
Abstract
Antipsychotic-induced low availability of group II metabotropic glutamate receptors (including mGlu2R and mGlu3R) in brains of schizophrenia patients may explain the limited efficacy of mGlu2/3R ligands in clinical trials. Studies evaluating mGlu2/3R levels in well-designed, large postmortem brain cohorts are needed to address this issue. Postmortem samples from the dorsolateral prefrontal cortex of 96 schizophrenia subjects and matched controls were collected. Toxicological analyses identified cases who were (AP+) or were not (AP-) receiving antipsychotic treatment near the time of death. Protein and mRNA levels of mGlu2R and mGlu3R, as well as GRM2 and GRM3 promoter-attached histone posttranslational modifications, were quantified. Experimental animal models were used to compare with data obtained in human tissues. Compared to matched controls, schizophrenia cortical samples had lower mGlu2R protein amounts, regardless of antipsychotic medication. Downregulation of mGlu3R was observed in AP- schizophrenia subjects only. Greater predicted occupancy values of dopamine D2 and serotonin 5HT2A receptors correlated with higher density of mGlu3R, but not mGlu2R. Clozapine treatment and maternal immune activation in rodents mimicked the mGlu2R, but not mGlu3R regulation observed in schizophrenia brains. mGlu2R and mGlu3R mRNA levels, and the epigenetic control mechanisms did not parallel the alterations at the protein level, and in some groups correlated inversely. Insufficient cortical availability of mGlu2R and mGlu3R may be associated with schizophrenia. Antipsychotic treatment may normalize mGlu3R, but not mGlu2R protein levels. A model in which epigenetic feedback mechanisms controlling mGlu3R expression are activated to counterbalance mGluR loss of function is described.
Similar content being viewed by others

Introduction
Randomized controlled trials of antipsychotic medications targeting metabotropic glutamate receptors (mGluRs) are plagued by failures of replication [1]. As understanding of the pathophysiology of schizophrenia improves, consideration of clinical features such as genetic background, age of onset, differences between acute and stable phases of illness, and the type and duration of treatment are beginning to be considered as contributors to variability in outcomes of clinical trials [2]. Assessing the integrity of glutamatergic neurotransmission in living humans remains challenging. Tissue levels of glutamate can be measured in living humans with magnetic resonance spectroscopy; these may indirectly provide an estimate of the level of glutamate specifically acting as a neurotransmitter [3]. Directly studying mGluRs in patients with schizophrenia using ligands suitable for positron emission tomography is not feasible at present, and multiple exposures to radioisotope ligands for specific receptors will always be limited by safety concerns. Studies in postmortem brain tissue provide a complementary approach that may help make future clinical trial design informative, with replicable results. Concerning clinical features, patients in large-scale trials are rarely treatment-naïve, and may have antipsychotic medications discontinued for a variable period of time or maintained for augmentation with the novel medication. All of these factors may influence the likelihood of consistency of response to a novel agent across trials.
A retrospective analysis of trials testing the antipsychotic efficacy of pomaglumetad methionil (LY2140023), a nonselective agonist of group II mGluRs (which includes mGlu2R and mGlu3R), provides an illustration of how confounding variables influence study outcomes. A patient history of antipsychotic drug treatment resulting in high occupancy of serotonin 5-HT2A (5HT2AR) as well as dopamine D2 (D2R) receptors was associated with poor response when compared with patients previously exposed only to predominant D2R antagonists [4]. Support for a mechanism related to these observations is provided by preclinical studies comparing the effects of first-(FGA) versus second-(SGA) generation antipsychotic drugs, with higher affinities for D2R or 5HT2AR, respectively [5, 6]. mGlu2R density and mRNA expression was downregulated in rodent brains following chronic clozapine (but not haloperidol) treatment, an effect mediated by histone deacetylase 2 (HDAC2) stimulation, and selective hypoacetylation of histones bound to the GRM2 gene promoter [7,8,9]. A contribution of previous antipsychotic drug treatment to downregulation of mGlu2R density may impair the capacity to respond to mGlu2/3R agonists.
The opportunity to investigate pre-existing treatment effects on multiple receptor types may be a value of postmortem studies. Antipsychotic drugs can be detected in postmortem brain tissue, allowing comparison of receptor amounts in groups of samples with or without antipsychotic drugs present, as well as with samples from individuals with no brain disorders. As well as detection, quantification of antipsychotic drugs allows modeling of the free and protein bound distribution of drugs [10]. With knowledge of the affinity of different antipsychotic drugs for their target receptors, estimates of free drug concentration can in turn provide estimates of receptor occupancy as predictors for effects on novel receptors [11].
Improving consistency across postmortem brain studies of group II mGluRs in schizophrenia (summarized in Supplemental Table S1) also requires attention. Design and technical differences such as selection of brain regions, use of radioligands that detect both mGlu2R and mGlu3R, or non-validated antibodies to quantify receptor protein levels, and the inconsistent correlation between protein and mRNA levels [12], may all contribute to inconsistencies. Of potentially greatest impact on clinical trial design is the role of previous or concurrent antipsychotic drug treatment, which is sometimes considered as a potential confound, but remains unstudied as a primary predictor of mGluR levels. Few studies addressed mGlu2/3R epigenetic regulation, which may mediate the effects of both neurodevelopmental risk factors and antipsychotic medication [13, 14].
The present study investigated the regulation of group II mGluRs in schizophrenia brain samples from a broad perspective. We quantified mGlu2R and mGlu3R protein immunodensities, mRNA levels, and the load of histone posttranslational modifications (HPTMs) at the GRM2 and GRM3 genes in a relatively large case-control postmortem brain cohort of well-characterized schizophrenia cases with qualitative and quantitative assessment of antipsychotic drugs in blood and brain tissue samples, respectively. We specifically evaluated changes in the dorsolateral prefrontal cortex (DLPFC) in the context of well-replicated clinical findings of dysfunction of this brain region in patients with schizophrenia [15,16,17,18]. For mGlu2R and mGlu3R immunodetection, only knockout (KO)-validated antibodies were used. The immunodensities of other G protein-coupled receptors (GPCRs), including the cannabinoid CB1 receptor (CB1R) and D2R, were also estimated to test the internal consistency across our prior studies using a different case-control cohort [19]. Animal studies were used to further explore the impact on brain GPCR densities of both chronic antipsychotic treatment and a neurodevelopmental insult that could lead to increased risk for schizophrenia. Finally, a model of mGlu2/3R regulation is proposed to explain the findings of the present and prior studies in the DLPFC of unmedicated and medicated subjects with schizophrenia.
