
Abstract
Since its rediscovery at the beginning of the 21st Century, memory reconsolidation has been proposed to be a therapeutic target for reducing the impact of emotional memories that can go awry in mental health disorders such as drug addiction (substance use disorder, SUD). Addiction can be conceptualised as a disorder of learning and memory, in which both pavlovian and instrumental learning systems become hijacked into supporting drug-seeking and drug-taking behaviours. The past two decades of research have characterised the details of the molecular pathways supporting the reconsolidation of pavlovian cue-drug memories, with more recent work indicating that the reconsolidation of instrumental drug-seeking memories also relies upon similar mechanisms. This narrative review considers what is known about the mechanisms underlying the reconsolidation of pavlovian and instrumental memories associated with drug use, how these approaches have translated to experimental medicine studies, and the challenges and opportunities for the clinical use of reconsolidation-based therapies.
Similar content being viewed by others


Introduction
Drug addiction, or substance use disorder (SUD) is a chronic relapsing disorder that is estimated to annually cost the UK National Health Service in excess of £30 billion [1, 2], and to have an economic impact in the United States in excess of $440 billion per year [3]. Individuals addicted to drugs show high motivation for the drug(s) of abuse, impaired control over drug use, and persistent use of drug despite adverse physical, psychological and social consequences of drug use [4].
Addiction is a complex disorder, involving the hijacking of neural circuitry that has evolved to support motivated behaviour relevant to survival of the individual and propagation of the species. A prominent view is that learning mechanisms, originally evolved to support foraging behaviour, become maladaptively recruited to drug-seeking and drug-taking, allowing addiction to be conceptualised as a disorder of maladaptive learning and memory [5, 6]. This involves the aberrant engagement of both pavlovian and instrumental learning mechanisms. The acts of drug-seeking and drug-taking are instrumental, initially being goal-directed but, in a subset of individuals [7, 8], ultimately becoming habitual and compulsive [9, 10]. However, drug-seeking and drug-taking occur in specific environments (contexts) and in the presence of people and paraphernalia (i.e. discrete cues) that become associated with the drug high in a pavlovian manner [5]. These pavlovian drug-associated conditioned stimuli (‘CS-drug memories’) become powerful precipitators of relapse in those trying to maintain abstinence [11,12,13,14,15,16,17,18,19,20,21,22,23,24], and in experimental animal models of addiction, can be used to promote drug-seeking behaviour [25,26,27,28,29].
Conceptualising addiction as a disorder of learning and memory raises the prospect that drug-associated memories, whether pavlovian or instrumental, might provide targets for new treatment development [30,31,32,33,34,35,36]. A similar conceptualisation of other ‘maladaptive memory disorders’ such as specific phobia [37, 38] led to the development of treatments such as ‘cue exposure’ or ‘prolonged exposure’ therapy. Prolonged exposure therapy involves the repeated re-exposure of individuals to pavlovian CSs, previously associated with motivationally relevant outcomes, in the absence of that outcome [37]. For example, a phobic stimulus (e.g. a spider, normally eliciting fear in an individual with arachnophobia) is repeatedly presented while the individual works through guided exercises to control their fear (e.g. relaxation exercises). These repeated exposures can become progressively more proximal in a process referred to as ‘systematic desensitisation’ [39]. On successful completion of the therapy, the individual should no longer show the pavlovian conditioned response (i.e. fear) to the stimulus. However, despite its success for disorders such as phobia [40], the efficacy of prolonged exposure therapy for addiction is limited and mixed, with meta-analyses suggesting little to no effect on treatment outcomes [41,42,43,44].
Prolonged exposure therapy may be less effective in the treatment of addiction because rather than ‘erasing’ or ‘overwriting’ the original memory, the mechanism underlying prolonged exposure creates a new, inhibitory ‘cue-no outcome’ extinction memory that competes with the original for behavioural expression [45, 46]. While extinction learning is effective for disorders such as phobia [47], there are issues around its contextual specificity [46] (though see ref. [48] for methods to minimise the impact of contextual influences). However, the CSs associated with drugs of abuse often develop conditioned reinforcing properties that make them remarkably resistant to extinction [49, 50]. Conditioned reinforcement refers to the capacity of a pavlovian CS to acquire affective properties related to the primary reinforcer, and subsequently to support responding for the CS in its own right, allowing the cue to bridge long delays to primary reinforcement [51]. The fact that the CS becomes reinforcing in its own right is a major challenge to therapies based upon extinction learning.
Targeting the consolidation of memories underlying addiction – their initial storage within the brain, hypothesised to rely upon synaptic plasticity changes [52] and the formation of memory traces or ‘engrams’ [53, 54] – is not a feasible therapeutic strategy. Many who use drugs of abuse will ultimately not become addicted [55, 56] and thus, any globally-applied treatment strategy would target a large proportion of the drug-using population who will never develop SUD. Furthermore, even for those who would benefit from disrupting the consolidation of drug-associated memories, the window for treatment opportunity is very small, encompassing only a few hours (typically thought to be around 6 h, and certainly within 24 h - see ref. [57] for review). Although memory consolidation presents a challenging target for disrupting drug-associated memories, the rediscovery of memory reconsolidation at the beginning of the 21st Century reignited interest in memory-based treatments for addiction.
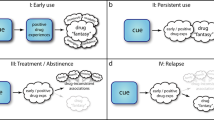
Reconsolidation is hypothesised to be the process by which memories can be updated under certain conditions of retrieval, and almost since its rediscovery in 2000 [58] it has been proposed as a means by which old, well-established (and potentially maladaptive) memories could be targeted for disruption [31, 33, 59, 60]. By reactivating the maladaptive memories relevant to drug-seeking, it should be possible to make them once again susceptible to disruption through administration of amnestic agents, or behavioural interference. Reactivation (or, more mechanistically, destabilisation) of the memory can be achieved in different ways, but typically involves brief re-exposure to either the CS [61, 62] or the US [63, 64] for pavlovian memories (referred to as ‘CS-based’ and ‘US-based’ reactivation respectively), and a change in the reinforcement contingency for instrumental memories [65, 66]. Importantly, reactivation of the memory occurs under conditions in which there is a ‘mismatch’ between what is expected and what actually occurs [61], although the relationship between memory retrieval, memory destabilisation/reconsolidation and extinction learning is not straightforward [67] and likely interacts with prior learning history [68]. Thus, although reconsolidation-based approaches are simple in principle – induce lability of a well-established memory and administer an amnestic agent or behavioural interference procedure to disrupt its reconsolidation, thereby weakening it in the long-term - the practicalities of disrupting pavlovian and instrumental drug-associated memories has presented multiple challenges, including identification of the optimal associations to target for disruption (Fig. 1). In principle, these associations can be dissociated experimentally and could be independently targeted for disruption under the appropriate conditions of memory reactivation. In practice, these associations act simultaneously in the real world to support ongoing drug-seeking behaviour and to promote relapse during abstinence [31]. Therefore targeting multiple associations, whether simultaneously through reactivating by re-exposure to the drug US [63, 64], or sequentially by reactivating different CSs individually, may be the optimal approach for reconsolidation-based therapies.

Associations are made between initially neutral stimuli (both discrete and contextual) that ultimately become associated with the drug of abuse, acting as an unconditioned stimulus, in a pavlovian manner. These associations can be affective or predictive, leading to actions (reflexive motor responses or more flexible instrumental behaviour). Additionally, instrumental associations are acquired between the action and the outcome (goal-directed responses) where individuals will work for the unconditioned stimulus if they are motivated to do so, and there is an instrumental contingency between the response and the unconditioned stimulus. Instrumental responding can also be supported by habitual, stimulus-response associations between conditioned stimuli and the action. Magenta lines represent learned associations.
The past two decades have seen a concerted research effort to determine whether reconsolidation-based approaches could be used to treat SUD, primarily in animal models, but also in a growing number of studies in humans. This review will focus upon treatments aiming to disrupt the reconsolidation of pavlovian memories (which should reduce the capacity of drug-associated CSs and contexts to precipitate relapse) and the disruption of instrumental memories (which should reduce drug-seeking behaviour directly) in both conditioned place preference (CPP) and self-administration models, and also consider how these proof-of-principle approaches have translated to clinical populations.
The reconsolidation of pavlovian drug-associated memories
Drug-associated CSs are powerful precipitators of relapse in individuals trying to maintain abstinence [13,14,15, 17] with imaging studies showing that activation of the limbic corticostriatal circuitry in response to drug CSs is predictive of subsequent relapse for individuals addicted to alcohol [11, 12], nicotine [19,20,21], opiates [22] and psychostimulants [23, 24]. Disrupting the reconsolidation of drug-associated CSs therefore presents a potential treatment target for reducing the impact of these CSs on relapse behaviour in the long term.
Numerous studies have investigated the impact of disrupting the reconsolidation of pavlovian drug-associated cues, in both self-administration and conditioned place preference (CPP; see below) procedures. Consistent with the activation of the limbic corticostriatal circuitry by drug-associated cues [13,14,15, 17], the reconsolidation of pavlovian CS-drug memories relies upon plasticity mechanisms within regions such as the basolateral amygdala (BLA), hippocampus and nucleus accumbens [69] (Fig. 2). The specific regions involved depend upon whether the memory being targeted for disruption is required for the association between contextual or discrete CSs and the drug outcome (unconditioned stimulus, US), and whether specific psychological processes (e.g. conditioned reinforcement) or the contribution of multiple psychological processes are being targeted. Indeed, even within regions, different behavioural procedures recruit specific ensembles that can be distinguished within the same animals using engram-labelling techniques [70]. However, for all these associations, the necessity of plasticity processes including activation of the NMDA subtype of glutamate receptor (NMDAR), protein kinase activation and protein synthesis has been demonstrated.

a Sagittal schematic of the rat brain, showing key regions of the corticolimbic striatal circuitry implicated in drug memory reconsolidation. b Connection of key brain regions implicated in drug memory reconsolidation, denoting associations supported by each structure and the type of memory reactivation session required to recruit it. Abbreviation: Amy amygdala, DH dorsal hippocampus, DS dorsal striatum, NAcb, nucleus accumbens. Reproduced, with permission, from ref. [69].
Disrupting context-drug memories
Studies of the impact of contextual cues on drug-seeking, and whether reconsolidation of these memories could be a target for reducing that impact, have relied primarily upon the conditioned place preference (CPP) procedure, or have explicitly assessed the impact of context on the self-administration of drugs of abuse (e.g. refs. [71,72,73,74]). The CPP procedure involves training animals to associate a specific context with the effects of an experimenter-administered drug, and subsequently manifests as a preference for that context over an alternative context paired with a control injection [75]. By contrast, context-induced reinstatement involves training animals to self-administer drug (typically intravenously, but orally in the case of alcohol) in a specific context, and testing the capacity of that context to support recovery of drug-seeking compared to control contexts [76]. The memory representing the association of the context and the drug outcome is likely supported by the BLA [77,78,79,80], as it is for discrete cues [81,82,83]. However, the context representation itself depends upon the dorsal hippocampus, similar to the circuitry supporting contextual fear conditioning [84,85,86].
