
Abstract
Cognitive impairment is a predictor of disability across different neuropsychiatric conditions, and cognitive abilities are also strongly related to educational attainment and indices of life success in the general population. Previous attempts at drug development for cognitive enhancement have commonly attempted to remedy defects in transmitters systems putatively associated with the conditions of interest such as the glutamate system in schizophrenia. Recent studies of the genomics of cognitive performance have suggested influences that are common in the general population and in different neuropsychiatric conditions. Thus, it seems possible that transmitter systems that are implicated for cognition across neuropsychiatric conditions and the general population would be a viable treatment target. We review the scientific data on cognition and the muscarinic cholinergic receptor system (M1 and M4) across different diagnoses, in aging, and in the general population. We suggest that there is evidence suggesting potential beneficial impacts of stimulation of critical muscarinic receptors for the enhancement of cognition in a broad manner, as well as the treatment of psychotic symptoms. Recent developments make stimulation of the M1 receptor more tolerable, and we identify the potential benefits of M1 and M4 receptor stimulation as a trans-diagnostic treatment model.
Similar content being viewed by others
Introduction
In 2016, schizophrenia was determined to be the 12th most disabling disorder globally in terms of healthy years of life lost [1]. Schizophrenia poses tremendous societal costs, including direct costs for treatment and drugs, but also indirect costs such as lost productivity, mortality, family impact, and criminal justice system costs; while affecting 0.7% of the population, schizophrenia typically accounts for 1.5–3% of all nations’ total health care expenditures [2]. Previous studies have reported that, as schizophrenia significantly impairs the ability of patients to perform social, vocational, and everyday living tasks [3], more than 70% of people with schizophrenia are challenged with living independently and finding employment [4, 5]. Despite the significant healthcare and societal resources dedicated to schizophrenia, adult-onset schizophrenia has only a 13.5% recovery rate [6], with recovery rates even lower for childhood onset.
While schizophrenia is defined diagnostically by positive symptoms such as hallucinations and delusions, cognitive deficits are much greater determinants of long-term disability and poor functional prognosis [7, 8]. Despite the considerable impact of neurocognitive deficits on function, current treatments for schizophrenia do not adequately address these dimensions. Given the magnitude of disability caused by the cognitive impacts of schizophrenia, treatments that may address these domains should be pursued. Accordingly, other targets beyond the dopamine antagonism used to treat psychosis seem critical to pursue. There are multiple other transmitter systems, as well as elements of brain structure and function, that may determine cognitive and functional impairments. Efforts to target these systems have also not been successful, for reasons addressed by the authors of those earlier reviews. One reason for concern, as we describe below, is that pharmacological treatment efforts aimed at cognition in schizophrenia have typically been aimed at neurotransmitter systems that have been implicated as having abnormalities in schizophrenia, generally related to the origin of psychotic symptoms. However, considerable genomic evidence from very large-scale Genome Wide Association studies (GWAS) has suggested that cognitive deficits in schizophrenia (and other serious mental illnesses) share genomic variance with polygenic scores (PS) for cognition (as well as intelligence and educational attainment) in the general population, possibly more than with PS for schizophrenia [9]. Also, PS for cognition in other serious mental illnesses, such as bipolar illness, shares variance with PS for cognition in the general population and in schizophrenia [10]. Thus, impairments in cognition in serious mental illness (SMI) may be best conceptualized as originating from factors that are operative in the general population in addition to disease specific processes across neuropsychiatric conditions.
These findings implicate several possible new strategies for cognitive enhancement on a pharmacological level in serious mental illness. If the genomics of cognition is similar in SMI and the general population, why should pharmacological treatment targets be diagnosis specific? Cognitive impairment seems to be more considerably more similar across DSM-based entities than symptom severity, questioning the need to consider cognitive impairments differentially across diagnostic entities. Cognitive impairments are modestly correlated with symptom severity on both cross-sectional and longitudinal bases in schizophrenia and bipolar disorder, as evidenced by the persistence of cognitive impairments in individuals with both diagnoses who are clinically stable [e.g [11, 12]]. The correlation between symptomatic status in bipolar disorder and cognition appears modest based on multiple studies of impairments in euthymic participants with bipolar illness and their generally unaffected relatives [13].
Finally, large scale studies of the similarity of cognitive deficits in these two conditions are beginning to appear. In the largest study to date focusing on this issue (n = 10,160), we [14] found that analyses of the factor structure of cognition across diagnoses manifested the best fit to the data when characterized as a single domain. These findings were substantiated by the results of genomic analyses like those described above [15], which found that a common latent trait for cognition across the two diagnoses was related to previous PS for cognition, intelligence, and educational attainment in general population samples [16,17,18]. There was no evidence for diagnosis specific profiles of cognitive impairment or differential genomic correlates of cognitive profiles across diagnoses, with greater differences in PS correlates based on racial status than on diagnosis. Multiple studies have suggested that cognition in schizophrenia is multifactorial, but the larger the study, the more likely that a unifactorial solution will be found. For example, in a large-scale study of cognition in participants with schizophrenia, bipolar disorder, and their unaffected relatives, as well as healthy controls (n = 2066) [19], cognitive performance measured with the Brief Assessment of Cognition in Schizophrenia (BACS) [20] was best defined as a unitary latent trait across all the different participant groups. A unifactorial solution based on neuropsychological tests does not mean that cognition has a single origin, but the similarity of findings of cognitive deficits on a cross diagnostic basis (using the tests that are strongly correlated with functional disability) suggests that cross-diagnostic therapeutic efforts may be equally as reasonable as diagnosis specific studies.