Materials and methods
Postmortem human brain samples
Grey matter specimens from the DLPFC (approximating Brodmann area 9), were collected at autopsies in the Basque Institute of Legal Medicine as described elsewhere [20], and immediately stored at −80°C until assayed. Sample collection met all legal and ethical requirements of Spanish legislation; the UPV/EHU Ethical Committee Board for Human Research reviewed all procedures described herein (CEISH-M10/2018/283). For the present study, two case-control cohorts were recruited using identical inclusion/exclusion criteria (see Supplemental Methods). Subjects’ demographic and toxicological features are summarized in Table 1 (collective data) and Supplemental Table S2 (individual characteristics). Samples from Cohort 1 (N = 21 case-control pairs) and Cohort 2 (N = 27 case-control pairs) were used in all Western blot (WB) assays. Due to limitations in tissue availability, other exploratory experiments used either Cohort 1 or 2. Antemortem schizophrenia diagnoses were performed by psychiatrists according to DSM-IV or ICD-10 criteria, as recorded in medical histories. Each schizophrenia case was paired with a comparison (control) subject without evidence of mental or neurological disorders in their medical records, and similar sex, age, postmortem interval (PMI), and storage time. Routine toxicological assessments in plasma samples detected antipsychotic drugs in 30 schizophrenia cases (AP + ), whereas 18 were antipsychotic-free (AP-) at the time of death. Of note, SGAs (associated with high 5HT2AR occupancy) were present in 28 of 30 AP+ subjects (Supplemental Table S2), which did not allow comparisons between FGA and SGA effects. Further quantitative toxicological assessments in cerebellum were performed as previously described [21], using available samples from mainly Cohort 2 (Supplemental Table S2). These measures of total drug concentration were used to model predicted free drug in brain, allowing an estimate of D2R and 5HT2AR occupancy to be made (see Supplemental Methods).
Animals and drug treatments
Generation of mGlu2R (Grm2−/−) and CB1R (Cnr1−/−) knockout mice and wildtype littermates was described elsewhere [22, 23]. Chronic treatment of Sprague-Dawley rats (N = 10 per group) with haloperidol (1 mg/kg/day), risperidone (1 mg/kg/day) and clozapine (10 mg/kg/day) lasted 21 consecutive days, as reported previously [24]. The maternal immune activation (MIA) model was induced by administration of polyinosinic-polycytidylic acid [poly(I:C), or PIC; 5 mg/kg, i.p.] to pregnant C57BL/6 J mice at gestational day (GD) 9.5, as described before [25, 26]. Dams were maintained undisturbed while breeding, and the offspring (N = 7–9 per group) were sacrificed at postnatal day (PND) 84. Pharmacological approaches were done in rats for the complete characterization of the treatment conditions with antipsychotic drugs necessary to obtain clinically comparable occupancy values [27], whereas mice were selected for the MIA modeling to be consistent with our previous work. Cerebral cortices were dissected and immediately frozen to −80°C. All protocols were approved by the UPV/EHU Ethical Committee Board for Animal Research (CEEA - M20/2018/284). Further details on rodent procedures and treatments are provided in the Supplemental Methods.
Immunoblotting
Preparation of both total homogenates and the pre- and postsynaptic terminal enriched fractions from human and/or rodent cortical samples, electrophoretic separation and transference to nitrocellulose membranes, and quantitative WB procedures were performed as previously described [28, 29], and further detailed in Supplemental Methods. All primary antibodies used are listed in Supplemental Table S3.
Quantitative PCR
Expression levels of select GPCR mRNA were quantified by reverse transcription PCR (RT-qPCR) analyses as previously described [20]. Standard procedures for mRNA extraction from postmortem human brain tissues from Cohort 1 and conversion into cDNA were used (Supplemental Methods). Forward and reverse primers for mGluRs amplification, as well as the TaqMan probes for GAPDH and RPS13 housekeeping gene expression, are listed in Supplemental Table S4.
Chromatin immunoprecipitation
Isolation of cell nuclei from postmortem brain samples and chromatin immunoprecipitation (ChIP) assays were done as recently reported [20]. A battery of primary antibodies against trimethylated (Me3) or acetylated (Ac) lysines (K) at histones H3 and H4 (see Supplemental Table S5) was used to immunoprecipitate GRM2 and GRM3 gene-attached histones carrying permissive (H3K4Me3, panacetylated H3 [H3panAc], H3K9Ac, H3K27Ac, H4K5Ac, H4K16Ac) or repressive (H3K27Me3) HPTMs. ChIP was followed by amplification of GRM2 and GRM3 promoter regions with the primers listed in Supplemental Table S4.
Data analysis and statistics
WB datasets were first standardized to the mean of the corresponding control group before merging the neurochemical results from both brain cohorts. The distribution of all datasets was tested with the Shapiro-Wilk test. Only those variables estimating the predicted occupancies of D2R and 5HT2AR displayed non-Gaussian distributions. A multivariate analysis was initially carried out to survey for possible associations between neurochemical measures and potential confounding variables (i.e., age at death, sex, PMI, brain pH, storage time, presence of psychotropic drugs, and/or ethanol). Comparisons between diagnostic groups were addressed following two parallel strategies. According to the case-control design of the study, the primary analysis was a paired t-test, either for all subjects together, or after stratifying subject pairs by cohort or by the presence/absence of antipsychotic drugs in blood samples. The secondary approach involved analyses of covariance (ANCOVA), with the neurochemical findings as dependent variables, schizophrenia diagnosis as independent variable, and cohort, sex, age, and PMI as covariates. In this approach, the medication effects were addressed in follow-up analyses replacing the independent variable with a term stratifying subjects in three groups: controls, AP-, and AP+ schizophrenia subjects. Spearman’s correlation analyses initially tested the possible associations between D2R or 5HT2AR predicted occupancies, and the GPCR immunodensities. Occupancy estimates were further used as independent variables in follow-up, sex-, age-, and PMI-controlled regression models predicting mGluR immunodensities. In animal studies, comparisons between groups were performed using Student’s t-test or one-way analysis of variance (ANOVA), followed by Dunnet’s test. All tests were two-tailed, and the statistical significance was set to p < 0.05. False discovery rate (FDR; Benjamini–Hochberg method) adjustment was applied whenever appropriate. Data were analyzed with JMP17 (SAS Institute, Cary, NC, USA), and plotted with Prism 9 (GraphPad, La Jolla, CA, USA).
