoxic treatment led to an increase in PASMC growth, whereas early miR-135a-5p mimic treatment, which started before hypoxic exposure, significantly decreased the growth rate of the PASMCs compared to that of the PASMCs treated with hypoxia alone. However, miR-135a-5p inhibitor showed no effect on PASMC growth under hypoxic treatment (Fig. 2a). The results from the [3H]-leucine incorporation assay, CCK-8 assay, and PCNA gene and protein expression revealed that early miR-135a-5p mimic treatment was able to inhibit the proliferation of PASMCs induced by hypoxia, but miR-135a-5p inhibitor had no effect at all on PASMC proliferation (Fig. 2b–d).

The effect of early or late miR-135a-5p mimic or inhibitor treatment on hypoxia-induced proliferation of PASMCs. a–c After PASMCs were exposed to 21% O2 (normoxia) or 3% O2 (hypoxia) for 48 h together with a miR-135a-5p mimic or inhibitor treatment that started at the beginning of the hypoxic treatment, the cell viability of the PASMCs was determined by CCK-8 assays (a), cell proliferation was determined by [3H]-leucine incorporation (b) and the expression of the PCNA gene and protein (c, d) were determined in PASMCs. e–g PASMCs were exposed to 21% O2 (normoxia) or 3% O2 (hypoxia) for 48 h together with a miR-135a-5p mimic or inhibitor treatment that started after 12 h of hypoxic treatment, and the cell viability and proliferation of the PASMCs were determined by CCK-8 assays (e) and by the mRNA expression of the PCNA gene and protein (f, g) in PASMCs. Data are presented as the mean ± s.d., and n = 3 independent experiments for each panel. *P < 0.05, **P < 0.01 vs. normoxia; ##P < 0.01 vs. hypoxia
Interestingly, there was no statistical difference between PASMC proliferation in the miR-135a-5p mimic or inhibitor treatments after 12 h hypoxic exposure (Fig. 2e–g). The results above indicated that early downregulation of miR-135a-5p plays a critical role in the initiation of PAH development.
Early miR-135a-5p agomir treatment protects PAH rats from increased mPAP, RVSP, and pulmonary vascular remodeling induced by MCT
To further explore the roles of early miR-135a-5p downregulation in the process of PAH development in vivo, miR-135a-5p agomir was administered to PAH rats every 3 days for 3 constitutive weeks after MCT treatment. MCT injection led to PAH characterized by increases of RVSP from (25.6 ± 1.4) mmHg to (54.5 ± 6.3) mmHg (control vs. MCT model, P < 0.01) and mPAP from (17.2 ± 1.9) mmHg to (34.1 ± 5.3) mmHg (control vs. MCT model, P < 0.01), while early miR-135a-5p agomir treatment reversed the elevated RVSP and mPAP levels caused by MCT treatment (MCT model vs. agomir, P < 0.01) (Fig. 3a, b). HE staining showed that MCT treatment resulted in major pulmonary vascular remodeling indicated by a significantly increased medial wall thickness, while early miR-135a-5p agomir treatment attenuated pulmonary vascular remodeling and WA% (control vs. MCT model, P < 0.01; MCT model vs. agomir, P < 0.01) (Fig. 3c).