As a result, it may be important to consider treatment targets that may be relevant to cognition across psychiatric and neuropsychiatric conditions, as well as in the general population. There are several reasons that this may be important, as reviewed above. First, the results of genomic studies and large-scale assessment-based studies suggest that the differences in performance across neuropsychiatric conditions are generally limited to differences in the severity of impairment that could be related to differences in premorbid functioning. Cognitive performance across diagnoses has similar correlational structures, and similar genomic correlates. Second, it is widely understood that symptomatic variables (psychosis, current mood state, negative symptoms) in schizophrenia and bipolar disorder (and to an extent in major depression [21]) are minimally correlated with concurrent cognitive impairment. Thus, for the rest of this paper we will review the importance of a neurotransmitter system where evidence has suggested that there is potential cross-diagnostic importance in cognition: the muscarinic cholinergic receptor system, with discussion of receptor subtypes and comparison with the nicotinic cholinergic system.
Aside from SMI, there are other diagnosed conditions where cognitive impairments are central features of the illness, including Alzheimer’s disease (AD) and related amnestic mild cognitive impairment (aMCI). Interesting and convincing evidence that AD was associated with reduced muscarinic cholinergic functioning [22] lead to the pursuit of treatments targeting this mechanism. Both muscarinic M1 agonists [23] and acetylcholinesterase inhibitors (ACHI) [24,25,26] were explored beginning two or more decades ago as treatments for cognitive impairments in AD and aMCI. These same treatments were also explored in healthy aging populations, albeit in small sample studies [27], focusing on improvements in episodic memory. Thus, it could be argued that the muscarinic cholinergic transmitter systems are relevant to cognitive functioning across diagnoses and that interventions aimed at improving the functioning of the system can lead to benefits in the domains targeted.
There is considerable evidence of the importance of the muscarinic cholinergic system in schizophrenia [28]. Muscarinic agonists were discovered to have antipsychotic-like activity in animal models [29], although it is not clear if this is due to M1 or M4 agonist effects [30]. Cognitive functioning in schizophrenia, like in AD, was reported to be improved by treatment with the M1/M4 agonist xanomeline [31]. Thus, these data suggest that manipulations affecting other neurotransmitter systems, not typically targeted in the treatment of schizophrenia, may exert beneficial effects on cognition across apparently very different (neurodegenerative vs. not; aging related vs. not) classes of neuropsychiatric conditions.
This is not a new concept. When amphetamine treatments were introduced for attention deficit disorders, younger and older healthy comparison participants commonly manifested the same degree of improvements in performance observed in the ADHD treatment samples [32]. Evidence from multiple studies has suggested that stimulant administration, at the correct doses, even in unimpaired samples, leads to improvements in test performance [33]. Thus, a baseline deficit in monoamine functioning is not required to detect an improvement in cognitive performance after dosing with an augmenting agent. Further, cognitive test performance [34] and response to cognitive training [35] are improved in participants with schizophrenia when treated with stimulants, despite concurrent treatment with antipsychotic medications. Thus, it is entirely possible that cross-diagnostic facilitation of cognition can be achieved with a single treatment.
Xanomeline, an M1/M4 muscarinic receptor agonist, has sparked renewed interest in its utility as a possible treatment for schizophrenia. Compared to dopamine antagonists, which rely on the dopamine D2 receptor blockade and preferentially treat positive symptoms, muscarinic agonists such as xanomeline may offer a different level of transmitter interaction in terms of reduction of psychosis. They also offer potential to address cognitive symptomology in both AD and schizophrenia [36]. While xanomeline was initially investigated in the 1990s, trials were discontinued due to intolerable peripheral cholinergic side effects. In a new formulation, xanomeline is combined with trospium chloride, a pan-muscarinic receptor antagonist that does not cross the blood-brain barrier, thus limiting the effects of xanomeline to the central nervous system (CNS) and potentially improving its side effect profile [37]. We will next examine the neuroscience underpinnings of this general treatment strategy and evaluate the role of agents affecting both M1 receptors and M4 receptors in the treatment of psychosis and other neuropsychiatric pathologies.
Cholinergic systems
Acetylcholine was the first neurotransmitter discovered. In the CNS, the cholinergic system has been implicated in numerous processes including sensory perception, motor function, cognitive processing, memory, attention, mood, and psychosis [38, 39]. Of note, the cholinergic system is believed to be the primary regulator of learning, memory, and attention functions [40], in both human [41] and animal models [42]. M1 receptor blockade produces reliable, dose and time course dependent, cognitive impairments in healthy people, younger and older [43], which can be reversed by agents that stimulate the cholinergic system [44]. Drugs that enhance cholinergic functioning produce cognitive improvements in healthy people who were selected for the absence of any cognitive impairments [27]. Accordingly, degeneration of the central cholinergic system is considered a driving force of Alzheimer’s dementia (AD) and even normal age-related memory changes [45]. Similarly, cholinergic functioning has been implicated in the pathophysiology of reduced cognition in schizophrenia, as studies have shown that neurocognition decreases in patients with schizophrenia after exposure to anticholinergic medications [46,47,48]. This association is particularly problematic given that some of the first and second-generation antipsychotics, the standard treatment for schizophrenia, have anticholinergic properties [49]. More imoprtantly, some of these medications cause extra-pyramidal symptoms, which are themselves treated with anticholinergic medications. Thus, current treatments for schizophrenia may worsen neurocognitive dysfunction through increasing the need for anticholinergic treatments.