Results
Immunodetection of mGlu2, mGlu3, CB1, and D2 receptors in human DLPFC and effects of potentially confounding variables
All antibodies used in quantitative immunoblotting experiments were previously validated in brain samples from knockout animals lacking the target receptor (Fig. 1A and Supplemental Fig. S1A) (see also refs. [30, 31]). While the present study mainly focused in the monomeric ~95-kDa mGlu2R, ~110-kDa mGlu3R, ~50-kDa CB1R, and ~75-kDa D2R species, other receptor forms (further referred to as mGlu3Rolig and D2R100k; Fig. 1A) were also quantified. Synaptosome fractionation experiments showed the preferential location of mGlu2R, mGlu3R, and CB1R at the presynaptic terminals, while ~75 and ~100 kDa D2R species displayed selective postsynaptic localization (Fig. 1B). Further considerations concerning antibody selectivity and pre- versus postsynaptic enrichment receptor distribution are reported in the Supplemental Results.

A Characterization of the four antibodies selected for quantification of mGlu2 (ab15672), mGlu3 (ab166608), CB1 (ab23703), and D2 (AB5084P) receptors (see Supplemental Table S3) in human and rodent brains, and validation in Grm2 (mGlu2R−/−) and Cnr1 (CB1R−/−) knockout mice. Images show representative immunoblots of human (HB), and wildtype (WT) or knockout mouse (MB) cortical samples loaded in duplicate onto SDS gels, and resolved by standard SDS-PAGE, followed by immunoprobing with the above commercial antibodies. Closed arrowheads indicate those immunoreactive bands considered selective; i.e., bands approaching the theoretical molecular size of the protomeric form of each GPCR, and absent in target knockout mice [knockout validation of anti-mGlu3R and -D2R antibodies was published elsewhere [30, 31]]. Open arrowheads indicate other bands putatively corresponding to oligomeric (mGlu3Rolig) or glycosylated (D2R100k) species of the receptors, and were also considered for quantitative assays based on previous validation studies [30, 31]. All membranes were stripped and reprobed with anti-β-actin antibody as a loading control. Molecular masses (in kDa) of SDS-PAGE prestained standards are shown on the left. B Horizontal bar plots (left) and immunoblots (right) depicting the distribution of the indicated protein species across the pre-(Pre; blue) and post-(Post; red) synaptic synaptosomal fractions isolated from postmortem human DLPFC of three different control subjects (HB1/2/3). The selective (or preferential) detection of synaptophysin (SYP) and synaptosomal-associated protein of 25 kDa (SNAP25) at the presynaptic fraction, and postsynaptic density 95 (PSD95) at the postsynaptic fraction, account for the purity of the subcellular compartments. Closed and open red arrowheads indicate the same as in A. The red asterisk in D2R immunoblot points at a putative presynaptic D2R species of ~150 kDa not consistently observed in crude homogenates, and was not further evaluated. C Heatmap representing Pearson’s r-coefficients of the pairwise associations between the potentially confounding variables of the study (displayed along the x-axis; AD antidepressants, AP antipsychotics, BZD benzodiazepines, PMI postmortem interval, THC tetrahydrocannabinol) and the studied GPCR immunodensities (displayed along the y-axis) in postmortem samples of the DLPFC from subjects with schizophrenia (SZ) and controls, combined altogether (top panel) or segregated by diagnosis (middle and bottom panels). Color scale on the r-values is shown on the top-right corner. *p < 0.05.
The effect of potentially confounding variables on GPCR immunodensities is reviewed in detail in the Supplemental Results. The paired design of the study accounted for possible age, sex, PMI and storage time effects. Multivariate analyses detected possible effects of benzodiazepine medication on brain GPCR amounts in control subjects (Fig. 1C). However, complementary ANCOVAs adjusting for benzodiazepine presence discarded potential effects of benzodiazepine medication on the reported results (see Supplemental Results).
Immunodensities of mGlu2, mGlu3, CB1, and D2 receptors in schizophrenia DLPFC
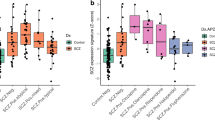
Cortical mGlu2R amounts were significantly lower in schizophrenia samples (−31%, p < 0.001), as compared to paired sex-, age-, and PMI-matched control samples (Fig. 2A, B). Lower mGlu2R immunoreactivity was observed in both AP- (−29%, p < 0.05) and AP+ (−33%, p < 0.01) schizophrenia subgroups. Direct comparison between AP- and AP+ cases did not yield statistically significant differences (Supplemental Fig. S2A and Table S6). These data suggest that schizophrenia is associated with lower cortical expression of mGlu2R protein, and antipsychotic medication has no detectable effect on mGlu2R levels.

A Box plots representing β-actin-normalized mGlu2R, mGlu3R, CB1R, and D2R immunodensities in the DLPFC of age-, sex- and PMI-matched pairs of schizophrenia (SZ) cases and controls, either altogether (All) or stratified by the absence (AP-) or presence (AP + ) of antipsychotic drugs in the blood sample of the SZ subject pair at the time of death. Paired t-tests detected significant differences (*p < 0.05, **p < 0.01, ***p < 0.001) between the diagnostic groups for mGlu2R (All, t1,47 = 5.66; AP-, t1,17 = 2.63; AP + , t1,29 = 5.41), mGlu3R (AP-, t1,17 = 2.27), and CB1R (All, t1,47 = 2.98; AP + , t1,29 = 2.70). B Representative mGlu2R, mGlu3R, CB1R, and D2R immunoblots depicting samples from all diagnosis (Dx) and toxicological (AP− and AP + ) comparison groups. Molecular mass (in kDa) of the most proximal prestained protein marker (Bio-Rad) to the target proteins is indicated on the left. C Scatterplots depicting pairwise associations between the predicted occupancy values of D2R and 5HT2AR, as estimated from drug concentrations in cerebellum samples, and the immunodensities of mGlu2R, mGlu3R, CB1R, and D2R in the DLPFC of SZ subjects. Fit line, as well as rho- and p-values of the Spearman correlation test are shown for each analysis.
While schizophrenia and matched control samples did not differ in amounts of monomeric mGlu3R, AP- (but not AP + ) schizophrenia cases displayed significantly lower mGlu3R immunodensities (−21%, p < 0.05), compared to their corresponding control pairs (Fig. 2A). Complementary ANCOVA tests demonstrated a difference between AP- and AP+ subgroups (−34%, p < 0.01) (Supplemental Fig. S2A and Table S6). Cortical immunodensities of mGlu3Rolig species did not differ across the diagnostic groups and subgroups (Supplemental Fig. S2A). These observations indicate that schizophrenia is associated with lower expression of mGlu3R monomers in the DLPFC, and antipsychotic medication may ameliorate this deficit.