The protective effects of early miR-135a-5p agomir treatment on PAH rats. miRNA-135a-5p agomir was intravenously given to the PAH rat model every 3 days for 3 consecutive weeks that started after MCT administration. At the 4th week after MCT treatment, right-ventricular systolic pressure (RVSP) (a) and mean pulmonary arterial pressure (mPAP) (b) were determined by a polyvinyl catheter. The histological structures of the left lung were determined by hematoxylin-eosin (HE) staining (c) (scale bars = 20 μm). WA% was calculated using the following formula: WA (%) = (Total vessel area - Lumen area)/Total vessel area × 100. Control: vehicle treatment group; model: MCT-treated group; Agomir: MCT plus miRNA-135a-5p agomir administration; NC: MCT plus agomir negative control sequence administration. n = 4–6 rats in each group. Data are presented as the mean ± s.e.m. **P < 0.01 vs. control; ##P < 0.01 vs. model
TRPC1 is a direct target gene of miR-135a-5p
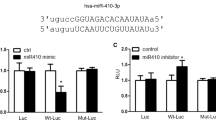
To further explore the potential mechanisms of miR-135a-5p in the process of PAH, we used the publicly available algorithms (TargetScan, PicTar, and miRanda database) to analyze the potential targets of miR-135a-5p. Among the potential targets, the 3′-UTR of TRPC1, the gene that has been reported to promote pulmonary arterial remodeling in hypoxic pulmonary hypertension [15,16,17], contains a highly conserved binding site for miR-135a-5p (Fig. 4a). We further used primary PASMCs transfected with miR-135a-5p mimic and inhibitor treated with hypoxia to determine the protein expression of TRPC1. Western blotting showed that miR-135a-5p inhibited TRPC1 protein expression after hypoxic exposure (Fig. 4b), which was consistent with the finding in kidney tissue [18]. To determine whether miR-135a-5p directly binds to the 3′-UTR sequence of rat TRPC1 mRNA and affects its expression, the 3′-UTR cDNA fragment of TRPC1 containing the putative binding site of miR-135a-5p was cloned into the Firefly-Renilla dual reporter vector of pmiR-REPORTTM. The constructed vector (WT or mutant) was then cotransfected with either the miR-135a-5p mimics or negative oligonucleotides into PASMCs. The luciferase activity was inhibited by transfection with the miR-135a-5p mimics but not by the negative oligonucleotides. Furthermore, the mutated miR-135a-5p binding site in the 3′-UTR of TRPC1 caused the greatest loss of the inhibitory effects of miR-135a-5p on luciferase activity (Fig. 4c). To clarify the effects of TRPC1 in promoting the proliferation of PASMCs were regulated by miR-135a-5p, isolated PASMCs were infected with the coding sequence of TRPC1 including or lacking the 3′-UTR. The results showed that the overexpression of TRPC1 improved PASMC proliferation and that miR-135a-5p mimic inhibited the role of TRPC1-3′-UTR. As expected, the miR-135a-5p mimic failed to decrease PASMC proliferation when the 3′-UTR lacking-TRPC1 was infected into PASMCs (Fig. 4d). These results suggested that TRPC1 is a direct target gene of miR-135a-5p in PASMCs in the process of PAH development.

TRPC1 is a direct target of miR-135a-5p in PASMCs. a Schematic representation of the TRPC1-3′-UTR with putative sites targeted by miR-135a-5p via Targetscan database. b PASMCs were treated as described in Fig. 2, and the protein expression of TRPC1 and β-actin was determined by Western blotting. c The combination between miR-135a-5p and the 3′-UTR sequence of rat TRPC1 mRNA was verified in PASMCs by luciferase reporter assay. d The effect of TRPC1 promoting the proliferation of PASMCs regulated by miR-135a-5p was determined by infection with TRPC1 including or lacking the 3′-UTR. Data are presented as the mean ± s.d. Normalized by the internal control, β-actin. n = 3 independent experiments. **P < 0.01 vs. normoxia in b, vs. wild type in c; *P < 0.05 vs. 3’-UTR-TRPC1 in d; ##P < 0.01 vs. hypoxia
Discussion
In the present study, we demonstrated the expression profile of miR-135a-5p in the process of PAH development. Our results showed that early downregulation of miRNA-135a-5p expression was responsible for PASMC viability and proliferation under hypoxic exposure in vitro and induced by MCT or hypoxic exposure in vivo, and we also found that it is critical for PAH initiation and progression. Mechanistically, TRPC1 is a potential target of miRNA-135a-5p in PASMCs, consistent with a previous finding in kidney tissue [18]. However, the role and mechanism of miRNA-135a-5p overexpression in the late stage of the in vivo and in vitro PAH models needs further investigation.
Several recent reviews have discussed the role of miRNAs in the development of PAH [19,20,21,22] and indicated that miRNAs might be potential targets for PAH therapeutics. A previous study discovered dynamic changes in lung miRNA profiles during the development of pulmonary hypertension in rats and showed that miR-22, miR-30, and let-7f were downregulated, whereas miR-322 and miR-451 were upregulated significantly during the development of PAH in both the monocrotaline model and the chronic hypoxia model. In addition, miR-322 was elevated at the beginning but downregulated at the late stage of PAH development [23]. By contrast, our results revealed that miRNA-135a-5p was downregulated at the initial stage but overexpressed at the late stage of PAH development in both in vivo MCT treatment or hypoxic exposure and in vitro hypoxic exposure. This result indicated that miRNA-135a-5p might be critical in several pathogeneses of PAH clinically, such as hypoxia, respiratory disease, chronic thrombotic disease, and left heart disease.