Disrupting the contextual memories underlying drug-conditioned place preference
The memories underlying drug CPP can be disrupted by targeting biochemical pathways that ultimately lead to immediate early gene expression and protein synthesis (Fig. 3). These manipulations produce amnesia more reliably when the reactivation session is reinforced with an injection of the drug of abuse (i.e. ‘US-based’ reactivation is used). Protein synthesis inhibitors disrupt both cocaine [87] and morphine [87, 88] CPP when administered systemically in conjunction with reinforced re-exposure to the drug-paired context, as do local infusions of protein synthesis inhibitors intracerebroventricularly [89] or within the BLA [88] (though see ref. [90]), central amygdala [91], dorsal hippocampus [88] and nucleus accumbens core [88]. Upstream of protein synthesis, the reconsolidation of drug CPP memories depends upon the activation of transcriptional and translational regulators including eIFα [92], circTmeff-1 [93] and mTORC1 [94], and protein kinases including ERK [87], p70S6 kinase [94] and GSK3β [95, 96]. Protein phosphatases, such as protein phosphatase 1, are also necessary for the reconsolidation of cocaine CPP memories [97]. Furthermore, the knockdown of the immediate early gene zif268 in the BLA or the nucleus accumbens disrupts the reconsolidation of the memories underlying cocaine CPP [98]. However, some proteases are also necessary for the reconsolidation of CPP memories, as the inhibition of the calcium-dependent cysteine protease calpain in the nucleus accumbens core prevents the reconsolidation of the memories underlying cocaine CPP by preventing its interaction with the scaffolding protein GRIP1 [99]. Epigenetic regulation is also critical for drug memory reconsolidation, as activation of the histone demethylase KDM6B in the medial prefrontal cortex [100] and Tet3 in the dorsal hippocampus [101] are both required for the reconsolidation of the memories underlying cocaine CPP.

Drug memory reconsolidation is modulated at the cell-surface, intracellular and nuclear levels, with crosstalk between the individual signalling molecules that have been investigated to date. Solid lines represent activation/enhancement of signalling; dashed lines represent inhibition. Abbreviations: AC adenylyl cyclase, AMP adenosine monophosphate, AMPAR α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid subtype of glutamate receptor, βAR β-adrenergic receptor, cAMP cyclic adenosine monophosphate, CaM calmodulin, CaMKs calcium-calmodulin dependent kinases, CaN calcineurin, D1R D1 dopamine receptor, D3R D3 dopamine receptor, eIFα eukaryotic initiation factor α, Epac exchange protein activated by cAMP, ERK extracellular signal-regulated kinase, GRIP1 glutamate receptor interacting protein 1, GSK3β glycogen synthase kinase β, IEG immediate early gene, KDM6B lysine demethylase 6B, mTORC1 mammalian target of rapamycin complex 1, NMDAR N-methyl-D-aspartate subtype of glutamate receptor, nNOS neuronal nitric oxide synthase, NO nitric oxide, p70S6K p70-S6 kinase, PKA protein kinase A, SO2 sulphur dioxide, SFKs src-family kinases, Tet3 tet methylcytosine dioxygenase 3.
At the level of cell surface receptors, catecholaminergic signalling appears necessary for the reconsolidation of the memories underlying drug CPP. Mice with a constitutive genetic knockout of the dopamine D3 receptor show impaired reconsolidation of cocaine CPP memories [102], as do wild type mice administered with the D3 receptor antagonist PG01037 [102] or the D1 receptor antagonist SCH23390 [103]. Furthermore, the enhancement of dopaminergic signalling with amphetamine facilitates the reconsolidation of the memories underlying morphine CPP [104]. Another catecholamine, noradrenaline, is also required for drug memory reconsolidation. Agonising α2 receptors with clonidine disrupts cocaine CPP [105], and antagonising β-adrenergic receptors with systemic propranolol during a single CPP memory reactivation session impairs subsequent CPP for cocaine [106], morphine [107] and nicotine [108], although not alcohol [109]. However, propranolol appears less effective when the drug CPP memory is strong, particularly if the memory has been recently acquired [110]. This boundary condition relating to memory strength may account for why some studies required multiple reactivation sessions under propranolol to observe subsequent amnesia [111]. Where propranolol is effective, it appears to exert its effects on memory via central noradrenergic signalling mechanisms. Administration of nadolol, a β-adrenergic receptor antagonist that does not cross the blood-brain barrier, does not disrupt the reconsolidation of the memories underlying morphine CPP under reactivation conditions for which propranolol is effective [107]. Furthermore, direct targeting of antagonists against α1 and β2 adrenergic receptors in the BLA disrupts the reconsolidation of the memories underlying cocaine CPP [112], as do both propranolol and nadolol when administered directly to the BLA [113]. β1 receptors in the central nucleus of the amygdala are also necessary for the reconsolidation of the memories underlying cocaine CPP [91]. Furthermore, administration of propranolol directly to the prelimbic cortex produces effects on both the retrieval and reconsolidation of cocaine CPP memories [113], perhaps indicating that some of the apparent discrepancies produced by studies using systemic propranolol can be attributed to effects in different neural structures.
The reconsolidation of the memories underlying drug CPP also depends upon activation at the NMDA subtype of glutamate receptor. Administration of the non-competitive NMDA receptor (NMDAR) antagonist MK-801 (dizocilpine) disrupts the reconsolidation of memories underlying cocaine CPP, usually with a single session of memory reactivation [114,115,116], but sometimes requiring multiple reactivation sessions [117]. Ketamine, another non-competitive NMDAR antagonist, disrupts the reconsolidation of memories underlying morphine CPP [118]. Amongst their many intracellular actions (Fig. 3), signalling at NMDARs leads to activation of neuronal nitric oxide synthase (nNOS) [119] and inhibition of nNOS also disrupts the reconsolidation of memories underlying cocaine CPP [115, 120]. Sulphur dioxide, which acts on nNOS pathways and affects neuronal function [121], impairs the reconsolidation of the memories underlying both cocaine and morphine CPP [122]. Finally, astrocytic lactate signalling, which likely exerts effects on synaptic plasticity via NMDARs [123], is also necessary for the reconsolidation of the memories underlying cocaine CPP [124, 125].
There has been great interest in whether behavioural interventions, aimed at exploiting the updating function of reconsolidation [126, 127], might reduce the strength of context-drug memories. Adapting the ‘retrieval-extinction’ procedure first used for updating fear memories by Monfils and colleagues [128] – in which a brief memory reactivation session, followed after a short delay by extinction training led to long-term reductions in fear memory – several studies have investigated whether reactivation of the drug-conditioned place preference memory can return it to a state in which it is susceptible to updating with the information that the context is no longer associated with drug. Xue and colleagues [129] reported reductions in both cocaine CPP and morphine CPP when CS-based reactivation of the CPP memory was followed by extinction training, though it has been reported that the initial reductions in place preference produced by the retrieval-extinction procedure can subsequently recover [130]. Differences in training history (e.g. the strength of conditioning) appear to alter the boundary conditions relevant to retrieval-extinction [131], allowing behaviour to be reduced either by a reconsolidation-dependent mechanism, or by facilitation of extinction. (Note that these two mechanisms are difficult to distinguish behaviourally [132]). Although the apparent discrepancies in the persistence of reduced preference for the drug-paired context following retrieval-extinction are unlikely due to the strength of conditioning (due to similar training protocols across studies), there may have been more subtle individual differences underlying the difference in treatment outcome [133]. An alternative updating approach, using counterconditioning (pairing a previously reward-associated CS to an aversive, rather than appetitive outcome) rather than extinction during the reconsolidation window, led to persistent reductions in both cocaine CPP [134] and alcohol CPP [135].
Disrupting the contextual memories influencing drug self-administration
In addition to supporting a preference for contexts paired with non-contingently administered drug, contextual cues also influence the propensity to seek drugs and to relapse following abstinence in individuals that have acquired drug self-administration [76]. Consistent with CPP studies, the reconsolidation of the memories underlying the influence of the drug-paired context on instrumental drug-seeking behaviour also relies upon protein synthesis [136], supported by intracellular signalling pathways activated by signalling at cell surface receptors (Fig. 3).
Consistent with its well-established role in representing contexts, much research into contextual influences on drug self-administration has focused upon the dorsal hippocampus. The projections between the dorsal hippocampus and basolateral amygdala are critical for the reconsolidation of memories underlying contextual influences on behaviour [72, 137], and disconnection of these structures during memory reactivation impairs subsequent context-induced reinstatement of drug-seeking [72]. Dorsal hippocampal activity is necessary for the reconsolidation of context-drug memories, as pharmacological inactivation with tetradotoxin [71] or optogenetic inhibition [138] of the dorsal hippocampus at reactivation impairs subsequent context-induced recovery of drug-seeking. However, inhibition of dorsal hippocampal protein synthesis with anisomycin at reactivation does not affect the capacity of the context to support the reinstatement of drug-seeking [71], although preventing signalling at GluN2A-containing NMDARs and activation of Src-family kinases does [139], suggesting engagement of some synaptic plasticity processes. As context-induced reinstatement of drug-seeking is subsequent impaired when anisomycin is delivered to the BLA [136] at reactivation, this has led to the hypothesis that the dorsal hippocampus modulates the reconsolidation of the context-drug memory, which may be stored within the BLA [138].
The plasticity mechanisms underlying the reconsolidation of the context memory within the BLA are like those described for the reconsolidation of the memories underlying CPP (Fig. 3). In addition to the requirement for protein synthesis [136], protein kinases including ERK [140] and PKA [141] (though not CaMKII [141]) are necessary. In contrast to studies of CPP, which have focused primarily upon monoaminergic and glutamatergic signalling at the cell surface level, studies of context-induced reinstatement have considered more extensively the requirement for glucocorticoid-mediated signalling and endocannabinoids. The glucocorticoid receptor antagonist, mifepristone, enhanced the reconsolidation of a context-cocaine memory [142] when administered directly to the BLA, similar to the effect of administering corticotropin-releasing factor (CRF) in female, but not male, rats [73]. CRF signalling in the BLA appears necessary for reconsolidation of the drug-context memory in both sexes, as administration of the CRF-1 receptor antagonist antalarmin at reactivation reduced subsequent context-induced cocaine-seeking in both males and females [73]. This activation of the HPA axis appears to interact with signalling at endocannabinoid receptors within the BLA. Systemic antagonism of CB1 receptors prior to, but not after, memory reactivation impairs the reconsolidation of the drug-context memory, and reduces the expression of immediate early genes including zif268 in the BLA [143]. Furthermore, while direct antagonism of CB1 receptors within the BLA leaves drug-context memory reconsolidation unimpaired (suggesting that systemic administration of CB1 receptor antagonists exerts amnestic effects via structures other than the BLA), administration of the CB1 receptor agonist AM251 during memory reactivation facilitates subsequent context-induced cocaine-seeking [74], and increases plasma corticosterone levels in a manner comparable to return to the drug-associated context [74]. This may indicate that CB1 receptors modulate the degree of HPA axis activation during drug-associated memory reconsolidation.