We also found downregulation of CB1R (−17%, p < 0.01) in the DLPFC samples of schizophrenia subjects, as compared to matched controls (Fig. 2A, B). While this difference was mainly attributed to AP+ schizophrenia subjects (−19%, p < 0.05), subgroup ANCOVA analyses did not detect significant differences between AP- and AP+ cases (Supplemental Fig. S2A and Table S6). Finally, cortical immunodensities of D2R species were similar in all groups.
When analyzed separately, Cohorts 1 and 2 displayed a very similar pattern of results to those reported above in terms of GPCR immunodensities across the diagnostic groups and subgroups (Supplemental Fig. S2B), although the statistical significance for some comparisons in these smaller groups was lost. Finally, among AP+ cases, cortical immunodensities of mGlu2/3R, CB1R, and D2R were similar in subjects who committed suicide, compared to those who died from other causes (data not shown).
Associations between D2R and 5HT2AR occupancy estimates and cortical immunodensities of target GPCRs in subjects with schizophrenia
Toxicological findings were highly consistent, as all drugs detected in blood were also found in brain samples. Conversely, some drugs detected in brain were not found in blood, likely due to their high liposolubility. For example, two cases initially classified as AP- according to blood toxicology, displayed detectable brain levels of paliperidone (see Fig. 2C), a particularly lipophilic compound. Control subjects were confirmed as being free from antipsychotic drugs, and were therefore excluded from the analyses to avoid the potential confounds of illness-associated variations in the GPCR targets.
To unmask the potential association between D2R and/or 5HT2AR occupancy and drug-induced alterations of target GPCR amounts in the DLPFC of schizophrenia cases, we estimated D2R and 5HT2AR occupancies from brain tissue concentrations of drugs with significant affinities for these receptors. Initial Spearman’s correlation analyses revealed that higher occupancy values of both D2R and 5HT2AR were associated with greater amounts of mGlu3R protein (but not other GPCRs) in schizophrenia DLPFC samples (Fig. 2C). More detailed sex-, age-, and PMI-controlled models testing the association between receptor occupancy and mGluR protein levels showed an effect of 5HT2AR (but not D2R) occupancy on mGlu3R (but not mGlu2R) immunodensities (Supplemental Table S7). Although the improvement in variance explained in the model when adding both 5HT2AR and D2R occupancy estimates suggests a minor contribution from D2R occupancy as well. These data suggest that antipsychotic-induced blockade primarily of 5HT2AR contributes to the upregulation of monomeric mGlu3R density in the DLPFC of subjects with schizophrenia.
Chronic effect of antipsychotic drugs on target GPCR immunodensities in rat cortex
The potential effects of chronic exposure to antipsychotic drugs on cortical amounts of target GPCRs was further addressed in rats chronically treated with haloperidol, risperidone, or clozapine (Fig. 3A). Notably, clozapine treatment (−31%, p < 0.05), and possibly haloperidol as well (−25%, p > 0.05), downregulated cortical amounts of mGlu2R, but not mGlu3R, as compared to saline-treated animals (Fig. 3B). Nonsignificant downregulations of CB1R (22–29%, p > 0.05) were also observed in the cerebral cortex of rats exposed to all three antipsychotic drugs, while D2R immunodensities remained unchanged in the same brain samples (Fig. 3B).

A Cartoon illustrating chronic treatment procedures with saline (SAL), haloperidol (HAL), risperidone (RIS), and clozapine (CLO) in rats and further Western blot (WB) analyses. B Effects of antipsychotic chronic treatments on cortical immunodensities of mGlu2R, mGlu3R, CB1R and D2R. Bars represent mean ± SEM values of each treatment group. One-way ANOVA detected significant differences between treatment groups for mGlu2R densities (F3,36 = 3.29). *p < 0.05, ANOVA followed by Dunnett’s post hoc test. C Cartoon illustrating the polyinosinic–polycytidylic acid- (PIC) induced maternal immune activation (MIA) procedure in pregnant dams, as compared to saline (SAL) administration, and further WB analyses in the adult offspring. D Effects of prenatal exposure to PIC-induced MIA on cortical immunodensities of mGlu2R, mGlu3R, CB1R and D2R in adult mice. Bars represent mean ± SEM values of SAL- or PIC-exposed groups of mice. Student t-test detected significant differences between treatment groups for mGlu2R densities (t1,14 = 2.27; *p < 0.05).
Effect of prenatal exposure to poly(I:C) on cortical immunodensities of target GPCRs
We used a poly(I:C)-induced murine model of maternal immune activation (Fig. 3C) to test the possibility that the observed glutamatergic alterations in schizophrenia brains might be associated with complications during neurodevelopment, which are well-known risk factors for schizophrenia [32]. Cortical samples of mice prenatally exposed to poly(I:C) displayed lower amounts of mGlu2R (−38%, p < 0.05), compared to those in saline-exposed animals (Fig. 3D). In contrast, the immunodensities of all other GPCRs studied remained unaltered.
Gene expression and epigenetic control of mGluRs in the DLPFC of schizophrenia subjects
Further exploratory assays were performed in DLPFC tissue samples from Cohort 1 subjects to address the possibility of an association between alterations in mGluR protein levels and dysregulated (epi)genetic mechanisms controlling the GRM2 and/or GRM3 genes. First, RT-qPCR assays were carried out with selective mGlu2R and mGlu3R mRNA probes (Supplemental Table S3) to estimate GRM2 and GRM3 gene expression, respectively (Fig. 4A). Despite the robust reduction of mGlu2R immunoreactivity in schizophrenia brains, no statistically significant differences in cortical mGlu2R mRNA expression were observed across the diagnostic groups and subgroups. Likewise, mGlu3R mRNA levels were similar in both schizophrenia cases (altogether) and controls. Surprisingly, AP-, but not AP+ schizophrenia subjects displayed greater mGlu3R mRNA levels (+64%, p < 0.05), as compared to matched controls (Fig. 4A). These observations largely contrasted with the above findings on mGlu2/3R immunodensities. Correlation analyses comparing mRNA and protein expression levels of these receptors in the same brain samples were not statistically significant (Fig. 4B).