In the periphery, the cholinergic system is responsible for the parasympathetic nervous system and regulates various functions including gastrointestinal motility, sweat production, smooth muscle activity, and heart rate. Accordingly, pharmaceuticals that act as cholinergic agonists in the periphery are prone to cause related side effects, including diarrhea, excessive sweating and salivation, and bradycardia. While muscarinic agonists such as xanomeline have shown promise in improving cognition through CNS cholinergic activation, dosing has ultimately been limited due to these peripheral side effects [50].
The cholinergic system contains two families of acetylcholine receptors: muscarinic receptors and nicotinic receptors. Nicotinic receptors are ligand-gated ion channels composed of 5 subunits; when acetylcholine binds to the receptor, an influx of sodium ions causes a rapid response [51]. Interest in development of nicotinic agents was spurred by the high prevalence of smoking on the part of people with schizophrenia and suggestions of alterations of genomic alterations associated with nicotinic cholinergic receptors [52, 53]. Proof of concept studies showed that nicotinic alpha-7 agonists could exert some benefits on cognition in schizophrenia [54], but a phase 2 trial was unsuccessful [55]. There appear to be considerable differences across these compounds in their receptor affinities and binding durations and other compounds such as EVP-6124 (encenicline), which targets the alpha-7 nicotinic receptor and showed preliminary promise in improving cognitive function in patients with schizophrenia and AD [56, 57], but did not separate from placebo in a phase 3 clinical trial. The development program was stopped and possible beneficial features of encenicline on psychotic symptoms coming from basic science studies have not been tested in humans [58]. Another alpha-7 nicotinic partial agonist was tested in phase 2B studies and did not separate from placebo in either smoking or nonsmoking participants with schizophrenia [59, 60] with the fact remaining that no alpha-7 nicotinic receptor has led to benefits on cognition that separated from placebo in an FDA-endorsed phase 3 clinical trial.
In contrast to nicotinic receptors, muscarinic receptors are G-protein coupled receptors which impact intracellular messengers and thus have a less rapid onset but longer lasting effect on cholinergic transmission than stimulation of nicotinic receptors [61]. 5 different subtypes of muscarinic receptors have been identified, and all have been found in the CNS at varying concentrations [62, 63]. Studies conducted with muscarinic receptor knockout mice suggest that the M1 receptor is primarily involved in neurotransmission for learning and memory, while the M4 receptor is largely involved in dopamine regulation [64]. M2 and M3 receptors, while sparingly present in the CNS, appear to primarily control the peripheral parasympathetic nervous system, with large concentrations in the heart and gastrointestinal smooth muscle [65]. Finally, M5 receptors in the periphery appear responsible for dilation of cerebral arteries, while their role in the CNS is less clear [62]. Accordingly, M1 and M4 have become targets for the treatment of psychosis and cognition in schizophrenia and Alzheimer’s disease; however, concurrent activation of M2 and M3 must be avoided to prevent peripheral side effects. Of note, while M1 is primarily expressed in the CNS, it is also expressed in lesser quantities in the periphery, which means that M1 activation may also result in peripheral effects.
M4 receptors and dopamine regulation
M4 is expressed preferentially in the substantia nigra, with lesser expression in the cortex and hippocampus [63]. M4 knockout mice exhibit hyperdopaminergic activity [65, 66], suggesting that M4 is responsible for downregulating dopamine release. It has been proposed that activation of the M4 muscarinic receptor inhibits dopamine release through cholinergic projections to the substantia nigra [67, 68]. This mechanism is supported by findings that M4 agonists can reverse D1 agonist-induced hyperlocomotion [69]. That same study reported that M4 agonism may concurrently enhance dopamine release in the hippocampus and cortex, thus increasing neurocognitive functioning. Importantly, while M4 agonists prevent D1 activation in the substantia nigra, they do not inhibit D1 function in the cortex or hippocampus, suggesting that M4 agonists may be capable of selectively inhibiting dopamine release in a way that dopamine antagonists, or antipsychotics, are not.
Prior imaging studies have found that patients with schizophrenia have reduced M4 receptors in the caudate-putamen and hippocampus [70, 71], which may correlate with the presence of both the positive and negative symptoms of schizophrenia. Further, recent studies report that M4 gene receptor polymorphisms are associated with schizophrenia [70, 72], thus further implicating M4 disfunction in the pathogenesis of schizophrenia. A recent study found that M4 agonism cannot inhibit dopamine release without coactivation of the mGlu1 receptor; [73] this finding is supported by data indicating that loss of function mutations in the gene encoding mGlu1 receptor are associated with schizophrenia [74, 75]. Thus, mutations in genes encoding both M4 and mGlu1could increase the risk of schizophrenia through the same pathway.