A small number of studies have investigated the impact of behavioural interventions on the reconsolidation of drug-context memories. As for CPP [134, 135], aversive counterconditioning following a memory reactivation session can subsequently reduce context-induced alcohol-seeking [135]. Post-retrieval extinction produces a similar reduction in alcohol-seeking [144], although as a similar outcome is observed when extinction precedes memory reactivation, it is not clear that this effect depends upon memory reconsolidation rather than a facilitation of extinction learning [132, 133, 144].
Disrupting discrete CS-drug memories influencing drug self-administration
Discrete pavlovian CS-drug memories interact with the instrumental acts of drug-seeking and drug-taking in multiple psychologically and neurobiologically dissociable ways [31]. Three such processes that have been studied in the context of reconsolidation are conditioned reinforcement, conditioned approach, and conditioned motivation. Pavlovian conditioned approach can be observed in different individuals as ‘sign-tracking’ or ‘goal-tracking’, reflecting a tendency to approach the pavlovian CS or the location of US delivery when the CS is presented, respectively (see ref. [145] for review). Conditioned motivation is usually assessed through ‘pavlovian-instrumental transfer’ (PIT) procedures, and describes the capacity of reward-associated cues to influence instrumental behaviour associated with the same or different rewards (see refs. [146, 147] for review). These processes can be studied in isolation using specific behavioural procedures [148], though in reality drug-seeking will be influenced by all of these processes. This can be addressed experimentally with the use of reinstatement procedures [149], which have also been used to interrogate the mechanisms underlying the reconsolidation of pavlovian CS-drug memories.
The memories underlying the conditioned reinforcing properties of cocaine-associated cues, like those underlying CPP (Fig. 3), depend upon protein synthesis [62] and the expression of immediate early genes including zif268 [62]. However, unlike CPP, the reconsolidation of the memory underlying conditioned reinforcement requires Zif268 expression in the BLA, but not the nucleus accumbens core [98]. Upstream of gene expression, the reconsolidation of the conditioned reinforcement memory depends upon PKA [150] and the activation of NMDA receptors [151] and β-adrenergic receptors (for both cocaine cues [152] and alcohol cues [153]), with the administration of the adrenergic prodrug dipivefrine enhancing reconsolidation of a CS-alcohol memory when given in conjunction with memory reactivation [153].
Less is known about the mechanisms underlying the reconsolidation of conditioned approach and conditioned motivation memories for drug-associated cues, which have been more extensively studied with natural (food) reinforcers [154,155,156]. However, it has been shown that the reconsolidation of pavlovian conditioned approach memories for CSs associated with alcohol [157] and sucrose [155, 156, 158,159,160] depend upon NMDAR activation, for both sign-tracking and goal-tracking responses. Although early studies suggested the memories underlying the goal-tracking responses were resistant to reconsolidation blockade [154], it has subsequently been shown that goal-tracking memories will reconsolidate under specific conditions [155, 158], and that the destabilisation of these memories relies upon dopaminergic signalling from the ventral tegmental area [159]. β-adrenergic receptor antagonism at reactivation has produced mixed results on the reconsolidation of conditioned approach memories, in contrast to its effects on the memories underlying conditioned reinforcement. Propranolol did not disrupt the reconsolidation of the memories underlying conditioned approach for either alcohol [157] or sucrose [156], and also did not disrupt the reconsolidation of memories underlying pavlovian-instrumental transfer under reactivation conditions in which NMDAR antagonism produced amnesia [156, 157]. Even when propranolol did disrupt the reconsolidation of a conditioned approach memory, only sign-tracking, and not goal-tracking, was impaired [161]. This has led to speculation that propranolol may impair the emotional component of the CS-US memory, without affecting the predictive value of CS [161]. This view would be consistent with studies of propranolol in fear-conditioned humans, in which automatic reflexive behaviours produced by an emotional CS were reduced by propranolol administration at reactivation, but the expectation that the CS would be associated with shock was unaffected [162].
Most studies assessing the mechanisms underlying the reconsolidation of CS-drug memories have used more translationally relevant procedures, in which the instrumental self-administration response for the drug produces both the drug reinforcer and the drug-associated CS, and the same response is tested following the manipulation aiming to disrupt reconsolidation [149, 163]. Reinstatement can subsequently be assessed through spontaneous recovery of previously acquired drug-seeking, or CS-induced or drug-induced reinstatement [163]. Like the memories underlying CPP, CS-drug memories acquired during self-administration depend upon protein synthesis [70, 164,165,166] and expression of the immediate early gene zif268 in the basolateral amygdala [167]. Prevention of transcription by inhibiting the dephosphorylation of eIFα also disrupts CS-cocaine memory reconsolidation [92]. Epigenetic changes occur during the reconsolidation of CS-drug memories, as inhibition of DNA methyltransferase (DNMT) during reactivation impairs subsequently both CS-induced and drug-induced reinstatement for heroin [168] and cocaine [169]. Administration of garcinol, which reduces histone acetylation, impairs subsequent CS-induced reinstatement and the expression of the immediate early genes arc and zif268 when administered into the lateral amygdala in conjunction with CS-cocaine memory reactivation [170]. Furthermore, garcinol can disrupt simultaneously the reconsolidation of multiple CS-cocaine memories when it is administered in conjunction with US-based reactivation [64].
Numerous protein kinases are required for the reconsolidation of CS-drug memories, including glycogen synthase kinase 3β (GSK3β), inhibition of which within the BLA, but not the CeN, impairs both subsequent CS-induced and drug-induced reinstatement [171]. Systemic administration of rapamycin, which inhibits mTOR signalling, similarly impairs the reconsolidation of CS-cocaine memories [172], and CaMKII inhibition not only impairs CS-cocaine memory reconsolidation, but also appears to facilitate its extinction [173]. Inhibition of PKA with Rp-cAMPS impairs CS-cocaine memory reconsolidation [150], as does activation of the exchange protein directed activated by cAMP (Epac) [174]. As for CPP, protein phosphatases are required for CS-drug memory reconsolidation, with inhibition of calpain in the nucleus accumbens core at reactivation reducing subsequent CS-induced cocaine-seeking [99]. Activation of the protein phosphatase calcineurin not only appears to disrupt the reconsolidation of CS-drug memory, but also appears to facilitate its extinction [175], similar to the effects of inhibiting CaMKII [173].
Monoaminergic signalling is also required for drug memory reconsolidation. Compared to CPP, dopaminergic signalling has been much less studied using self-administration procedures, but both D1 and D3-mediated signalling are necessary for the reconsolidation of CS-cocaine memories in mice [102, 176]. By contrast, noradrenergic signalling has been more extensively investigated, but has produced some apparently conflicting results in the literature that remain to be reconciled. Despite only being necessary for one of the three ‘routes to relapse’ [31] when CS-based reactivation procedures are used [152, 156, 157], signalling at β-adrenergic receptors is more effective in impairing CS-drug memory reconsolidation in reinstatement procedures when administered in conjunction with US-based reactivation. Propranolol administration in conjunction with a CS-based memory reactivation session gave mixed effects on the subsequent reinstatement of drug-seeking, producing no effect on the reinstatement of cocaine-seeking after enforced abstinence [177] or extinction of cocaine-seeking responses [164], but reducing subsequent alcohol-seeking after enforced abstinence [178] (with repeated rounds of propranolol administration and reactivation), and heroin-seeking in an extinction-reinstatement procedure [179]. Whether this reflects a difference in the noradrenergic mechanisms underlying the reconsolidation of memories associated with psychostimulants and depressants, or is related to the specific behavioural procedures used, warrants further investigation. However, when exposure to the drug US was used to reactivate the memory, β1 adrenergic receptor antagonism within the CeN impaired the reinstatement of cocaine-seeking after instrumental extinction [91], and administration of propranolol directly to the BLA reduced subsequent alcohol self-administration when alcohol reinforcers were available during the reactivation session [180]. The key adrenergic innervation supporting reconsolidation within the BLA appears to come from the nucleus of the solitary tract (NST) rather than the locus coeruleus (LC), as reactivation of CS-morphine memories led to increases in activation (measured through immediate early gene expression) in the NST and BLA, but not the LC [181]. Moreover, chemogenetic activation of NST-BLA projections (but not LC-BLA projections) led to greater sensitivity of the CS-drug memory to protein synthesis inhibition, while inhibition of these projections prevented the amnestic effect usually produced by protein synthesis inhibition [181]. However, these latter data are more consistent with noradrenaline from the NST supporting the destabilisation of the CS-drug memory, in contrast to the effects on restabilisation of the CS-drug memory reported above (see also refs. [113, 182]). They also stand in contrast to findings from the fear memory reconsolidation literature, where inhibiting noradrenergic signalling via LC-BLA projections rescued sensitivity to protein synthesis inhibition in a strong fear memory that was otherwise resistant [183]. Whether the source of noradrenaline input determines whether noradrenergic signalling influences memory destabilisation or restabilisation remains a question for future research.
Glutamatergic signalling is critical for cue-drug memory reconsolidation, and silent synapses within the nucleus accumbens appear to be unsilenced during drug memory reconsolidation, via a mechanism dependent upon the intracellular signalling molecule Rac1 [184]. Systemic antagonism of NMDARs at reactivation impairs cue-alcohol [166, 185, 186] and cue-cocaine [151] memory reconsolidation, as does antagonism of NMDARs within the BLA (which also reduces the expression of Zif268 in a reactivation-dependent manner) [151] and antagonism of GluN2A-containing NMDARs in the infralimbic cortex [187]. Systemic enhancement of NMDAR-mediated signalling with the partial agonist D-cycloserine enhances the reconsolidation of CS-cocaine memories, and increases Zif268 expression within the BLA [188]. Inhibition of lactate signalling at reactivation also reduces subsequent cocaine-seeking [125], potentially via effects on NMDARs.
As for CPP procedures, there has been interest in non-pharmacological interventions targeting cue-drug memory reconsolidation, including retrieval-extinction. As for CPP, the data have been mixed and may have been influenced by subtle differences in procedure (previous reviews have considered extensively procedural differences and their impact on retrieval-extinction outcomes for seeking behaviour for natural [189] and drug-associated [190] reinforcers) or individual differences in boundary conditions [133]. While the first demonstration of reduced recovery of drug-seeking following retrieval-extinction was effective for both opiate and cocaine memories [129], there have also been reports that a retrieval-extinction procedure that successfully impairs CS-nicotine memories is ineffective for CS-cocaine memories [191]. More research is needed to determine whether the retrieval-extinction procedure is supported by reconsolidation or extinction mechanisms [133].