A Box plots representing housekeeping gene-normalized levels of mGlu2R and mGlu3R mRNA (in arbitrary units [a.u.]). Paired t-tests detected significant differences (*p < 0.05) between the diagnosis groups for mGlu3R mRNA (AP-, t1,11 = 2.49), (B) Scatterplots depicting pairwise associations between mRNA levels and the corresponding immunodensities of mGlu2R or mGlu3R in the same DLPFC samples of control (Con) and SZ subjects. Fit line, as well as r- and p-values of the Pearson’s correlation test are shown for each analysis. C Bar plots representing the load of trimethylated (Me3) and/or acetylated (Ac; including pan-acetylated, panAc) lysine (K) residues of histones H3 (top plots) and H4 (bottom plots) attached to the promoter regions of the mGlu2R (left bars) or mGlu3R (right bars) coding genes (estimated by ChIP followed by qPCR assays [ChIP-PCR], and represented in percent from input values). Paired t-tests detected significant differences (*p < 0.05) between the diagnosis groups for the load of H3K4Me3 (All, t1,18 = 2.69), H3panAc (All, t1,19 = 2.13), H3K27Ac (AP + , t1,9 = 2.81), H4K5Ac (All, t1,18 = 2.17; AP + , t1,9 = 2.71), and H4K16Ac (AP + , t1,9 = 2.68) attached to the mGlu3R (but not mGlu2R) gene. No overall similarities were observed between the load of HPTMs at the mGlu2/3R and GAPDH (used for housekeeping purposes [20]) genes, which may account for the specificity of the current results. D Heatmap representing pairwise associations between protein or mRNA levels of mGlu2R or mGlu3R (on the x-axis), and the amounts of HPTMs at histones H3 or H4 associated with the mGlu2R or mGlu3R gene promoters (on the y-axis) in the same DLPFC samples of control and SZ subjects altogether (Con+SZ|All), or stratified by the absence (Con+SZ|AP-) or presence (Con+SZ|AP+) of antipsychotic drugs. Color scale on the r-values is shown on the top-right corner. *FDR-adjusted p < 0.05.
ChIP analyses evaluated the amounts of key HPTMs at promoter-bound histones in the mGlu2R and mGlu3R coding genes. Consistent with mRNA expression levels, none of the permissive or repressive GRM2-associated HPTMs was altered in schizophrenia DLPFC samples (Fig. 4C). In contrast, schizophrenia samples displayed an overload of GRM3-associated HPTMs favoring gene expression, including H3K4Me3, H3panAc, H3K27Ac, H4K5Ac, and/or H4K16Ac (+61–110%, p < 0.05), with no alterations in the repressive mark studied (Fig. 4C). These alterations were mainly attributed to AP+ cases, especially histone H4-PTMs.
Overall, greater amounts of HPTMs in histones H3 and H4 at the mGlu2R and mGlu3R coding genes correlated with lower protein densities and/or greater mRNA levels of these receptors in postmortem human brain samples (Fig. 4D). These associations were more robust in case-control pairs that included the AP- subjects only (Fig. 4D, middle panel), in contrast with lack of correlation in the control-AP+ subgroup (Fig. 4D, right panel).
Discussion
The present study explored potential alterations in molecular mechanisms regulating the expression of group II mGluRs in the DLPFC in schizophrenia, with a focus on associations with antipsychotic drugs detected in brain tissue. The main results indicate that schizophrenia is associated with downregulated protein expression of mGlu2R and mGlu3R, and antipsychotic medication may normalize mGlu3R, but not mGlu2R density. Regulation of mGlu2R and mGlu3R gene expression (i.e., mRNA levels) and epigenetic control (i.e., load of HPTMs at the mGlu2/3R gene promoters) did not parallel the observed protein alterations in the same brain samples, but rather showed opposite associations. Illness- and antipsychotic medication-associated alterations in mGlu2R (but not mGlu3R) densities were mimicked in rodent experiments.
While the preferential presynaptic location of mGlu2R was expected, previous studies reported a postsynaptic location of mGlu3R in nonhuman primates and murine brains [33,34,35,36]. Recent data suggested a greater axonal (and presynaptic) location of mGlu3R in layers III and V of rat medial PFC [37]. Differences across species, or in antibody specificity may account for these contrasting reports. In the context of the high abundance of group II mGluRs at presynaptic terminals in human DLPFC, reconciling differences between protein densities and gene expression levels requires consideration of the different origins of neurons contributing to each measure. The presynaptic receptor proteins originate from both local neurons and anatomically distant neurons in subcortical or distributed cortical sites projecting to the DLPFC. In contrast, the mRNA is exclusively local in origin (Fig. 5A). Remarkably, thalamocortical projections exhibit extensive innervation of layer III within the DLPFC [38], wherein the most prominent punctate immunoreactivity of mGlu3R is localized [39, 40]. Stimulation of layer-III synaptic activity is indispensable to resolve working memory-dependent tasks [41], a cognitive domain largely impaired in schizophrenia patients. Of note, the sole study of mGlu2R and mGlu3R mRNA in thalamus showed no differences in expression between schizophrenia and control samples [42, 43] (see also Supplemental Table S1). Future immunohistochemical studies could evaluate the layer-specific manifestation of mGlu2/3R depletion in schizophrenia postmortem brains. This approach may elucidate the cellular origins of mGlu2/3R dysregulation and unraveling the clinical ramifications associated with their deficiency. Since mGlu3R is also expressed in astrocytes [44], we cannot discard that the observed alterations in schizophrenia samples may have a glial origin.

A Cartoon illustrating a possible explanation of the observed changes in mGlu2/3R mRNA and protein cortical amounts from a neuroanatomical perspective. B Schematic flow diagram depicting the potential molecular mechanisms involved in the regulation of mGlu2/3R protein and mRNA expression in the DLPFC of schizophrenia subjects, and possible effects of antipsychotic (AP) medication on the regulatory pathway via 5HT2AR and/or D2R. Numbered circles correspond to the hypothetical, stepwise changes described in C. HAT histone acetyl transferase, HDAC histone deacetylase, HDM histone demethylase, HMT histone methyl transferase, miRNA microRNA. C Hypothetical model illustrating cortical changes in protein (red lines) and mRNA (yellow lines) expression, as well as epigenetic regulation at histone posttranslational modifications (PTMs; purple lines), of mGlu2R (top plot) and mGlu3R (bottom plot) over the course of schizophrenia development and illness progression, including effects of AP medication (dotted lines), following first-episode psychosis (FEP).