Medications that augment the M4 receptor have shown efficacy as antipsychotics, likely by downregulating dopamine release. VU0152100, an M4 positive allosteric modulator, has been shown to treat both psychotic symptoms and cognitive disturbances in rodent models of schizophrenia [76]. The same study also revealed that VU0152100, which acts at only M4, was able to reverse amphetamine-induced cognitive deficits, but did not improve cognition from baseline. A very recent phase I study in schizophrenia patients [77], tested the M4 allosteric modulator emraclidine, reporting suitable tolerability across a wide dose range. This study is being followed with placebo-controlled efficacy studies targeting psychotic symptoms with a monotherapy approach [78].
Xanomeline, a muscarinic agonist, is relatively selective for M1 and M4 receptors. Xanomeline has been shown to increase dopamine release in the medial prefrontal cortex [79], where dopamine release is typically lessened in patients with schizophrenia, which may be implicated in the origin of negative symptomology [80]. Xanomeline has also been shown to decrease dopamine release in the midbrain [30], where hyperdopaminergic activity has been correlated with schizophrenia [81]. A 2004 rodent student found that M4-knockout mice have decreased acetylcholine efflux and increased dopamine efflux in the midbrain, thus causing a hyperexcitable dopaminergic state [67]. In turn, by acting as an agonist at M4, xanomeline may be able to increase acetylcholine influx and decrease dopamine efflux in the midbrain, thus causing dopamine to remain intracellularly with reduced availability for neurotransmission. The mechanism by which xanomeline may increase dopamine release in the medial prefrontal cortex is less clear.
M1 receptors and cognition
Some studies have reported that patients with schizophrenia have reduced levels of M1 receptors in the dorsolateral prefrontal cortex [82, 83]. Other studies have also shown M1 receptor density in the striatum and hippocampus of patients with schizophrenia to be comparable to that of healthy controls [84] and postmortem studies have generally found that levels of cholinergic markers are within normal limits, even in very old samples [85]. When an M1 receptor (a G-protein coupled receptor) is activated by acetylcholine, phospholipase C is activated by the associated G-protein and intracellular calcium stores are released [86]. M1 has been primarily implicated in cognitive processes, as M1 receptors are able to potentiate N-methyl-D-aspartate (NMDA) signaling [87, 88], which is critical to synaptic plasticity and, accordingly, learning and memory. M1 knockout mice display decreased cognitive function and increased amyloid formation [89,90,91,92], suggesting that M1 dysfunction may play a two-factor role in cognitive decline in AD. Studies conducted in animal models have shown that M1 activation can mediate amyloid processing, thus decreasing amyloid formation and tau hyperphosphorylation [93, 94]. These findings suggest that agents acting at the M1 receptor may be able to act in a disease-modifying role for dementia processes.
The cholinergic hypothesis of AD states that loss of memory function occurs as the result of a progressive decrease in density of basal forebrain cholinergic neurons to the hippocampus and cortex [95]. Accordingly, acetylcholinesterase inhibitors, which increase the availability of acetylcholine in synapses, are used to alleviate symptoms in the dementia phase of AD by augmenting cholinergic function [96]. Additionally, several M1 agonists have been successful in increasing cognition in AD patients [97], but usage has been limited by concomitant activation of M2 and M3 receptors resulting in intolerable side effects or by the need to administer medication intravenously (e.g., physostigmine).
Interestingly, while acetylcholinesterase inhibitors can increase cognition and memory modestly in AD, they have been consistently unsuccessful in treating schizophrenia-related cognitive impairment [98, 99]. However, drugs that agonize M1, such as xanomeline, have yielded improvements in cognition in rodent models of schizophrenia [100] and in recent human studies [101]. This discrepancy may suggest minimal alterations in sensitivity of M1 cholinergic receptors in patients with schizophrenia, as increasing availability of acetylcholine in the synapse does not improve cognition but stimulating the M1 receptor through agonist activity (or potentially allosteric modulation) appears to have benefits. The findings regarding normal levels of M1 receptor density in schizophrenia may suggest that M1 agonists may improve cognitive functioning even if the origin of cognitive dysfunction is not due to pathological changes in M1 receptors. A parallel to this possibility is the findings studies of stimulant treatment in schizophrenia, whereby treatment with amphetamines improves cognition and response to cognitive training even in the presence of dopamine D2 blockade.
Alternative strategies for M1 regulation include allosteric modulators. Such compounds and strategies have been shown to reverse memory loss and slow progression in mouse models of prion disease [102]. These compounds have been reported to have reduced side effect potential, particularly peripheral effects, compared to M1 agonists [103]. Thus, this strategy has the potential for cognitive enhancement with reduced off-target side effects [104].