The Reconsolidation Of Instrumental Drug-Associated Memories
The acts of drug-seeking and drug-taking are supported by instrumental associations, and can be considered to be a form of aberrantly engaged, maladaptive foraging behaviour [5]. These associations are initially goal-directed (i.e. an association forms between the drug-seeking action and the drug outcome), but ultimately become habitual and elicited by environmental, pavlovian CSs (i.e. become supported by ‘stimulus-response’ associations) [10]. In a subpopulation of individuals, these drug-seeking habits ultimately become uncontrolled and compulsive in nature [9, 10]. Pavlovian and instrumental memories interact to support ongoing drug-seeking behaviour and to promote relapse during attempted abstinence (Fig. 1; see refs. [31, 32], for further detail) and many studies, including those discussed in the previous section, have measured reductions in the capacity of pavlovian CSs to influence instrumental responding following a disruption of pavlovian memory reconsolidation. Far fewer studies have attempted to disrupt the memories underlying the instrumental associations. However, disrupting the reconsolidation of the instrumental memories supporting drug-seeking and drug-taking behaviour would be an exciting therapeutic prospect.
It is likely that the relative paucity of studies of instrumental memory reconsolidation is at least partly due to an early study [192] in which an instrumental memory appeared not to undergo protein synthesis-dependent reconsolidation following retrieval. There may also be differences in the boundary conditions determining whether instrumental memories reconsolidate, as compared to pavlovian memories. Whereas pavlovian memories are susceptible to disruption following CS-based reactivation, instrumental memories may require a ‘mismatch’ in the type of reinforcement contingency experienced at reactivation [65, 66]. A predictable reinforcement schedule (i.e. additional sessions of training) did not render an instrumental memory underlying saccharin-seeking susceptible to disruption with cycloheximide [193] and studies investigating well-established cocaine-seeking memories in self-administering rats have indicated that a change in reinforcement schedule at reactivation, from a predictable ‘fixed-ratio’ schedule to a less predictable ‘variable ratio’ schedule of reinforcement, is required to induce susceptibility to disruption with the NMDAR antagonist MK-801, with non-reinforced memory reactivation sessions being insufficient to destabilise the instrumental memory [65, 66]. However, other studies of instrumental sucrose-seeking and nicotine-seeking have shown that non-reinforced memory reactivation sessions can lead to memory lability [194,195,196]. A detailed analysis of procedural differences that may have contributed to these apparent differences in boundary conditions is considered by Piva et al. [197], and may be related to the dose of amnestic agent administered, the potential engagement of metaplasticity mechanisms [194], the specific reinforcer, the strength of training, changes in reactivation context [198] or the passage of time between learning and reactivation [199]. It may be that a better understanding of the relationship between prior learning – which generates the expectations that determine whether a ‘mismatch’ or prediction error is detected [200] – and the reactivation session itself will be necessary to resolve these apparent differences [68].
If US-based reactivation session sessions are necessary to induce the destabilisation of instrumental memories, then this could present a challenge to clinical translation. Although prolonged exposure therapy often involves the handling of drug-associated paraphernalia [129] and, sometimes, limited self-administration of legal drugs such as nicotine (e.g. ref. [108]), a requirement to self-administer the drug of abuse would be challenging for ethical and safeguarding reasons in the case of illicit drugs. Whether non-contingent administration of the drug US (see Fig. 1), or use of pharmacological agents with similar neurochemical effects but reduced abuse liability (e.g. methylphenidate for psychostimulants [63] or methadone for opioids [201]), would be sufficient to destabilise the instrumental memory underlying drug-seeking remains an important outstanding question.
Reconsolidation In The Clinical Context
To what extent can the findings discussed above, primarily from animal models relevant to drug addiction, be extrapolated to humans and to the clinical context? In light of the more extensive animal literature on disrupting the reconsolidation of pavlovian drug-associated memories, the majority of studies in humans have focused upon reducing reactivity to drug-associated CSs following reconsolidation-based manipulations. Many of these have recruited participants using legal drugs (alcohol and nicotine) but a small number of studies and clinical trials have focused upon psychostimulant and opiate users.
As for animals, pavlovian CS-drug memory reconsolidation depends upon NMDA receptor-mediated signalling in humans. The engagement of plasticity mechanisms appears to be important, as simply reducing neuronal activation with intravenous lidocaine infusions during reactivation had no effect on subsequent CS-induced cocaine craving [202].The administration of the NMDA receptor antagonist ketamine, in conjunction with a memory reactivation session that engaged prediction error, reduced subsequent alcohol-seeking in a population of hazardous drinkers [203], as did administration of the non-competitive NMDA receptor inhibitor nitrous oxide [204]. By contrast, another non-competitive NMDA receptor antagonist, memantine, given at memory reactivation was ineffective at reducing subsequent cigarette intake in a population of smokers [205]. However, despite both being NMDA receptor antagonists, memantine and ketamine have distinct effects on NMDA receptors [206] that may explain this apparent discrepancy.
Antagonism at β-adrenergic receptors has been used to target reconsolidation of addictive drug memories in both small-scale human trials and larger clinical trials. Despite initial reports that propranolol was ineffective at disrupting the reconsolidation of drug-associated memories in smokers when smoking-related CSs were used to reactivate the memory [207], subsequent studies using US-based reactivation of the nicotine memory showed a subsequent reduction in cigarette use in the group that had received propranolol [108]. The requirement for US-based reactivation to destabilise the memory may be specific to the strong interoceptive cues associated with smoking [208, 209], as reactivation of cocaine-associated memories through exposure to cocaine CSs was sufficient to induce sensitivity to disruption with propranolol in cocaine users [210, 211].
Numerous studies have investigated whether behavioural interventions might be used to interfere or alter reconsolidation of drug memories in humans. The types of interventions used include extinction training following memory reactivation (the ‘retrieval-extinction’ procedures discussed previously), counterconditioning, and cognitive reappraisal. The first demonstration of retrieval-extinction in drug users was that of Xue et al. [129], who built upon their rodent work to show in outpatient heroin users that a brief re-exposure to heroin-associated CSs, followed by prolonged exposure therapy, was sufficient to reduce CS-induced craving for at least 6 months [129]. A subsequent study showed that retrieval-extinction was also effective at reducing self-reported cigarette consumption in a population of smokers [212], although there were no effects on physiological reactivity to smoking-related CSs. This lack of physiological modulation was also found in a recent study [213], which also failed to replicate the reduction in cigarettes consumed in the retrieval-extinction group. However, they found that administering a stressful treatment (the Montreal Imaging Stress Test) prior to extinction itself led to reduced cigarette consumption. This is consistent with stress facilitating extinction learning, as has been observed previously for fear memories [214].
Several studies have investigated whether post-reactivation counterconditioning might be used to reduce subsequent drug use. In a population of hazardous drinkers, re-exposure to alcohol-associated CSs combined with visual counterconditioning (exposure to unpleasant images from the International Affective Picture Scale [215]) and gustatory counterconditioning (exposure to solutions laced with Bitrex) was sufficient to reduce alcohol consumption [216] for at least 9 months follow-up [217]. The effects of counterconditioning appeared stronger than those of cognitive reappraisal following memory reactivation, which reduced scores on an alcohol fluency task, but not subsequent alcohol consumption or attentional bias towards alcohol-associated CSs [218]. However, this intervention was brief compared to the procedures usually used in cognitive therapy, and likely did not reproduce the therapeutic relationship necessary for the effectiveness of cognitive therapy (see ref. [219] for further discussion). More intensive and extensive cognitive therapy with cocaine use disorder patients showed enhanced efficacy of cognitive therapy when combined with memory reactivation procedures [220, 221]. These findings suggest that the addition of a brief CS reminder session to already established therapies, such as prolonged exposure and cognitive therapy, could markedly enhance patient outcomes, though more research is needed to test this in larger scale clinical populations.
Conclusions
The conceptualisation of addiction as a disorder of learning and memory, combined with the re-emergence of research into the mechanisms underlying memory reconsolidation, provides the potential for the development of novel therapeutic approaches for treating addiction. There are a multitude of maladaptive emotional memories that contribute to the persistence of drug-seeking and that promote relapse following a period of abstinence, including pavlovian (contextual and discrete CSs) and instrumental memories. While there are many reports that reactivating drug memories through presentation of the US (i.e. allowing the administration of a small dose of the drug of abuse) allows for widespread disruption within the drug-associated memory network, this approach would present the ethical challenge of administering an (often illegal) drug of abuse to a patient who is trying to maintain abstinence. Whether drugs with a similar pharmacological action could substitute for the drug of abuse in a ‘US-based’ reactivation session is worthy of further study.
The clinical trials of reconsolidation-based intervention for the treatment of addiction have been relatively small-scale, but overall there is cause for optimism. Future clinical studies will need to address a number of research questions, including the optimal reactivation procedure(s) to induce drug memory destabilisation (and ideally, this would include the development of methods to measure online and in real-time the induction of memory lability [68]), whether there are predictable individual differences in patients that could be used to tailor the reactivation procedure [222], and whether the effects seen to date generalise to larger patient populations. However, the encouraging data to date suggest that there is potential for reconsolidation-based interventions in providing a greater range of treatment options for patients.
Questions also remain within the preclinical literature, including whether behavioural interference approaches such as retrieval-extinction are acting via reconsolidation or extinction mechanisms [133] and whether reconsolidation-based interventions are effective in animal models of compulsive drug-seeking [7, 8]. Demonstrations that instrumental drug-seeking memories can be disrupted [65, 66, 223] raises the exciting prospect that it may be possible to return drug-seeking from being habitual to goal-directed, although whether individuals treated in this fashion would reacquire habitual and compulsive drug-seeking remains to be established. At the very least, it seems feasible that reconsolidation-based interventions would serve to reduce the pernicious effects of drug memories in eliciting automatic drug-seeking behaviours sufficiently to allow other, complementary therapies such as contingency management or cognitive behavioural therapy to have a greater chance of success.
References
Public Health England. An evidence review of the outcomes that can be expected of drug misuse treatment in England. (2017).
Public Health England. The public health burden of alcohol and the effectiveness and cost-effectiveness of alcohol control policies: an evidence review. (2018).
The Secretary General. Facing addiction in America: The Surgeon General’s spotlight on opioids. (2018).
American Psychiatric Association. The Diagnostic and Statistical Manual of Mental Disorders (5th ed.). (American Psychiatric Publishing, 2013).
Everitt BJ, Dickinson A, Robbins TW. The neuropsychological basis of addictive behaviour. Brain Res Rev. 2001;36:129–38.
Hyman SE. Addiction: a disease of learning and memory. Am J Psychiatry. 2005;162:1414–22.
Deroche-Gamonet V, Belin D, Piazza PV. Evidence for addiction-like behavior in the rat. Science. 2004;305:1014–7.
Pelloux Y, Everitt BJ, Dickinson A. Compulsive drug seeking by rats under punishment: effects of drug taking history. Psychopharmacology. 2007;194:127–37.
Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–9.
Everitt BJ, Robbins TW. Drug addiction: updating actions to habits to compulsions ten years on. Annu Rev Psychol. 2016;67:8.1–8.28.