Alternatively, mRNA and protein level discordance may arise from compensatory feedback mechanisms. A hypothetical model describing changes in mGlu2R and mGlu3R gene and protein expression over the course of illness is illustrated in Fig. 5B, C. The model proposes that cortical densities of group II mGluRs are downregulated during neurodevelopment (Fig. 5B, C; Step 1). While the specific cause of mGlu2/3R downregulation in schizophrenia brains is unknown, the observation of lower mGlu2R densities in cortical samples from rodents prenatally exposed to poly(I:C) is consistent with a developmental origin (see also [45, 46]). mGlu2/3R deficiency may activate compensatory feedback mechanisms to counterbalance mGlu2/3R loss of function, which may involve increased mGlu3R gene expression, via modulation of the epigenetic control mechanisms (Fig. 5B, C; Step 2). However, this feedback mechanism may be insufficient to normalize mGlu2/3R protein levels, possibly explaining the inverse correlation between mGlu3R protein amounts and permissive HPTM loads at the mGlu3R promoter in the DLPFC of AP- schizophrenia samples. Prolonged mGlu2/3R loss of function may contribute to abnormal development of glutamatergic circuits, and the excitatory/inhibitory imbalance observed in patients [47]. Glutamatergic dysregulation, perhaps combined with other genetic and environmental risk factors, may trigger schizophrenia-related symptoms in early adulthood. Later, chronic antipsychotic treatment may further stimulate the epigenetic control mechanisms generating greater load of permissive HPTMs at the mGlu3R gene (Fig. 5B, C; Step 3). Consequently, mGlu3R protein density may return to normal values (Fig. 5B, C; Step 4), possibly contributing to the therapeutic action of antipsychotic drugs. Since we did not detect elevated mGlu3R transcript levels in AP+ subjects, the antipsychotic-induced stimulation of mGlu3R gene expression may only occur transiently, and mRNA levels may return to basal values once normal mGlu3R function is recovered. Alternatively, antipsychotic-induced inhibition of microRNAs repressing mGlu3R translation could explain the normalization of mGlu3R protein amounts in AP+ subjects [48, 49]. In contrast, antipsychotic medication (clozapine at least) may contribute to further downregulate mGlu2R cortical density. Since we cannot ignore the likelihood that AP- subjects were exposed to antipsychotic drugs earlier in their lives, it is possible that the prior mGlu2/3R imbalance is restored following treatment cessation (Fig. 5B, C; Step 5).
The model predicts schizophrenia DLPFC samples may display normal or low mGlu2/3R protein amounts, depending on the stage of illness at the time of death, and adherence with antipsychotic treatment. These effects, as well as those noted in the Introduction, may contribute to the large diversity of the reported results across the case-control studies analyzing mGlu2/3R in schizophrenia postmortem brains (Supplemental Table S1). Most of these studies did not report toxicological assessments, and the influence of antipsychotic drugs on the reported findings may have been overlooked. A recent postmortem brain study reporting toxicological assessments in a large schizophrenia case-control cohort did not find illness- or treatment-related alterations in mGlu3R immunodensities in the temporal lobe [31], indicating the brain region specificity of the abnormalities underlying schizophrenia.
A major goal of the present study was to evaluate the effect of antipsychotic treatment. Chronic blockade of 5HT2AR with clozapine was reported to reduce mGlu2/3R binding and mGlu2R mRNA levels via HDAC2 stimulation and selective hypoacetylation of the mGlu2R, but not mGlu3R gene promoter [7]. In contrast, our data suggests that in schizophrenia brains, antipsychotics may induce hyperacetylation of the mGlu3R, but not mGlu2R gene promoter and, consequently, increase mGlu3R immunodensity over that in AP- subjects. Since the vast majority of AP+ subjects were on SGAs proximate to death, no direct comparisons between FGA- and SGA-induced modulation of mGlu2/3R were possible. As alternatives, two complementary strategies were deployed. First, we compared mGlu2/3R cortical densities with predicted D2R and 5HT2AR occupancies. Greater 5HT2AR occupancy predicted higher densities of mGlu3R, but not mGlu2R, suggesting that stimulation of the epigenetic mechanisms facilitating mGlu3R gene expression may be associated with SGA-induced 5HT2AR blockade. The second approach evaluated mGlu2/3R cortical levels in rats treated with antipsychotics with low (i.e., haloperidol) or high (i.e., risperidone and clozapine) serotonergic/dopaminergic affinity ratios. In these experiments, chronic treatment with clozapine (but not haloperidol or risperidone) reduced the cortical density of mGlu2R, which may be compatible with the robust downregulation of this receptor in AP+ subjects. However, AP+ and AP- schizophrenia subgroups did not differ significantly in terms of their mGlu2R cortical densities. In turn, none of the antipsychotic treatments replicated the mGlu3R upregulation observed in schizophrenia AP+ subjects. Antipsychotic drug effects in schizophrenia patients with downregulated mGlu2/3R protein levels, may differ from those observed in ‘healthy’ rats. Differences in the brain regions studied (DLPFC versus whole cerebral cortex) may also explain the inconsistencies observed across species.
We also evaluated potential differences in the antipsychotic-induced regulation of mGlu2/3R in suicide and non-suicide schizophrenia cases. Arguably, subjects with schizophrenia who were on antipsychotic medication by the time of death and committed suicide would likely be classified as treatment-resistant patients. Since similar mGlu2/3R immunodensities were observed in suicide and non-suicide AP+ cases, regulation of these receptors may not be directly related to the efficacy of the antipsychotic treatment.
CB1R and D2R were evaluated to test the robustness of findings across studies. In agreement with our prior work [19], no changes were found in D2R between diagnostic groups. CB1R density was lower in AP+ schizophrenia brains, consistent with an overall consensus of downregulated CB1R expression in schizophrenia brains [50].
In conclusion, the present findings support a role for group II mGluRs in schizophrenia. Lower densities of mGlu2/3R may limit the efficacy of ligands targeting these receptors directly. The apparent normalization of mGlu3R by 5HT2AR/D2R antagonists may merit more investigation and consideration in clinical trial design, especially of mGlu3R agonists. Development of PET ligands for group II mGluRs may help resolve the time course of changes in the amounts of these receptors after initiation or withdrawal of antipsychotic treatment. Finally, development of larger samples of postmortem brain tissues from patients with schizophrenia may allow analyses of the effects of genetic variation on receptors, and provide a bridge to clinical trial design and interpretation.