Peripheral effects of muscarinic agonism
While M1 is preferentially expressed in the CNS, it is also present in gastric and salivary glands and enteric ganglia [105]. Accordingly, while M1 agonism can improve cognition, it can also cause peripheral side effects such as increased secretions and increased gastric motility. In a randomized control trial of xanomeline in AD patients [23], more than half of patients receiving high-dose xanomeline (225 mg per day) discontinued use due to side effects, including excessive sweating, nausea, vomiting, increased salivation, diarrhea, and fecal incontinence. Of particular concern, 12.6% of the high-dose patients suffered syncope while taking the medication.
While xanomeline has been shown to be functionally selective for M1 and M4 receptors, it is capable of binding to all muscarinic receptors, including M2 and M3 [106, 107]. M2 is largely expressed in the heart and smooth muscle, and M2 agonism can result in bradycardia and increased gastrointestinal motility [108]. Similarly, M3 is widely expressed in smooth muscle and glands; M3 agonism can result in increased salivation, urinary urgency, and incontinence, and increased gastrointestinal motility [109]. As discussed above, M1, while preferentially expressed in the CNS, is also present in salivary glands and the enteric ganglion. As xanomeline functionally selects for M1 and M4 but may have some off-target activity at M2 and M3, it is plausible that the intolerable side effects of xanomeline are primarily caused by peripheral M1 agonism in the salivary glands and enteric ganglia but may also result from lesser agonism of M2 and M3.
Xanomeline-trospium
Trospium chloride is a pan-muscarinic antagonist; its structure contains a tertiary amine, rendering it highly polar and unable to cross the blood brain barrier and enter the CNS [110]. It is approved for used for the treatment of overactive bladder. In a new formulation, xanomeline is combined with trospium with the intent of blocking xanomeline’s peripheral agonism to minimize side effects. A recent phase 2 trial of xanomeline-trospium found that, by adding trospium, the side effects of xanomeline are drastically reduced, with 20% of patients receiving xanomeline-trospium discontinuing the drug compared to 21% of patients receiving placebo [37]. 54% of xanomeline-trospium patients reported adverse events, as compared with 43% of placebo patients. Interestingly, two of the most reported adverse events in the xanomeline-trospium group were constipation and dry mouth, which is suggestive of a highly effective peripheral muscarinic blockade, as the side effects of xanomeline alone include diarrhea and excessive salivation. Importantly, while the earlier trial of xanomeline alone in patients with AD reported a high incidence of syncope [23], there were no episodes of syncope reported in patients taking xanomeline-trospium.
While combining xanomeline with trospium appears to lower rates of cholinergic side effects, xanomeline remains effective for schizophrenia in this combination. In the same phase 2 trial, patients taking xanomeline-trospium received a significant benefit over placebo on scales evaluating both positive and negative symptomatology. Patients in the xanomeline-trospium group also showed significant benefit over placebo on the Clinical Global Impression-Severity scale (CGI-S) [111], which is intended to serve as a measure of overall clinical status. Accordingly, xanomeline-trospium shows potential as a novel treatment for schizophrenia with a greatly improved side effect profile over xanomeline alone.
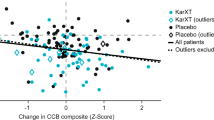
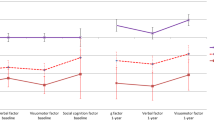
Importantly for the arguments presented in this paper, a recent publication based on data from the same phase 2 study reported statistically significant cognitive benefits from xanomeline-trospium compared to placebo [101]. After excluding participants who had invalid baseline scores on the computerized cognitive battery, participants receiving active treatment improved by approximately 0.3 SD more than the participants receiving placebo. This is a level of improvement consistent with that seen in previous phase 2 studies of other cognitive enhancing agents. Importantly, when participants whose cognitive performance was in the normal range were censored from the analysis, the separation between active and placebo treatment increased to 0.71 SD.
Increasing interest in adverse impact of anticholinergic medications?
Several very recent studies have revisited the impact of anticholinergic burden with a focus on serious mental illness. For instance, Joshi et al. [112] reported on 1120 participants with schizophrenia or schizoaffective disorder who participated in a large-scale genomic study, COGS-2 [113]. The authors reported that all cognitive domains were affected by current anticholinergic burden and that demographic factors were not a contributor. In two related studies, Eum et al. [114] reported that adverse impacts of anticholinergic burden were preferentially detected in participants with schizophrenia, and not in schizoaffective or bipolar participants. In a GWAS follow-up of this sample [115], the authors reported that they identified five variants that were significantly associated with global cognitive performance, with these variants located at the chromosome 3p21.1 locus, with the top SNP in the inter-alpha-trypsin inhibitor heavy chain 1 (ITIH1) gene (P = 3.25 × E-9). The inclusion of anticholinergic burden as a predictor improved association models between the loci and cognitive performance (P < 0.001) and additional significant SNPs were identified. This genomic locus (3p21.1) was previously reported to be associated with both risk for psychosis and with cognitive performance in the general population [see Davies et al., 17]. Further, this association was replicated by the same investigators in a largely treatment naïve first episode sample, suggesting that the findings were not related to prior medication exposure. Later research could easily be directed at genomic correlates of adverse response to anticholinergics examining potentially moderating effects of genomic factors with greater adverse impacts of anticholinergics.
Other potential uses for muscarinic agonists: broader applicability?