Grüsser SM, Wrase J, Klein S, Hermann D, Smolka MN, Ruf M, et al. Cue-induced activation of the striatum and medial prefrontal cortex is associated with subsequent relapse in abstinent alcoholics. Psychopharmacology. 2004;175:296–302.
Seo D, Lacadie CM, Tuit K, Hong KI, Constable RT, Sinha R. Disrupted ventromedial prefrontal function, alcohol craving, and subsequent relapse risk. JAMA Psychiatry. 2013;70:727–39.
Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999;156:11–18.
Gawin, FH & Kleber, HD in Clinician’s Guide to Cocaine Addiction: Theory, Research and Treatment, pp. 33–52 (The Guildford Press, 1992).
Grant S, London ED, Newlin DB, Villemagne VL, Liu X, Contoreggi C, et al. Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci. 1996;93:12040–5.
O’Brien CP, Testa T, O’Brien TJ, Greenstein R. Conditioning in human opiate addicts. Pavlovian J Biol Sci. 1976;11:195–202.
O’Brien CP, Childress AR, McClellan TA, Ehrman R. Classical conditioning in drug dependent humans. Ann N. Y Acad Sci. 1992;654:400–15.
Ehrman RN, Robbins SJ, Childress AR, O’Brien CP. Conditioned responses to cocaine-related stimuli in cocaine abuse patients. Psychopharmacology. 1992;107:523–9.
Janes AC, Pizzagalli DA, Richardt S, Frederick BD, Chuzi S, Pachas G, et al. Brain reactivity to smoking cues prior to smoking cessation predicts ability to maintain tobacco abstinence. Biol Psychiatry. 2010;67:722–9.
McClernon FJ, Hiott FB, Liu J, Salley AN, Behm FM, Rose JE. Selectively reduced respones to smoking cues in amygdala following extinction-based smoking cessation: results of a preliminary functional magnetic resonance imaging study. Addiction Biol. 2007;12:503–12.
Versace F, Engelmann JM, Robinson JD, Jackson EF, Green CE, Lam CY, et al. Prequit fMRI responses to pleasant cues and cigarette-related cues predict smoking cessation outcome. Nicotine Tob Res. 2014;16:697–708.
Li Q, Li W, Wang H, Wang Y, Zhang Y, Zhu J, et al. Predicting subsequent relapse by drug-related cue-induced brain activation in heroin addiction: an event-related functional magnetic resonance imaging study. Addiction Biol. 2015;20:968–78.
Kosten TR, Scanley BE, Tucker KA, Oliveto A, Prince C, Sinha R, et al. Cue-induced brain activity changes and relapse in cocaine-dependent patients. Neuropsychopharmacology. 2006;31:644–50.
Regier PS, Jagannathan K, Franklin TR, Wetherill RR, Langleben DD, Gawyrsiak M, et al. Sustained brain response to repeated drug cues is associated with poor drug-use outcomes. Addict Biol. 2021;26:e13028.
de Wit H, Stewart J. Reinstatement of cocaine-reinforced responding in the rat. Psychopharmacology. 1981;75:134–43.
Arroyo M, Markou A, Robbins TW, Everitt BJ. Acquisition, maintenance and reinstatement of intravenous cocaine self-administration under a second-order schedule of reinforcement in rats: effects of conditioned cues and continuous access to cocaine. Psychopharmacology. 1998;140:331–44.
Crombag HS, Shaham Y. Renewal of drug seeking by contextual cues after prolonged extinction in rats. Behav Neurosci. 2002;116:169–73.
Ito R, Dalley JW, Robbins TW, Everitt BJ. Dopamine release in the dorsal striatum during cocaine-seeking behavior under the control of a drug-associated cue. J Neurosci. 2002;22:6247–53.
Feltenstein MW, See RE. Potentiation of cue-induced reinstatement of cocaine-seeking in rats by the anxiogenic drug yohimbine. Behav Brain Res. 2006;174:1–8.
Lee JLC, Everitt BJ. In Memories: Molecules and Circuits Research and Perspectives in Neurosciences (eds B Bontempi, AJ Silva, & Y Christen) 83–98 (Springer-Verlag, 2007).
Milton AL, Everitt BJ. The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur J Neurosci. 2010;31:2308–19.
Milton AL, Everitt BJ. The persistence of maladaptive memory: addiction, drug memories and anti-relapse treatments. Neurosci Biobehav Rev. 2012;36:1119–39.
Torregrossa MM, Corlett PR, Taylor JR. Aberrant learning and memory in addiction. Neurobiol Learn Mem. 2011;96:609–23.
Tronson NC, Taylor JR. Addiction: a drug-induced disorder of memory reconsolidation. Curr Opin Neurobiol. 2013;23:573–80.
Torregrossa MM, Taylor JR. Learning to forget: manipulating extinction and reconsolidation processes to treat addiction. Psychopharmacology. 2013;226:659–72.
Dunbar AB, Taylor JR. Reconsolidation and psychopathology: moving towards reconsolidation-based treatments. Neurobiol Learn Mem. 2016;142:162–71.
Watson JP, Gaind R, Marks IM. Prolonged exposure: a rapid treatment for phobias. Br Med J. 1971;1:13–15.
Wolitzky-Taylor KB, Horowitz JD, Powers MB, Telch MJ. Psychological approaches in the treatment of specific phobias: a meta-analysis. Clin Psychol Rev. 2008;28:1021–37.
Wolpe J. Psychotherapy by reciprocal inhibition. Cond Reflex. 1968;3:234–40.
Holmes EA, Craske MG, Graybiel AM. A call for mental-health science. Nature. 2014;511:287–9.
Mellentin AI, Skøt L, Nielsen B, Schippers GM, Nielsen AS, Stenager E, et al. Cue exposure therapy for the treatment of alcohol use disorders: a meta-analytic review. Clin Psychol Rev. 2017;57:195–207.
Kiyak C, Simonetti ME, Norton S, Deluca P. The efficacy of cue exposure therapy on alcohol use disorders: a quantitative meta-analysis and systematic review. Addict Behav. 2022;139:107578.
Conklin CA, Tiffany ST. Applying extinction research and theory to cue-exposure addiction treatments. Addiction. 2002;97:155–67.
Mayet, S, Farrell, M, Ferri, M, Amato, L & Davoli, M Psychosocial treatment for opiate abuse and dependence. Cochrane Database Syst Rev 4 (2004).
Bouton ME, Swartzentruber D. Sources of relapse after extinction in pavlovian and instrumental learning. Clin Psychol Rev. 1991;11:123–40.
Bouton ME. Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol Psychiatry. 2002;52:976–86.
Wechsler TF, Kümpers F, Mühlberger A. Inferiority or even superiority of virtual reality exposure therapy in phobias? - A systematic review and quantitative meta-analysis on randomized controlled trials specifically comparing the efficacy of virtual reality exposure to gold standard in vivo exposure in agoraphobia, specific phobia, and social phobia. Front Psychol. 2019;10:1758.
Craske MG, Treanor M, Conway CC, Zbozinek T, Vervliet B. Maximizing exposure therapy: an inhibitory learning approach. Behav Res Ther. 2014;58:10–23.
Di Ciano P, Everitt BJ. Conditioned reinforcing properties of stimuli paired with self-adminstered cocaine, heroin or sucrose: implications for the persistence of addictive behavior. Neuropharmacology. 2004;47:202–13.
Tunstall BJ, Kearns DN. Cocaine can generate a stronger conditioned reinforcer than food despite being a weaker primary reinforcer. Addict Biol. 2016;21:282–93.
Mackintosh, NJ. The Psychology of Animal Learning. (Academic Press, 1974).
Martin SJ, Morris RGM. New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus. 2002;12:609–36.
Josselyn SA, Köhler S, Frankland PW. Finding the engram. Nat Rev Neurosci. 2015;16:521–34.
Tonegawa S, Pignatelli M, Roy DS, Ryan TJ. Memory engram storage and retrieval. Curr Opin Neurobiol. 2015;35:101–9.
Anthony JC, Warner LA, Kessler RC. Comparative epidemiology of dependence on tobacco, alcohol, controlled substances, and inhalants: findings from the National Comorbidity Survey. Exp Clin Psychopharmacol. 1994;166:244–68.
Schlag AK. Percentages of problem drug use and their implications for policy making: a review of the literature. Drug Sci, Policy Law. 2020;6:1–9.
McGaugh JL. Memory - a century of consolidation. Science. 2000;287:248–51.
Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–6.
Dębiec, J & Altemus, M Toward a new treatment for traumatic memories. Cerebrum. 2006:2–11.
Hernandez PJ, Kelley AE. Cracking addiction the second time around: reconsolidation of drug-related memories. Neuron. 2005;47:772–5.
Pedreira ME, Pérez-Cuesta LM, Maldonado H. Mismatch between what is expected and what actually occurs triggers memory reconsolidation or extinction. Learn Mem. 2004;11:579–85.
Lee JLC, Di Ciano P, Thomas KL, Everitt BJ. Disrupting reconsolidation of drug memories reduces cocaine seeking behavior. Neuron. 2005;47:795–801.
Luo YX, Xue YX, Liu JF, Shi HS, Jian M, Han Y, et al. A novel UCS memory retrieval-extinction procedure to inhibit relapse to drug seeking. Nat Commun. 2015;6:7675.
Dunbar AB, Taylor JR. Garcinol blocks the reconsolidation of multiple cocaine-paired cues after a single cocaine-reactivation session. Neuropsychopharmacology. 2017;42:1884–92.
Exton-McGuinness MTJ, Patton RC, Sacco LB, Lee JLC. Reconsolidation of a well-learned instrumental memory. Learn Mem. 2014;21:468–77.
Exton-McGuinness MTJ, Lee JLC. Reduction in responding for sucrose and cocaine reinforcement by disruption of memory reconsolidation. eNeuro. 2015;2:e0009–0015.2015.
Vaverková Z, Milton AL, Merlo E. Retrieval-dependent mechanisms affecting emotional memory persistence: reconsolidation, extinction, and the space in between. Front Behav Neurosci. 2020;14:574358.
Milton AL, Das RK, Merlo E. The challenge of memory destabilisation: from prediction error to prior expectations and biomarkers. Brain Res Bull. 2023;194:100–4.
Taujanskaitė U, Cahill EN, Milton AL. Targeting drug memory reconsolidation: a neural analysis. Curr Opin Pharmacol. 2021;56:7–12.
Xue YX, Chen YY, Zhang LB, Zhang LQ, Huang GD, Sun SC, et al. Selective inhibition of amygdala neuronal ensembles encoding nicotine-associated memories prevents nicotine seeking and relapse. Biol Psychiatry. 2017;82:781–93.
Ramirez DR, Bell GH, Lasseter HC, Xie X, Traina SA, Fuchs RA. Dorsal hippocampal regulation of memory reconsolidation processes that facilitate drug context-induced cocaine-seeking behavior in rats. Eur J Neurosci. 2009;30:901–12.