References
Dogra S, Conn PJ. Metabotropic glutamate receptors as emerging targets for the treatment of schizophrenia. Mol Pharmacol. 2022;101:275–85.
Krystal JH, Anticevic A. Toward illness phase-specific pharmacotherapy for schizophrenia. Biol Psychiatry. 2015;78:738–40.
Nakahara T, Tsugawa S, Noda Y, Ueno F, Honda S, Kinjo M, et al. Glutamatergic and GABAergic metabolite levels in schizophrenia-spectrum disorders: a meta-analysis of (1)H-magnetic resonance spectroscopy studies. Mol Psychiatry. 2022;27:744–57.
Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol Psychiatry. 2015;78:754–62.
Marek GJ. When is a Proof-of-Concept (POC) not a POC? Pomaglumetad (LY2140023) as a case study for antipsychotic efficacy. Curr Pharm Des. 2015;21:3788–96.
Muguruza C, Meana JJ, Callado LF. Group II metabotropic glutamate receptors as targets for novel antipsychotic drugs. Front Pharmacol. 2016;7:130.
Kurita M, Holloway T, GarcÃ- A-Bea A, Kozlenkov A, Friedman AK, Moreno JL, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–54.
Hideshima KS, Hojati A, Saunders JM, On DM, de la Fuente Revenga M, Shin JM, et al. Role of mGlu2 in the 5-HT(2A) receptor-dependent antipsychotic activity of clozapine in mice. Psychopharmacology. 2018;235:3149–65.
de la Fuente Revenga M, Ibi D, Cuddy T, Toneatti R, Kurita M, Ijaz MK, et al. Chronic clozapine treatment restrains via HDAC2 the performance of mGlu2 receptor agonism in a rodent model of antipsychotic activity. Neuropsychopharmacology. 2019;44:443–54.
Liu X, Van Natta K, Yeo H, Vilenski O, Weller PE, Worboys PD, et al. Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos. 2009;37:787–93.
Rivero G, Llorente J, McPherson J, Cooke A, Mundell SJ, McArdle CA, et al. Endomorphin-2: a biased agonist at the μ-opioid receptor. Mol Pharmacol. 2012;82:178–88.
Irmler M, Hartl D, Schmidt T, Schuchhardt J, Lach C, Meyer HE, et al. An approach to handling and interpretation of ambiguous data in transcriptome and proteome comparisons. Proteomics. 2008;8:1165–9.
Ibi D, González-Maeso J. Epigenetic signaling in schizophrenia. Cell Signal. 2015;27:2131–6.
Ibi D, De La Fuente Revenga M, Kezunovic N, Muguruza C, Saunders JM, Gaitonde SA, et al. Antipsychotic-induced Hdac2 transcription via NF-kappaB leads to synaptic and cognitive side effects. Nat Neurosci. 2017;20:1247–59.
Fuster JM. The prefrontal cortex-an update: time is of the essence. Neuron. 2001;30:319–33.
Honea R, Crow TJ, Passingham D, Mackay CE. Regional deficits in brain volume in schizophrenia: a meta-analysis of voxel-based morphometry studies. Am J Psychiatry. 2005;162:2233–45.
Camchong J, Dyckman KA, Austin BP, Clementz BA, McDowell JE. Common neural circuitry supporting volitional saccades and its disruption in schizophrenia patients and relatives. Biol Psychiatry. 2008;64:1042–50.
Yoon JH, Minzenberg MJ, Ursu S, Ryan Walter BS, Wendelken C, Ragland JD, et al. Association of dorsolateral prefrontal cortex dysfunction with disrupted coordinated brain activity in schizophrenia: relationship with impaired cognition, behavioral disorganization, and global function. Am J Psychiatry. 2008;165:1006–14.
Urigüen L, García-Fuster MJ, Callado LF, Morentin B, La Harpe R, Casadó V, et al. Immunodensity and mRNA expression of A2A adenosine, D2 dopamine, and CB1 cannabinoid receptors in postmortem frontal cortex of subjects with schizophrenia: effect of antipsychotic treatment. Psychopharmacology. 2009;206:313–24.
Brocos-Mosquera I, Miranda-Azpiazu P, Muguruza C, Corzo-Monje V, Morentin B, Meana JJ, et al. Differential brain ADRA2A and ADRA2C gene expression and epigenetic regulation in schizophrenia. Effect of antipsychotic drug treatment. Transl Psychiatry. 2021;11:643.
Sampedro MC, Unceta N, Gómez-Caballero A, Callado LF, Morentin B, Goicolea MA, et al. Screening and quantification of antipsychotic drugs in human brain tissue by liquid chromatography-tandem mass spectrometry: application to postmortem diagnostics of forensic interest. Forensic Sci Int. 2012;219:172–8.
Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakaguchi I, Katsuura G, et al. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science (80-). 1996;273:645–7.
Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–4.
Barakauskas VE, Beasley CL, Barr AM, Ypsilanti AR, Li H-Y, Thornton AE, et al. A novel mechanism and treatment target for presynaptic abnormalities in specific striatal regions in schizophrenia. Neuropsychopharmacology. 2010;35:1226–38.
MacDowell KS, Munarriz-Cuezva E, Caso JR, Madrigal JLM, Zabala A, Meana JJ, et al. Paliperidone reverts Toll-like receptor 3 signaling pathway activation and cognitive deficits in a maternal immune activation mouse model of schizophrenia. Neuropharmacology. 2017;116:196–207.
Prades R, Munarriz-Cuezva E, Urigüen L, Gil-Pisa I, Gómez L, Mendieta L, et al. The prolyl oligopeptidase inhibitor IPR19 ameliorates cognitive deficits in mouse models of schizophrenia. Eur Neuropsychopharmacol. 2017;27:180–91.
Kapur S, VanderSpek SC, Brownlee BA, Nobrega JN. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharmacol Exp Ther. 2003;305:625–31.
Ramos-Miguel A, Beasley CL, Dwork AJ, Mann JJ, Rosoklija G, Barr AM, et al. Increased SNARE protein-protein interactions in orbitofrontal and anterior cingulate cortices in schizophrenia. Biol Psychiatry. 2015;78:361–73.
Erdozain AM, Brocos-Mosquera I, Gabilondo AM, Meana JJ, Callado LF. Differential α(2A)- and α(2C)-adrenoceptor protein expression in presynaptic and postsynaptic density fractions of postmortem human prefrontal cortex. J Psychopharmacol. 2019;33:244–9.