Given the findings that M1 and M4 muscarinic receptors have different functions, treatments that target these receptors may have several different uses. While previous studies have shown that xanomeline had efficacy for cognition and psychosis in both AD and schizophrenia, there are other conditions that share cognitive impairments. Further, for individuals whose psychotic symptoms are refractory to antipsychotic treatments, alternative strategies for down-regulation of dopamine activity could be considered as an intervention. These uses can include cognitive targets, as well as symptomatic targets that may be related to excessive striatal dopaminergic D2 activity. Although there are clear interactions with muscarinic agonists and neurotransmitter systems implicated more clearly in schizophrenia than other conditions (e.g., NMDA, D2), the impacts of these compounds may be more broadly applicable, even in the absence of a very strong signal for abnormal activity of dopamine or glutamate.
Age-related cognitive changes
It has been found since the advent of standardized cognitive testing that there are normative age-related changes in cognitive functioning. These changes are greatest for what are referred to as fluid cognitive abilities, including the ability to process novel information, solve new problems, as well as concentration, attention, and processing speed [116]. Even in individuals with no evidence of gross changes in cognitive performance, some abilities are performed at levels 30–50% less efficient in the ninth decade of life than in the third. These normative differences are larger in individuals with lower levels of performance, such that individuals whose performance at age 25 was at the 50th percentile have relatively greater differences in their performance on indices of fluid cognition across the lifespan than individuals with higher levels of performance.
Crystallized knowledge includes vocabulary and other fact-based information. These indices do not change with age and may increase slightly over time within individuals [117]. Thus, the situation where individuals notice that they do not seem as “sharp” with aging is associated with objectively reduced performance in certain domains compared to earlier years and an increased risk of non-normative cognitive decline even in the absence of a diagnosable condition [118]. The data reviewed above regarding the potential efficacy of M1 agents for treatment of cognitive impairment in AD would likely also be applicable to the treatment of mild cognitive impairment (MCI), which in many cases is an early stage of AD. The advent of PET scan-based and blood-based AD biomarkers has suggested that MCI is commonly associated with amyloid deposition and this deposition is a risk factor for increased conversion to dementia [119]. Further, reductions in M1 activity are also shown to correlate with increased deposition of amyloid [120], suggesting that early treatment with M1 agonists may have the potential to improve cognitive functioning and to have a potentially disease modifying influence.
A subset of aging individuals reports more substantial cognitive declines, referred to as “subjective cognitive declines”. The presence of subjective memory complaints, even in the context of unimpaired performance on traditional screening measures, has been reported to be correlated with development of dementia at a 3-year follow-up interval [121]. In that study, participants with memory complaints and unimpaired Mini-Mental State Exam (MMSE) scores were 2.7 times as likely to be diagnosed with AD at the follow-up compared to the unimpaired cases without memory complaints. These individuals can be detected with validated rating scales, such as the Alzheimer’s Disease Cooperative Study Prevention Instrument Project – Mail-In Cognitive Function Screening Instrument (ADCS-MCFSI). This rating scale assesses subjective cognitive decline across domains that are relevant to the early stages of MCI. Recent studies have suggested that individuals identified on this rating scale as having definite subjective cognitive decline were more impaired on an array of cognitive measures and performance assessments of everyday functional skills [122].
The subjective cognitive decline population has been targeted for pharmacological and cognitive training interventions. For instance, a recent trial randomized participants with subjective cognitive decline (limited to 1.0 SD below normative standards on structured testing) to computerized cognitive training combined with either the novel antidepressant medication vortioxetine or placebo [123]. The authors reported that the combined therapy was more effective at improving targeted assessment of fluid cognition than monotherapy with cognitive training. As the objective performance changes seen in subjective cognitive decline are consistent with both the early signs of mild cognitive impairment and are responsive to treatment with plasticity-focused interventions, these conditions might be an additional target for treatment with M1 agonists. A further use of these M1 agonists might be as a safer strategy for pharmacologically augmented cognitive training (PACT [124]) than the stimulant strategies described above for schizophrenia.
Partial treatment response in schizophrenia
Although many patients with schizophrenia have a good response to antipsychotic treatment and achieve remission, there are a proportion of cases who manifest a partial or negligible response even with confirmed adherence [125]. The presence of residual symptoms is commonplace, even when clozapine is prescribed; these cases are designated as ultra-treatment resistant [126]. Even in cases who are adherent to long-acting antipsychotic treatment, rates of partial response (defined as failure to achieve remission) can approximate 60% [127]. Further, relapse can occur even during confirmed continuous treatment with long-acting injectable medications, with a recent study suggesting a relapse rate slightly higher than 20% per year [128]. The relapse rate was only 15% in cases who had achieved full remission.
These data suggest a need to develop additional strategies to address failures to achieve or sustain remission in schizophrenia. The data reviewed above regarding direct downregulation of dopaminergic activity in critical subcortical areas suggests that M4 agonists may have the potential to provide an additive benefit in terms of dopamine regulation compared to D2 antagonists. Further, the finding that dopamine release in the cortex in critical regions relevant to cognition is possibly even upregulated by M4 agonists suggests that this may be an additional beneficial strategy. Other compounds can downregulate dopamine activity without direct dopamine antagonism, such as trace amine-associated receptor 1 (TAAR1) antagonists that also appear to operate by downregulating dopaminergic activity in critical brain regions, such as the ventral tegmental area [129]. Alternative strategies to downregulate dopaminergic activity without D2 antagonism may also reduce risks for side effects such as extrapyramidal symptoms (EPS) and tardive dyskinesia (TD) while offering incremental benefits for control of psychotic symptoms.