It is well-known that the pathogenesis of PAH has several fundamental similarities to that of cancer, as PASMCs have many cancer-like characteristics, such as enhanced proliferation and suppressed apoptosis [24]. Several findings have uncovered different roles of the indicated miRNAs in regulating PASMC proliferation and pulmonary arterial remodeling. For example, miR-17, miR-20a, miR-210, and miR-145 have been found to promote PASMC proliferation, whereas miR-21, miR-206, and miR-204 are known to inhibit PASMC proliferation or growth [21]. In this study, we found that the phenomenon of miR-135a-5p downregulation at the early stage of PAH development induced by MCT treatment or hypoxic exposure plays an important role in promoting PASMC proliferation, as early miR-135a-5p mimic administration could reverse the PASMC proliferation induced by MCT administration or hypoxic exposure. Interestingly, neither miR-135a-5p mimics nor inhibitor treatment showed protection at the late stage of PAH development. The finding that miR-135a-5p regulates cell proliferation was consistent with previous reports in the cancer field. It has been reported that miR-135a-5p promotes proliferation in bladder cancer [25, 26], liver cancer [27], and colorectal cancer [28]. However, several other studies also showed that miR-135a-5p could inhibit cancer cell proliferation, such as gastric cancer [29] and lung cancer [30, 31]. Due to this dual role of miR-135a-5p in regulating cell proliferation, further studies addressing the precise feedback in the signaling pathway involving miR-135a-5p in the process of PAH is of great significance.
Mechanistically, we found that the potential target of miR-135a-5p in PASMCs was TRPC1. TRPC1 is a plasma membrane cationic channel that has been detected in many cell and tissue types of mice, rats, and humans [32]. Its function is relevant to various cellular events, particularly calcium signaling through store-operated channels (SOCs). For example, TRPC1 is regarded as a key component of responses to hypoxia in breast cancer cells, and its expression was also significantly prognostic for basal breast cancers [33]. Furthermore, TRPC1 plays essential roles in PASMC biology and pulmonary vascular emodelling [34]. It has been shown that TRPC1 is more abundant than other TRPC isoforms in mouse distal PASMCs [35]. Researchers have confirmed that the expression of TRPC1 was increased in cultured myocytes from rat distal pulmonary arteries after chronic hypoxic exposure [17]. Ca2+ signaling in PASMCs plays a central role in the pathogenesis of PAH because of its involvement in both vasoconstriction and vascular remodeling through its stimulatory effect on PASMC proliferation. Increased TRPC1 expression led to enhanced capacitative Ca2+ entry through SOCs and might contribute to hypoxic pulmonary hypertension by increasing basal [Ca2+]I in PASMCs [17]. Downregulation of TRPC1 expression could inhibit the proliferation of cultured pulmonary artery smooth muscle cells [32]. Antisense DNA targeting the TRPC1 mRNA attenuated store-operated Ca2+ entry signals in cultured human PASMCs, whereas overexpression of human TRPC1 enhanced contractile responses to cyclopiazonic acid in rat pulmonary arterial rings [35]. It has been reported that miR-135a regulated the expression of TRPC1 in renal fibrosis of diabetic nephropathy [18]. MiR-135a-5p was predicted to target the site located at position 112–118 in the TRPC1-3′-UTR. We found that early miR-135a-5p mimic treatment could reverse the increased TRPC1 protein level that was induced by hypoxic exposure in PASMCs. This result indicated that TPRC1 was one of the targets of miR-135a-5p in PASMCs. It is worth noting that miR-135a-5p inhibitor did not show any effect on the protein level of TRPC1, consistent with the data shown in renal tissue [18]. This lack of effect might also be the reason that miR-135a-5p inhibitor showed no protective effect on PASMC proliferation after hypoxic exposure. This result suggested that the role of miR-135a-5p in the process of PAH development is related to TRPC1.
In conclusion, dysregulation of miR-135a-5p in PASMCs was associated with abnormal PASMC proliferation and PAH development. Enhanced miR-135a-5p expression at the early stage of PAH is likely a valid new avenue to prevent PAH development.