Stojanovic T, Orlova M, Sialana FJ, Höger H, Stuchlik S, Milenkovic I, et al. Validation of dopamine receptor DRD1 and DRD2 antibodies using receptor deficient mice. Amino Acids. 2017;49:1101–9.
García-Bea A, Walker MA, Hyde TM, Kleinman JE, Harrison PJ, Lane TA. Metabotropic glutamate receptor 3 (mGlu3; mGluR3; GRM3) in schizophrenia: antibody characterisation and a semi-quantitative western blot study. Schizophr Res. 2016;177:18–27.
Meyer U. Prenatal Poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry. 2014;75:307–15.
Neki A, Ohishi H, Kaneko T, Shigemoto R, Nakanishi S, Mizuno N. Pre- and postsynaptic localization of a metabotropic glutamate receptor, mGluR2, in the rat brain: an immunohistochemical study with a monoclonal antibody. Neurosci Lett. 1996;202:197–200.
Jin LE, Wang M, Yang S-T, Yang Y, Galvin VC, Lightbourne TC, et al. mGluR2/3 mechanisms in primate dorsolateral prefrontal cortex: evidence for both presynaptic and postsynaptic actions. Mol Psychiatry. 2017;22:1615–25.
Jin LE, Wang M, Galvin VC, Lightbourne TC, Conn PJ, Arnsten AFT, et al. mGluR2 versus mGluR3 metabotropic glutamate receptors in primate dorsolateral prefrontal cortex: postsynaptic mglur3 strengthen working memory networks. Cereb Cortex. 2018;28:974–87.
Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503.
Woo E, Datta D, Arnsten AFT. Glutamate metabotropic receptor type 3 (mGlu3) localization in the rat prelimbic medial prefrontal cortex. Front Neuroanat. 2022;16:849937.
Opris I, Casanova MF. Prefrontal cortical minicolumn: from executive control to disrupted cognitive processing. Brain. 2014;137:1863–75.
Crook JM, Akil M, Law BCW, Hyde TM, Kleinman JE. Comparative analysis of group II metabotropic glutamate receptor immunoreactivity in Brodmann’s area 46 of the dorsolateral prefrontal cortex from patients with schizophrenia and normal subjects. Mol Psychiatry. 2002;7:157–64.
Matosin N, Fernandez-Enright F, Frank E, Deng C, Wong J, Huang X-F, et al. Metabotropic glutamate receptor mGluR2/3 and mGluR5 binding in the anterior cingulate cortex in psychotic and nonpsychotic depression, bipolar disorder and schizophrenia: implications for novel mGluR-based therapeutics. J Psychiatry Neurosci. 2014;39:407–16.
Finn ES, Huber L, Jangraw DC, Molfese PJ, Bandettini PA. Layer-dependent activity in human prefrontal cortex during working memory. Nat Neurosci. 2019;22:1687–95.
Benes FM. Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacol Publ Am Coll Neuropsychopharmacol. 2010;35:239–57.
Levitt JJ, Nestor PG, Levin L, Pelavin P, Lin P, Kubicki M, et al. Reduced structural connectivity in frontostriatal white matter tracts in the associative loop in schizophrenia. Am J Psychiatry. 2017;174:1102–11.
Copeland CS, Salt TE. The role of thalamic group II mGlu receptors in health and disease. Neuronal Signal. 2022;6:NS20210058.
Moreno JL, Kurita M, Holloway T, López J, Cadagan R, Martínez-Sobrido L, et al. Maternal influenza viral infection causes schizophrenia-like alterations of 5-HT2A and mGlu2 receptors in the adult offspring. J Neurosci J Soc Neurosci. 2011;31:1863–72.
Holloway T, Moreno JL, Umali A, Rayannavar V, Hodes GE, Russo SJ, et al. Prenatal stress induces schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors in the adult offspring: role of maternal immune system. J Neurosci J Soc Neurosci. 2013;33:1088–98.
Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry. 2012;69:776–86.
Santarelli DM, Liu B, Duncan CE, Beveridge NJ, Tooney PA, Schofield PR, et al. Gene-microRNA interactions associated with antipsychotic mechanisms and the metabolic side effects of olanzapine. Psychopharmacology. 2013;227:67–78.
Miller BH, Zeier Z, Xi L, Lanz TA, Deng S, Strathmann J, et al. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci USA. 2012;109:3125–30.
Borgan F, Kokkinou M, Howes O. The Cannabinoid CB(1) Receptor in Schizophrenia. Biol Psychiatry Cogn Neurosci Neuroimaging. 2021;6:646–59.
Acknowledgements
WGH was supported by the Jack Bell Chair in Schizophrenia. The authors would like to thank the staff members of the Basque Institute of Legal Medicine for their cooperation in the study. The study was supported by grants PID2022-137848OB-I00 (to AR-M) and RTI2018-094414-A-I00 (to AR-M and AME) from the Spanish Ministry of Science, Innovation and Universities and the European Regional Development Fund (AEI/MCIU/ERDF), BIO22/ALZ/002 from EiTB-Maratoia/BIOEF (to LFC and AR-M) and IT1512/22 (to LFC) from the Basque Government. AR-M is a ‘Ramón y Cajal’ Researcher (grant RYC-2016-19282), and is a member of the ExoPsyCog Consortium, funded by the IKUR-Neurobiosciences Strategy (Basque Government).
Author information
Authors and Affiliations
Contributions
AR-M and AME conceived and, together with GR, designed the study. BM and LFC collected human postmortem brain specimens, reviewed medical records, and designed Cohorts 1 and 2. JDC-B, AME, and IB-M processed postmortem brain tissues and, together with OM-P, GR. PM-A, and JAS-M performed all WB, qPCR, and ChIP analyses. OM-P, EM-C, PM-A, and RD-A conducted experiments in rodents. WGH produced anti-SYP and anti-SNAP25 monoclonal antibodies. JDC-B, AME and AR-M collected all study data and conducted the statistical analyses. AR-M and WGH wrote the first draft of the manuscript, and all authors reviewed and approved its final version.
Corresponding author
Ethics declarations
Competing interests
WGH is a consultant with AbbVie, Newron, and Translational Life Sciences. Other authors do not have any financial disclosures to report.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
DelaCuesta-Barrutia, J., Martínez-Peula, O., Rivero, G. et al. Effect of antipsychotic drugs on group II metabotropic glutamate receptor expression and epigenetic control in postmortem brains of schizophrenia subjects. Transl Psychiatry 14, 113 (2024). https://doi.org/10.1038/s41398-024-02832-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-024-02832-z