Conclusions
In this paper we argue that there are alternatives to attempts to reduce cognitive deficits through development of disease specific pharmacological interventions. Cognitive functioning in serious mental illness has considerable cross-diagnostic overlap and considerable genomic overlap with cognition, intelligence, and educational attainment in the general population. Thus, identification of pharmacological targets that have a broad influence on cognition action across the general and pathological populations appears to be a viable strategy. The muscarinic cholinergic system appears to be one such candidate. This system has attracted attention for its role in aging related cognitive changes, in the development of dementia, and in schizophrenia. The muscarinic system interacts with several different transmitters that are also affected in schizophrenia, including dopamine and glutamate, but its importance clearly transcends a single condition.
Previous intervention attempts targeting this system were handicapped by fact that peripheral activity of acetylcholine includes gastric and cardiac functions which were notably impacted by M1 agonists with both peripheral and central effects. New developments in medicinal chemistry have led to the development of a compound with an anticholinergic compound that does not enter the CNS and preliminary results suggest increased safety. Further developments include focusing on the development of positive allosteric modulators that may have reduced off target effects in the periphery. The muscarinic system also regulates dopamine at certain receptor subtypes and compounds are in development to target both M1 and M4 receptors to improve cognition and reduce dopamine activity respectably.
Targeting the muscarinic system has some parallels to the use of stimulant medication, which has also been shown to have cross-diagnostic benefits on cognition as well as potential to augment the effects of other training interventions. It is possible that muscarinic treatments directly targeting cognition and or used as an adjunct to other interventions could have improved safety potential compared to amphetamine and related treatments. Further, the broad age range of cholinergic effects on cognition and the fact that fluid cognition commonly changes for the worse with aging suggests intervention potential that is broader than would be present with stimulant medications.
The eventual application of muscarinic strategies will be dependent on upcoming information on safety and tolerability over longer time periods. Recent conference presentations have suggested similar tolerability and efficacy in a large-scale phase 3 study, although those data have not been reviewed by the FDA or published in a peer-reviewed journal [130]. Pending that information, it seems plausible that muscarinic agonist therapy has broad applicability for use in a wide range of potential uses.
References
Vos T, Amanuel Alemu A, Kalkidan Hassen A, Abbafati C, Abbas KM, Abd-Allah F, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet. 2017;390:1211–59.
Knapp M, Mangalore R, Simon J. The global costs of schizophrenia. Schizophr Bull. 2004;30:279–93.
Harvey PD. Cognitive functioning and disability in schizophrenia. Curr Dir Psychol Sci. 2010;19:249–54.
Lee RSC, Hermens DF, Naismith SL, Lagopoulos J, Jones A, Scott J, et al. Neuropsychological and functional outcomes in recent-onset major depression, bipolar disorder and schizophrenia-spectrum disorders: a longitudinal cohort study. Trans Psychiatry. 2015;5:e555–e555.
Leung WW, Bowie CR, Harvey PD. Functional implications of neuropsychological normality and symptom remission in older outpatients diagnosed with schizophrenia: a cross-sectional study. JINS. 2008;14:479–88.
Jääskeläinen E, Juola P, Hirvonen N, McGrath JJ, Saha S, Isohanni M, et al. A systematic review and meta-analysis of recovery in schizophrenia. Schizophr Bull. 2013;39:1296–306.
Keefe RSE, Harvey PD. Cognitive impairment in schizophrenia. Handb Exp Pharmacolog. 2012;213:11–37.
Bowie CR, Harvey PD. Cognition in schizophrenia: impairments, determinants, and functional importance. Psychiatr Clin North Am. 2005;28:613–26.
Richards AL, Pardinas AF, Frizzati A, Tansey KE, Lynham AJ, Holmans P, et al. The relationship between polygenic risk scores and cognition in schizophrenia. Schizophr Bull. 2020;46:336–44.
Smeland OB, Bahrami S, Frei O, Shain A, O’Connell K, Savage J, et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol Psychiatry. 2019;25:1–10.
Buckley PF, Harvey PD, Bowie CR, Loebel A. The relationship between symptomatic remission and neuropsychological improvement in schizophrenia patients switched to treatment with ziprasidone. Schizophr Res. 2007;94:99–106.
Altshuler LL, Ventura J, van Gorp WG, Green MF, Theberge DC, Mintz J. Neurocognitive function in clinically stable men with bipolar I disorder or schizophrenia and normal control subjects. Biol Psychiatry. 2004;56:560–9.
Hill SK, Reilly JL, Keefe RSE, Gold JM, Bishop JR, Gershon ES, et al. Neuropsychological impairments in schizophrenia and psychotic bipolar disorder: findings from the bipolar-schizophrenia network on intermediate phenotypes (B-SNIP) study. Am J Psychiatry. 2013;170:1275–84.