Wells AM, Lasseter HC, Xie X, Cowhey KE, Reittinger AM, Fuchs RA. Interaction between the basolateral amygdala and dorsal hippocampus is critical for cocaine memory reconsolidation and subsequent drug context-induced cocaine-seeking behavior in rats. Learn Mem. 2011;18:693–702.
Ritchie JL, Walters JL, Galliou JM, Christian RJ, Qi S, Savenkova MI, et al. Basolateral amygdala corticotropin-releasing factor type 1 regulates context-cocaine memory strength during reconsolidation in a sex-dependent manner. Neuropharmacology. 2021;200:108819.
Higginbotham JA, Jones NM, Wang R, Christian RJ, Ritchie JL, McLaughlin RJ, et al. Basolateral amygdala CB1 receptors gate HPA axis activation and context-cocaine memory strength during reconsolidation. Neuropsychopharmacology. 2021;46:1554–64.
Schechter MD, Calcagnetti DJ. Trends in place preference conditioning with a cross-indexed bibliography; 1957-1991. Neurosci Biobehav Rev. 1993;17:21–41.
Crombag HS, Bossert JM, Koya E, Shaham Y. Context-induced relapse to drug seeking: a review. Philos Trans R Soc B. 2008;363:3233–43.
Everitt BJ, Morris KA, O’Brien A, Robbins TW. The basolateral amygdala-ventral striatal system and conditioned place preference: further evidence of limbic-striatal interactions underlying reward-related processes. Neuroscience. 1991;42:1–18.
White NM, McDonald RJ. Acquisition of a spatial conditioned place preference is impaired by amygdala lesions and improved by fornix lesions. Behav Brain Res. 1993;55:269–81.
Brown EE, Fibiger HC. Differential effects of excitotoxic lesions of the amygdala on cocaine-induced conditioned locomotion and conditioned place preference. Psychopharmacology. 1993;113:123–30.
Fuchs RA, Weber SM, Rice HJ, Neisewander JL. Effects of excitotoxic lesions of the basolateral amygdala on cocaine-seeking behavior and cocaine conditioned place preference in rats. Brain Res. 2002;929:15–25.
Everitt, BJ, Cardinal, RN, Hall, J, Parkinson, JA, & Robbins, TW. Differential involvement of amygdala subsystems in appetitive conditioning and drug addiction. In The Amygdala: A Functional Analysis (2nd Ed.) pp. 353–90 (Oxford University Press, 2000).
Burns LH, Everitt BJ, Robbins TW. Effects of excitotoxic lesions of the basolateral amygdala on conditional discrimination learning with primary and conditioned reinforcement. Behav Brain Res. 1999;100:123–33.
Whitelaw RB, Markou A, Robbins TW, Everitt BJ. Excitotoxic lesions of the basolateral amygdala impair the acquisition of cocaine-seeking behaviour under a second-order schedule of reinforcement. Psychopharmacology. 1996;127:213–24.
Selden NRW, Everitt BJ, Jarrard LE, Robbins TW. Complementary roles for the amygdala and hippocampus in aversive conditioning to explicit and contextual cues. Neuroscience. 1991;42:335–50.
Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–85.
Ito R, Robbins TW, McNaughton BL, Everitt BJ. Selective excitotoxic lesions of the hippocampus and basolateral amygdala have dissociable effects on appetitive cue and place conditioning based on path integration in a novel Y-maze procedure. Eur J Neurosci. 2006;23:3071–80.
Valjent E, Corbillé AG, Bertran-Gonzelez J, Hervé D, Girault JA. Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc Natl Acad Sci. 2006;103:2932–7.
Milekic MH, Brown SD, Castellini C, Alberini CM. Persistent disruption of an established morphine conditioned place preference. J Neurosci. 2006;26:3010–20.
Robinson MJF, Franklin KBJ. Effects of anisomycin on consolidation and reconsolidation of a morphine-conditioned place preference. Behav Brain Res. 2007;178:146–53.
Yim AJ, Moraes CRG, Ferreira TL, Oliveira MGM. Protein synthesis inhibition in the basolateral amygdala following retrieval does not impair expression of morphine-associated conditioned place preference. Behav Brain Res. 2006;171:162–9.
Zhu H, Zhou Y, Liu Z, Chen X, Li Y, Liu X, et al. β1-adrenoceptor in the central amygdala is required for unconditioned stimulus-induced drug memory reconsolidation. Int J Neuropsychopharmacol. 2018;21:267–80.
Jian M, Luo YX, Xue YX, Han Y, Shi HS, Liu JF, et al. eIF2α dephosphorylation in basolateral amygdala mediates reconsolidation of drug memory. J Neurosci. 2014;34:10010–21.
Shen Q, Xie B, Galaj E, Yu H, Li X, Lu Y, et al. CircTmeff-1 in the nucleus accumbens regulates the reconsolidation of cocaine-associated memory. Brain Res Bull. 2022;185:64–73.
Shi X, von Weltin E, Fitzsimmons E, Do C, Caban Rivera C, Chen C, et al. Reactivation of cocaine contextual memory engages mechanistic target of rapamycin/S6 kinase 1 signaling. Front Pharmacol. 2022;13:976932.
Shi X, Miller JS, Harper LJ, Poole RL, Gould TJ, Unterwald EM. Reactivation of cocaine reward memory engages the Akt/GSK3/mTOR signaling pathway and can be disrupted by GSK3 inhibition. Psychopharmacology. 2014;231:3109–18.
Wu P, Xue YX, Ding ZB, Xue LF, Xu CM, Lu L. Glycogen synthase kinase 3B in the basolateral amygdala is critical for the reconsolidation of cocaine reward memory. J Neurochem. 2011;118:113–25.
Shi X, von Weltin E, Barr JL, Unterwald EM. Activation of GSK3β induced by recall of cocaine reward memories is dependent on GluN2A/B NMDA receptor signaling. J Neurochem. 2019;151:91–102.
Théberge FR, Milton AL, Belin D, Lee JLC, Everitt BJ. The basolateral amygdala and nucleus accumbens core mediate dissociable aspects of drug memory reconsolidation. Learn Mem. 2010;17:444–53.
Liang J, Li JL, Han Y, Luo YX, Xue YX, Zhang Y, et al. Calpain-GRIP signaling in nucleus accumbens core mediates the reconsolidation of drug reward memory. J Neurosci. 2017;37:8938–51.
Zhang YX, Akumuo RC, España RA, Yan CX, Gao WJ, Li YC. The histone demethylase KDM6B in the medial prefrontal cortex epigenetically regulates cocaine reward memory. Neuropharmacology. 2018;141:113–25.
Liu C, Sun X, Wang Z, Le Q, Liu P, Jiang C, et al. Retrieval-induced upregulation of Tet3 in pyramidal neurons of the dorsal hippocampus mediates cocaine-associated memory reconsolidation. Int J Neuropsychopharmacol. 2018;21:255–66.
Yan Y, Kong H, Wu EJ, Newman AH, Xu M. Dopamine D3 receptors regulate reconsolidation of cocaine memory. Neuroscience. 2013;241:32–40.
Marion-Poll L, Besnard A, Longueville S, Valjent E, Engmann O, Caboche J, et al. Cocaine conditioned place preference: unexpected suppression of preference due to testing combined with strong conditioning. Addict Biol. 2019;24:364–75.
Blaiss CA, Janak PH. Post-training and post-reactivation administration of amphetamine enhances morphine conditioned place preference. Behav Brain Res. 2006;171:329–37.
Denny RR, Unterwald EM. Clonidine, an α2 receptor agonist, disrupts reconsolidation of a cocaine-paired environmental memory. Behav Pharmacol. 2019;30:529–33.
Bernardi RE, Lattal KM, Berger SP. Postretrieval propranolol disrupts a cocaine conditioned place preference. Neuroreport. 2006;17:1443–7.
Robinson MJF, Franklin KBJ. Central but not peripheral beta-adrenergic antagonism blocks reconsolidation for a morphine place preference. Behav Brain Res. 2007;182:129–34.
Xue YX, Deng JH, Chen YY, Zhang LB, Wu P, Huang GD, et al. Effect of selective inhibition of reactivated nicotine-associated memories with propranolol on nicotine craving. JAMA Psychiatry. 2017;74:224–32.
Font L, Cunningham CL. Post-retrieval propranolol treatment does not modulate reconsolidation or extinction of ethanol-induced conditioned place preference. Pharmacol Biochem Behav. 2012;101:222–30.
Robinson MJF, Franklin KBJ. Reconsolidation of a morphine place preference: impact of the strength and age of memory on disruption by propranolol and midazolam. Behav Brain Res. 2010;213:201–7.
Fricks-Gleason AN, Marshall JF. Post-retrieval b-adrenergic receptor blockade: effects on extinction and reconsolidation of cocaine-cue memories. Learn Mem. 2008;15:643–8.
Bernardi RE, Ryabinin AE, Berger SP, Lattal KM. Post-retrieval disruption of a cocaine conditioned place preference by systemic and intrabasolateral amygdala b2 and a1-adrenergic antagonists. Learn Mem. 2009;16:777–89.
Otis JM, Dashew KB, Mueller D. Neurobiological dissociation of retrieval and reconsolidation of cocaine-associated memory. J Neurosci. 2013;33:1271–81.
Kelley JB, Anderson KL, Itzhak Y. Long-term memory of cocaine-associated context: disruption and reinstatement. Neuroreport. 2007;18:777–80.
Itzhak Y. Role of the NMDA receptor and nitric oxide in memory reconsolidation of cocaine-induced conditioned place preference in mice. Ann N. Y Acad Sci. 2008;1139:350–7.
Brown TE, Lee BR, Sorg BA. The NMDA antagonist MK-801 disrupts reconsolidation of a cocaine-associated memory for conditioned place preference but not for self-administration in rats. Learn Mem. 2008;15:857–65.
Sadler R, Herzig V, Schmidt WJ. Repeated treatment with the NMDA antagonist MK-801 disrupts reconsolidation of memory for amphetamine-conditioned place preference. Behav Pharmacol. 2007;18:699–703.
Zhai H, Wu P, Chen S, Li F, Liu Y, Lu L. Effects of scopolamine and ketamine on reconsolidation of morphine conditioned place preference in rats. Behav Pharmacol. 2008;19:211–6.
Garthwaite J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991;14:60–67.
Itzhak Y, Anderson KL. Memory reconsolidation of cocaine-associated context requires nitric oxide signaling. Synapse. 2007;61:1002–5.
Yun Y, Yao G, Yue H, Guo L, Qin G, Li G, et al. SO2 inhalation causes synaptic injury in rat hippocampus via its derivatives in vivo. Chemosphere. 2013;93:2426–32.
Rulan D, Zhenbang Y, Yipu Z, Yuan G, Galaj E, Xiaorui S, et al. Exogenous SO2 donor treatment impairs reconsolidation of drug reward memory in mice. Eur J Pharmacol. 2021;896:173911.
Yang J, Ruchti E, Petit JM, Jourdain P, Grenningloh G, Allaman I, et al. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc Natl Acad Sci. 2014;111:12228–33.