References
Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-Hahnle K, et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4:306–22.
Hoeper MM, McLaughlin VV, Dalaan AMA, Satoh T, Galiè N. Treatment of pulmonary hypertension. Lancet Respir Med. 2016;4:323–36.
Bienertova-Vasku J, Novak J, Vasku A. MicroRNAs in pulmonary arterial hypertension: pathogenesis, diagnosis and treatment. J Am Soc Hypertens. 2015;9:221–34.
Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation. 2012;125:1520–32.
Okada M, Harada T, Kikuzuki R, Yamawaki H, Hara Y. Effects of telmisartan on right ventricular remodeling induced by monocrotaline in rats. J Pharmacol Sci. 2009;111:193–200.
Ma TT, Wang Y, Zhou XL, Jiang H, Guo R, Jia LN, et al. Research on rat models of hypobaric hypoxia-induced pulmonary hypertension. Eur Rev Med Pharmacol Sci. 2015;19:3723–30.
Yan L, Gao H, Li C, Han X, Qi X. Effect of miR-23a on anoxia-induced phenotypic transformation of smooth muscle cells of rat pulmonary arteries and regulatory mechanism. Oncol Lett. 2017;13:89–98.
Kunichika N, Yu Y, Remillard CV, Platoshyn O, Zhang S, Yuan JX. Overexpression of TRPC1 enhances pulmonary vasoconstriction induced by capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol. 2004;287:L962–9.
Deng Y, Zhang Q, Luo H, Chen X, Han Q, Wang F, et al. Sustained elevation of NF-kappaB activity sensitizes offspring of maternal inflammation to hypertension via impairing PGC-1alpha recovery. Sci Rep. 2016;6:32642.
Zhang Q, Deng Y, Lai W, Guan X, Sun X, Han Q, et al. Maternal inflammation activated ROS-p38 MAPK predisposes offspring to heart damages caused by isoproterenol via augmenting ROS generation. Sci Rep. 2016;6:30146.
Deng Y, Kerdiles Y, Chu J, Yuan S, Wang Y, Chen X, et al. Transcription factor foxo1 is a negative regulator of natural killer cell maturation and function. Immunity. 2015;42:457–70.
Zhu R, Bi L, Kong H, Xie W, Hong Y, Wang H. Ruscogenin exerts beneficial effects on monocrotaline-induced pulmonary hypertension by inhibiting NF-kappaB expression. Int J Clin Exp Pathol. 2015;8:12169–76.
Deng Y, Deng Y, He X, Chu J, Zhou J, Zhang Q, et al. Prenatal inflammation-induced NF-kappaB dyshomeostasis contributes to renin-angiotensin system over-activity resulting in prenatally programmed hypertension in offspring. Sci Rep. 2016;6:21692.
Xie X, Wang G, Zhang D, Zhang Y, Zhu Y, Li F, et al. Activation of peroxisome proliferator-activated receptor gamma ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. Biomed Rep. 2015;3:537–42.
Xia Y, Yang XR, Fu Z, Paudel O, Abramowitz J, Birnbaumer L, et al. Classical transient receptor potential 1 and 6 contribute to hypoxic pulmonary hypertension through differential regulation of pulmonary vascular functions. Hypertension. 2014;63:173–80.
Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, et al. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res. 2004;95:496–505.
Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res. 2006;98:1528–37.
He F, Peng F, Xia X, Zhao C, Luo Q, Guan W, et al. MiR-135a promotes renal fibrosis in diabetic nephropathy by regulating TRPC1. Diabetologia. 2014;57:1726–36.
Lee A, McLean D, Choi J, Kang H, Chang W, Kim J. Therapeutic implications of microRNAs in pulmonary arterial hypertension. BMB Rep. 2014;47:311–7.
Kim JD, Lee A, Choi J, Park Y, Kang H, Chang W, et al. Epigenetic modulation as a therapeutic approach for pulmonary arterial hypertension. Exp Mol Med. 2015;47:e175.
Meloche J, Paulin R, Provencher S, Bonnet S. Therapeutic potential of microRNA modulation in pulmonary arterial hypertension. Curr Vasc Pharmacol. 2015;13:331–40.