Boury-Jamot B, Carrard A, Martin JL, Halfon O, Magistretti PJ, Boutrel B. Disrupting astrocyte-neuron lactate transfer persistently reduces conditioned responses to cocaine. Mol Psychiatry. 2016;21:1070–6.
Zhang Y, Xue Y, Meng S, Luo Y, Liang J, Li J, et al. Inhibition of lactate transport erases drug memory and prevents drug relapse. Biol Psychiatry. 2016;79:928–39.
Lee JLC, Nader K, Schiller D. An update on memory reconsolidation updating. Trends Cogn Sci. 2017;21:531–45.
Lee JLC. Reconsolidation: maintaining memory relevance. Trends Neurosci. 2009;32:413–20.
Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–5.
Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C, et al. A memory retrieval-extinction procedure to prevent drug craving and relapse. Science. 2012;336:241–5.
Ma X, Zhang J-J, Yu L-C. Post-retrieval extinction training enhances or hinders the extinction of morphine-induced conditioned place preference in rats dependent on the retrieval-extinction interval. Psychopharmacology. 2012;221:19–26.
Chang H-A, Dai W, Hu SS-J. Dose-dependent effect of retrieval-extinction on preventing reinstatement of cocaine-associated memory in mice. Chin J Physiol. 2022;65:159–70.
Cahill EN, Milton AL. Neurochemical and molecular mechanisms underlying the retrieval-extinction effect. Psychopharmacology. 2019;236:111–32.
Kuijer, EJ, Ferragud, A & Milton, AL. Retrieval-extinction and relapse prevention: rewriting maladaptive drug memories? Front Behav Neurosci. 2020;14:23.
Goltseker K, Bolotin L, Barak S. Counterconditioning during reconsolidation prevents relapse of cocaine memories. Neuropsychopharmacology. 2017;42:716–26.
Goltseker, K, Handrus, H & Barak, S Disruption of relapse to alcohol seeking by aversive counterconditioning following memory retrieval. Addict Biol. 2020:e12935.
Fuchs RA, Bell GH, Ramirez DR, Eaddy JL, Su Z-I. Basolateral amygdala involvement in memory reconsolidation processes that facilitate drug context-induced cocaine seeking. Eur J Neurosci. 2009;30:889–900.
Mamiya N, Fukushima H, Suzuki A, Matsuyama Z, Homma S, Frankland PW, et al. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J Neurosci. 2009;29:402–13.
Qi S, Tan SM, Wang R, Higginbotham JA, Ritchie JL, Ibarra CK, et al. Optogenetic inhibition of the dorsal hippocampus CA3 region during early-stage cocaine-memory reconsolidation disrupts subsequent context-induced cocaine seeking in rats. Neuropsychopharmacology. 2022;47:1473–83.
Wells AM, Xie X, Higginbotham JA, Arguello AA, Healey KL, Blanton M, et al. Contribution of an SFK-mediated signaling pathway in the dorsal hippocampus to cocaine-memory reconsolidation in rats. Neuropsychopharmacology. 2016;41:675–85.
Wells AM, Arguello AA, Xie X, Blanton MA, Lasseter HC, Reittinger AM, et al. Extracellular signal-regulated kinase in the basolateral amygdala, but not the nucleus accumbens core, is critical for context-response-cocaine memory reconsolidation in rats. Neuropsychopharmacology. 2013;38:753–62.
Arguello AA, Hodges MA, Wells AM, Lara H, Xie X, Fuchs RA. Involvement of amygdalar protein kinase A, but not calcium/calmodulin-dependent protein kinase II, in the reconsolidation of cocaine-related contextual memories in rats. Psychopharmacology. 2014;231:55–65.
Stringfield SJ, Higginbotham JA, Wang R, Berger AL, McLaughlin RJ, Fuchs RA. Role of glucocorticoid receptor-mediated mechanisms in cocaine memory enhancement. Neuropharmacology. 2017;123:349–58.
Higginbotham JA, Wang R, Richardson BD, Shiina H, Tan SM, Presker MA, et al. CB1 receptor signaling modulates amygdalar plasticity during context-cocaine memory reconsolidation to promote subsequent cocaine seeking. J Neurosci. 2021;41:613–29.
Millan EZ, Milligan-Saville J, McNally GP. Memory retrieval, extinction, and reinstatement of alcohol seeking. Neurobiol Learn Mem. 2013;101:26–32.
Flagel SB, Akil H, Robinson TE. Individual differences in the attribution of incentive salience to reward-related cues: implications for addiction. Neuropharmacology. 2009;56:139–48.
Cartoni E, Balleine B, Baldassarre G. Appetitive pavlovian-instrumental transfer: a review. Neurosci Biobehav Rev. 2016;71:829–48.
Corbit LH, Balleine BW. Learning and motivational processes contributing to pavlovian-instrumental transfer and their neural bases: dopamine and beyond. Curr Top Behav Neurosci. 2015;27:259–89.
Rutherford LG, Milton AL. Deconstructing and reconstructing behaviour relevant to mental health disorders: the benefits of a psychological approach, with a focus on addiction. Neurosci Biobehav Rev. 2022;133:104514.
Reichel CM, Bevins RA. Forced abstinence model of relapse to study pharmacological treatments of substance use disorder. Curr Drug Abus Rev. 2009;2:184–94.
Sanchez H, Quinn JJ, Torregrossa MM, Taylor JR. Reconsolidation of a cocaine-associated stimulus requires amygdalar protein kinase A. J Neurosci. 2010;30:4401–7.
Milton AL, Lee JLC, Butler VJ, Gardner RJ, Everitt BJ. Intra-amygdala and systemic antagonism of NMDA receptors prevents the reconsolidation of drug-associated memory and impairs subsequently both novel and previously acquired drug-seeking behaviors. J Neurosci. 2008;28:8230–7.
Milton AL, Lee JLC, Everitt BJ. Reconsolidation of appetitive memories for both natural and drug reinforcement is dependent on β-adrenergic receptors. Learn Mem. 2008;15:88–92.
Schramm MJW, Everitt BJ, Milton AL. Bidirectional modulation of alcohol-associated memory reconsolidation through manipulation of adrenergic signaling. Neuropsychopharmacology. 2016;41:1103–11.
Blaiss CA, Janak PH. Post-training, but not post-reactivation, administration of amphetamine and anisomycin modulates pavlovian conditioned approach. Neurobiol Learn Mem. 2007;87:644–58.
Reichelt AC, Lee JLC. Appetitive Pavlovian goal-tracking memories reconsolidate only under specific conditions. Learn Mem. 2012;20:51–60.
Lee JLC, Everitt BJ. Reactivation-dependent amnesia in pavlovian approach and instrumental transfer. Learn Mem. 2008;15:597–602.
Milton AL, Schramm MJ, Wawrzynski JR, Gore F, Oikonomou-Mpegeti F, Wang NQ, et al. Antagonism at NMDA receptors, but not β-adrenergic receptors, disrupts the reconsolidation of pavlovian conditioned approach and instrumental transfer for ethanol-associated conditioned stimuli. Psychopharmacology. 2012;219:751–61.
Reichelt AC, Lee JLC. Over-expectation generated in a complex appetitive goal-tracking task is capable of inducing memory reconsolidation. Psychopharmacology. 2013;226:649–58.
Reichelt AC, Exton-McGuinness MT, Lee JLC. Ventral tegmental dopamine dysregulation prevents appetitive memory destabilization. J Neurosci. 2013;33:14205–10.
Drame ML, Balaet M, Lee JLC. Memory reconsolidation impairments in sign-tracking to an audiovisual compound stimulus. Behav Brain Res. 2020;393:112774.
Cogan ES, Shapses MA, Robinson TE, Tronson NC. Disrupting reconsolidation: memory erasure or blunting of emotional/motivational value? Neuropsychopharmacology. 2019;44:399–407.
Kindt M, Soeter M, Vervliet B. Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci. 2009;12:256–8.
Sanchis-Segura C, Spanagel R. Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol. 2008;11:2–38.
Dunbar AB, Taylor JR. Inhibition of protein synthesis but not β-adrenergic receptors blocks reconsolidation of a cocaine-associated cue memory. Learn Mem. 2016;23:391–8.
Sorg BA, Todd RP, Slaker M, Churchill L. Anisomycin in the medial prefrontal cortex reduces reconsolidation of cocaine-associated memories in the rat self-administration model. Neuropharmacology. 2015;92:25–33.
von der Goltz C, Vengeliene V, Bilbao A, Perreau-Lenz S, Pawlak CR, Kiefer F, et al. Cue-induced alcohol seeking behaviour is reduced by disrupting the reconsolidation of alcohol-related memories. Psychopharmacology. 2009;205:389–97.
Lee JLC, Milton AL, Everitt BJ. Cue-induced cocaine seeking and relapse are reduced by disruption of drug memory reconsolidation. J Neurosci. 2006;26:5881–7.
Qian S, Shi C, Huang S, Yang C, Luo Y. DNA methyltransferase activity in the basolateral amygdala is critical for reconsolidation of a heroin reward memory. Front Mol Neurosci. 2022;15:1002139.
Shi HS, Luo YX, Yin X, Wu HH, Xue G, Geng XH, et al. Reconsolidation of a cocaine associated memory requires DNA methyltransferase activity in the basolateral amygdala. Sci Rep. 2015;5:13327.
Monsey MS, Ruiz SG, Taylor JR. Regulation of garcinol on histone acetylation in the amygdala and on the reconsolidation of a cocaine-associated memory. Front Behav Neurosci. 2020;13:281.
Xie Y, Zhang Y, Hu T, Zhao Z, Liu Q, Li H. Inhibition of glycogen synthase kinase 3β activity in the basolateral amygdala disrupts reconsolidation and attenuates heroin relapse. Front Mol Neurosci. 2022;15:932939.
Zhang F, Huang S, Bu H, Zhou Y, Chen L, Kang Z, et al. Disrupting reconsolidation by systemic inhibition of mTOR kinase via rapamycin reduces cocaine-seeking behavior. Front Pharmacol. 2021;12:652865.
Rich MT, Abbott TB, Chung L, Gulcicek EE, Stone KL, Colangelo CM, et al. Phosphoproteomic analysis reveals a novel mechanism of CaMKIIα regulation inversely induced by cocaine memory extinction versus reconsolidation. J Neurosci. 2016;36:7613–27.
Wan X, Torregrossa MM, Sanchez H, Nairn AC, Taylor JR. Activation of exchange protein activated by cAMP in the rat basolateral amygdala impairs reconsolidation of a memory associated with self-administered cocaine. PLoS One. 2014;9:e107359.
Rich MT, Huang YH, Torregrossa MM. Calcineurin promotes neuroplastic changes in the amygdala associated with weakened cocaine-cue memories. J Neurosci. 2020;40:1344–54.
Yan Y, Newman AH, Xu M. Dopamine D1 and D3 receptors mediate reconsolidation of cocaine memories in mouse models of drug self-administration. Neuroscience. 2014;278:154–64.