Grant JS, White K, MacLean MR, Baker AH. MicroRNAs in pulmonary arterial remodeling. Cell Mol Life Sci. 2013;70:4479–94.
Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. 2010;30:716–23.
Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121:2045–66.
Yang X, Wang X, Nie F, Liu T, Yu X, Wang H, et al. miR-135 family members mediate podocyte injury through the activation of Wnt/beta-catenin signaling. Int J Mol Med. 2015;36:669–77.
Dang Z, Xu WH, Lu P, Wu N, Liu J, Ruan B, et al. MicroRNA-135a inhibits cell proliferation by targeting Bmi1 in pancreatic ductal adenocarcinoma. Int J Biol Sci. 2014;10:733–45.
Yao S, Tian C, Ding Y, Ye Q, Gao Y, Yang N, et al. Down-regulation of Kruppel-like factor-4 by microRNA-135a-5p promotes proliferation and metastasis in hepatocellular carcinoma by transforming growth factor-beta1. Oncotarget. 2016;7:42566–78.
Zhou W, Li X, Liu F, Xiao Z, He M, Shen S, et al. MiR-135a promotes growth and invasion of colorectal cancer via metastasis suppressor 1 in vitro. Acta Biochim Biophys Sin (Shanghai). 2012;44:838–46.
Wu H, Huang M, Cao P, Wang T, Shu Y, Liu P. MiR-135a targets JAK2 and inhibits gastric cancer cell proliferation. Cancer Biol Ther. 2012;13:281–8.
Zhao J, Li X, Zou M, He J, Han Y, Wu D, et al. miR-135a inhibition protects A549 cells from LPS-induced apoptosis by targeting Bcl-2. Biochem Biophys Res Commun. 2014;452:951–7.
Shi H, Ji Y, Zhang D, Liu Y, Fang P. MiR-135a inhibits migration and invasion and regulates EMT-related marker genes by targeting KLF8 in lung cancer cells. Biochem Biophys Res Commun. 2015;465:125–30.
Beech DJ, Xu SZ, McHugh D, Flemming R. TRPC1 store-operated cationic channel subunit. Cell Calcium. 2003;33:433–40.
Azimi I, Milevskiy MJG, Kaemmerer E, Turner D, KTDS Yapa, Brown MA, et al. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J Cell Sci. 2017;130:2292–305.
Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX. New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling. Am J Physiol Heart Circ Physiol. 2012;302:H1546–62.
Lu W, Wang J, Shimoda LA, Sylvester JT. Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2008;295:L104–13.
Acknowledgements
This work was supported by the Chinese National Major New Drugs Innovation Projects [2011ZXJ09105X to Y. Tang], National Natural Science Foundation of China [81503083 to Y. Deng, 81372272 to J. Yan, and 81273507 to X. Li] and Scientific and Technological Research Program of Chongqing Municipal Education Commission [KJ1601703 to N. Liao]. Thanks for You-cai DENG's language editing.
Author contributions
H.-m.L. and Y.J. designed the research and conducted the experiments; Y.-x.Z. performed the infection assay; J.Y. and N.L. analyzed the data and wrote the paper; X.-h.L. and Y.T. designed the research, supervised the study, and wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no other commercial or financial conflict of interest.
Rights and permissions
About this article

Cite this article
Liu, Hm., Jia, Y., Zhang, Yx. et al. Dysregulation of miR-135a-5p promotes the development of rat pulmonary arterial hypertension in vivo and in vitro. Acta Pharmacol Sin 40, 477–485 (2019). https://doi.org/10.1038/s41401-018-0076-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0076-9
Keywords
This article is cited by
-
Astragaloside IV restrains pyroptosis and fibrotic development of pulmonary artery smooth muscle cells to ameliorate pulmonary artery hypertension through the PHD2/HIF1α signaling pathway
BMC Pulmonary Medicine (2023)
-
Plasma metabolomics in the perioperative period of defect repair in patients with pulmonary arterial hypertension associated with congenital heart disease
Acta Pharmacologica Sinica (2022)
-
Extracellular vesicles in vascular remodeling
Acta Pharmacologica Sinica (2022)
-
Farnesoid X receptor activation inhibits TGFBR1/TAK1-mediated vascular inflammation and calcification via miR-135a-5p
Communications Biology (2020)