Milton AL, Everitt BJ. NMDA receptors and beta-adrenergic receptors as molecular targets for the prevention of relapse to drug-seeking. Eur Neuropsychopharmacol. 2009;19:S86–S87.
Wouda JA, Diergaarde L, Riga D, Mourik Y V, Schoffelmeer AN, De Vries TJ. Disruption of long-term alcohol-related memory reconsolidation: role of β-adrenoceptors and NMDA receptors. Frontiers in. Behav Neurosci. 2010;4:179.
Chen L, Huang S, Yang C, Wu F, Zheng Q, Yan H, et al. Blockade of β-adrenergic receptors by propranolol disrupts reconsolidation of drug memory and attenuates heroin seeking. Front Pharmacol. 2021;12:686845.
Chesworth R, Corbit LH. Noradrenergic β-receptor antagonism in the basolateral amygdala impairs reconsolidation, but not extinction, of alcohol self-administration. Neurobiol Learn Mem. 2018;151:59–70.
Zheng W, Wu C, Du WJ, Li Y, Shen F, Haghparast A, et al. Differential involvement of nucleus tractus solitarius projections and locus coeruleus projections to the basolateral amygdala in morphine-associated memory destabilization. Progress in Neuropsychopharmacology &. Biol Psychiatry. 2022;115:110496.
Otis JM, Werner CT, Mueller D. Noradrenergic regulation of fear and drug-associated memory reconsolidation. Neuropsychopharmacology. 2015;40:793–803.
Haubrich J, Bernabo M, Nader K. Noradrenergic projections from the locus coeruleus to the amygdala constrain fear memory reconsolidation. eLife. 2020;9:e57010.
Wright WJ, Graziane NM, Neumann PA, Hamilton PJ, Cates HM, Fuerst L, et al. Silent synapses dictate cocaine memory destabilization and reconsolidation. Nat Neurosci. 2020;23:32–46.
Puaud M, Ossowska Z, Barnard J, Milton AL. Saccharin fading is not required for the acquisition of alcohol self-administration, and can alter the dynamics of cue-alcohol memory reconsolidation. Psychopharmacology. 2018;235:1069–82.
Vengeliene V, Olevska A, Spanagel R. Long-lasting effect of NMDA receptor antagonist memantine on ethanol-cue association and relapse. J Neurochem. 2015;135:1080–5.
Hafenbreidel M, Rafa Todd C, Mueller D. Infralimbic GluN2A-containing NMDA receptors modulate reconsolidation of cocaine self-administration memory. Neuropsychopharmacology. 2017;42:1113–25.
Lee JLC, Gardner RJ, Butler VJ, Everitt BJ. D-cycloserine potentiates the reconsolidation of cocaine-associated memories. Learn Mem. 2009;16:82–85.
Kredlow MA, Unger LD, Otto MW. Harnessing reconsolidation to weaken fear and appetitive memories: a meta-analysis of post-retrieval extinction effects. Psychological Bull. 2016;142:314–36.
Hutton-Bedbrook K, McNally GP. The promises and pitfalls of retrieval-extinction procedures in preventing relapse to drug-seeking. Front Psychiatry. 2013;4:14.
Struik RF, De Vries TJ, Peters J. Detrimental effects of a retrieval-extinction procedure on nicotine seeking, but not cocaine seeking. Front Behav Neurosci. 2019;13:243.
Hernandez PJ, Kelley AE. Long-term memory for instrumental responses does not undergo protein synthesis-dependent reconsolidation upon retrieval. Learn Mem. 2004;11:748–54.
Mierzejewski P, Korkosz A, Rogowski A, Korkosz I, Kostowski W, Scinska A. Post-session injections of a protein synthesis inhibitor, cycloheximide do not alter saccharin self-administration. Prog Neuro-Psychopharmacol Biol Psychiatry. 2009;33:286–9.
Piva A, Gerace E, Di Chio M, Osanni L, Padovani L, Caffino L, et al. The metaplastic effects of NMDA receptors blockade on reactivation of instrumental memories in rats. Neurobiol Learn Mem. 2018;154:87–96.
Tedesco V, Mutti A, Auber A, Chiamulera C. Nicotine-seeking reinstatement is reduced by inhibition of instrumental memory reconsolidation. Behav Pharmacol. 2014;25:725–31.
Cahill EN, Vousden GH, Exton-McGuinness MT, Beh IR, Swerner CB, Macak M, et al. Knockdown of zif268 in the posterior dorsolateral striatum does not enduringly disrupt a response memory of a rewarded T-maze task. Neuroscience. 2018;370:112–20.
Piva A, Pintori N, Padovani L, Chiamulera C. Protocols for instrumental memory reconsolidation in rodents: a methodological review. J Neurosci Methods. 2020;342:108766.
Piva A, Gerace E, Di Chio M, Padovani L, Paolone G, Pellegrini-Giampietro DE, et al. Reconsolidation of sucrose instrumental memory in rats: the role of retrieval context. Brain Res. 2019;1714:193–201.
Cheng C, Exton-McGuinness MTJ, Lee JLC. Procedures between training and reactivation influence the destabilization of instrumental sucrose memory. Front Behav Neurosci. 2022;16:953629.
Gershman SJ, Monfils MH, Norman KA, Niv Y. The computational nature of memory modification. eLife. 2017;6:e23763.
Yue JL, Yuan K, Bao YP, Meng SQ, Shi L, Fang Q, et al. The effect of a methadone-initiated memory reconsolidation updating procedure in opioid use disorder: a translational study. eBioMedicine. 2022;85:104283.
Becker JE, Price JL, Leonard D, Suris A, Kandil E, Shaw M, et al. The efficacy of lidocaine in disrupting cocaine cue-induced memory reconsolidation. Drug Alcohol Depend. 2020;212:108062.
Das RK, Gale G, Walsh K, Hennessy VE, Iskandar G, Mordecai LA, et al. Ketamine can reduce harmful drinking by pharmacologically rewriting drinking memories. Nat Commun. 2019;10:5187.
Das RK, Walsh K, Hannaford J, Lazzarino AI, Kamboj SK. Nitrous oxide may interfere with the reconsolidation of drinking memories in hazardous drinkers in a prediction-error-dependent manner. Eur Neuropsychopharmacol. 2018;28:828–40.
Das RK, Hindocha C, Freeman TP, Lazzarino AI, Curran HV, Kamboj SK. Assessing the translational feasibility of pharmacological drug memory reconsolidation blockade with memantine in quitting smokers. Psychopharmacology. 2015;232:3363–74.
Glasgow NG, Povysheva N V, Azofeifa AM, Johnson JW. Memantine and ketamine differentially alter NMDA receptor desensitization. J Neurosci. 2017;37:9686–704.
Pachas GN, Gilman J, Orr SP, Hoeppner B, Carlini SV, Loebl T, et al. Single dose propranolol does not affect physiologic or emotional reactivity to smoking cues. Psychopharmacology. 2015;232:1619–28.
Murray JE, Bevins RA. Acquired appetitive responding to intravenous nicotine reflects a pavlovian conditioned association. Behav Neurosci. 2009;123:97–108.
Naqvi NH, Bechara A. The insula and drug addiction: an interoceptive view of pleasure, urges, and decision-making. Brain Struct Funct. 2010;214:435–50.
Saladin ME, Gray KM, McRae-Clark AL, LaRowe SD, Yeatts SD, Baker NL, et al. A double blind, placebo-controlled study of the effects of post-retrieval propranolol on reconsolidation of memory for craving and cue reactivity in cocaine dependent humans. Psychopharmacology. 2013;226:721–37.
Lonergan M, Saumier D, Tremblay J, Kieffer B, Brown TG, Brunet A. Reactivating addiction-related memories under propranolol to reduce craving: a pilot randomized controlled trial. J Behav Ther Exp Psychiatry. 2016;50:245–9.
Germeroth LJ, Carpenter MJ, Baker NL, Froeliger B, LaRowe SD, Saladin ME. Effect of a brief memory updating intervention on smoking behavior: a randomized clinical trial. JAMA Psychiatry. 2017;74:214–23.
Barnabe A, Gamache K, de Camargo JVP, Allen-Flanagan E, Rioux M, Pruessner J, et al. A novel stress-based intervention reduces cigarette use in non-treatment seeking smokers. Neuropsychopharmacology. 2023;48:308–16.
Meir Drexler S, Merz CJ, Wolf OT. Preextinction stress prevents context-related renewal of fear. Behav Ther. 2018;49:1008–19.
Bradley, MM & Lang, PJ The International Affective Picture System (IAPS) in the study of emotion and attention. In Handbook of emotion elicitation and assessment (eds JA Coan & JJB Allen), 29-46 (Oxford University Press, 2007).
Das RK, Lawn W, Kamboj SK. Rewriting the valuation and salience of alcohol-related stimuli via memory reconsolidation. Transl Psychiatry. 2015;5:e645.
Gale G, Walsh K, Hennessy VE, Stemerding LE, Ni KS, Thomas E, et al. Long-term behavioural rewriting of maladaptive drinking memories via reconsolidation-update mechanisms. Psychol Med. 2021;51:2875–85.
Hon T, Das RK, Kamboj SK. The effects of cognitive reappraisal following retrieval-procedures designed to destabilize alcohol memories in high-risk drinkers. Psychopharmacology. 2016;233:851–61.
Lane RD, Ryan L, Nadel L, Greenberg L. Memory reconsolidation, emotional arousal, and the process of change in psychotherapy: new insights from brain science. Behav Brain Sci. 2015;38:e1.
Marsden J, Goetz C, Meynen T, Mitcheson L, Stillwell G, Eastwood B, et al. Memory-Focused Cognitive Therapy for Cocaine Use Disorder: theory, procedures and preliminary evidence from an external pilot randomised controlled trial. EBioMedicine. 2018;29:177–89.
Goetz C, Meynen T, Mitcheson L, Grey N, Eastwood B, Strang J, et al. Does craving for cocaine mediate cocaine use? Analysis of a randomized controlled pilot trial of memory-focused cognitive therapy. J Exp Psychopathol. 2019;10:1–14.
Bui, UTD & Milton, AL. Making leaps and hitting boundaries in reconsolidation: overcoming boundary conditions to increase clinical translatability of reconsolidation-based therapies. Neuroscience. 2023;519:198–206.
Exton-McGuinness MTJ, Drame ML, Flavell CR, Lee JLC. On the resistance to relapse to cocaine-seeking following impairment of instrumental cocaine memory reconsolidation. Front Behav Neurosci. 2019;13:242.
Acknowledgements
ALM’s research is supported by UKRI (MR/N02530X/1 and BB/W001195/1). ALM is the Ferreras-Willetts Fellow at Downing College, University of Cambridge. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
ALM was responsible for the manuscript conceptualisation, drafting, critical editing, figure visualisation and acquisition of funding.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Milton, A.L. Drug memory reconsolidation: from molecular mechanisms to the clinical context. Transl Psychiatry 13, 370 (2023). https://doi.org/10.1038/s41398-023-02666-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-023-02666-1