Harvey PD, Aslan M, Du M, Zhao H, Siever LJ, Pulver A, et al. Factor structure of cognition and functional capacity in two studies of schizophrenia and bipolar disorder: implications for genomic studies. Neuropsychology. 2016;30:28–39.
Harvey PD, Sun N, Bigdeli TB, Fanous AH, Aslan M, Malhotra AK, et al. Genome‐wide association study of cognitive performance in U.S. veterans with schizophrenia or bipolar disorder. Am J Medical Gen Part B. 2020;183:181–94.
Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nature Gen. 2018;50:1112–21.
Davies G, Lam M, Trampush JW, Luciano M, Hagenaars SP, Ritchie SJ, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9:2098.
Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, Awasthi S, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nature Gen. 2018;50:912–9.
Hochberger WC, Hill SK, Nelson CLM, Reilly JL, Keefe RSE, Pearlson GD, et al. Unitary construct of generalized cognitive ability underlying BACS performance across psychotic disorders and in their first-degree relatives. Schizophr Res. 2015;170:156–61.
Keefe RSE, Goldberg TE, Harvey PD, Gold JM, Poe MP, Coughenour L. The brief assessment of cognition in schizophrenia: reliability, sensitivity, and comparison with a standard neurocognitive battery. Schizophr Res. 2004;68:283–97.
Pan Z, Park C, Brietzke E, Zuckerman H, Rong C, Mansur RB, et al. Cognitive impairment in major depressive disorder. CNS Spectrums. 2019;24:22–29.
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, DeLong MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–9.
Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–73.
Summers WK, Majovski LV, Marsh GM, Tachiki K, Kling A. Oral tetrahydroaminoacridine in long-term treatment of senile dementia, alzheimer type. NEJM. 1986;315:1241–5.
Rogers SL, Friedhoff LT. The efficacy and safety of donepezil in patients with alzheimer’s disease: results of a us multicentre, randomized, double-blind, placebo-controlled trial. Dement Geriatr Cogn Disord. 1996;7:293–303.
Tariot PN, Solomon PR, Morris JC, Kershaw P, Lilienfeld S, Ding C. A 5-month, randomized, placebo-controlled trial of galantamine in AD. Neurology. 2000;54:2269–76.
Furey ML, Pietrini P, Alexander GE, Schapiro MB, Horwitz B. Cholinergic enhancement improves performance on working memory by modulating the functional activity in distinct brain regions: a positron emission tomography regional cerebral blood flow study in healthy humans. Brain Res Bull. 2000;51:213–8.
Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2007;12:232–46.
Bymaster FP, Shannon HE, Rasmussen K, Delapp NW, Mitch CH, Ward JS, et al. Unexpected antipsychotic-like activity with the muscarinic receptor ligand (5 R,6 R)6-(3-propylthio-1,2,5-thiadiazol-4-yl)-1-azabicyclo[3.2.1]octane. Eur J Pharmacol. 1998;356:109–19.
Shannon HE, Rasmussen K, Bymaster FP, Hart JC, Peters SC, Swedberg MD, et al. Xanomeline, an M(1)/M(4) preferring muscarinic cholinergic receptor agonist, produces antipsychotic-like activity in rats and mice. Schizophr Res. 2000;42:249–59.
Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–9.
Rapoport JL, Buchsbaum MS, Weingartner H, Zahn TP, Ludlow C, Mikkelsen EJ. Dextroamphetamine: its cognitive and behavioral effects in normal and hyperactive boys and normal men. Arch Gen Psychiatry. 1980;37:933–43.
Sostek AJ, Buchsbaum MS, Rapoport JL. Effects of amphetamine on vigilance performance in normal and hyperactive children. J Abnorm Chlld Psychol. 1980;8:491–500.
Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophr Res. 2005;77:43–58.
Swerdlow NR, Bhakta SG, Talledo J, Benster L, Kotz J, Lavadia M, et al. Lessons learned by giving amphetamine to antipsychotic-medicated schizophrenia patients. Neuropsychopharmacology. 2019;44:2277–84.
Foster DJ, Choi DL, Jeffrey Conn P, Rook JM. Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsychiatr Dis Treat. 2014;10:183–91.
Brannan SK, Sawchak S, Miller AC, Lieberman JA, Paul SM, Breier A. Muscarinic cholinergic receptor agonist and peripheral antagonist for schizophrenia. NEJM. 2021;384:717–26.
Yeomans JS. Role of tegmental cholinergic neurons in dopaminergic activation, antimuscarinic psychosis and schizophrenia. Neuropsychopharmacology. 1995;12:3–16.
Furey ML. The prominent role of stimulus processing: Cholinergic function and dysfunction in cognition. Curr Opin Neurology. 2011;24:364–70.
Deutsch AJ. The cholinergic synapse and the site of memory. Science. 1971;174:788–94.
Furey ML, Pietrini P, Haxby JV, Alexander GE, Lee HC, Vanmeter J, et al. Cholinergic stimulation alters performance and task-specific regional cerebral blood flow during working memory. PNAS. 1997;94:6512–6.
Murray CL, Fibiger HC. Learning and memory deficits after lesions of the nucleus basalis magnocellularis: reversal by physostigmine. Neuroscience. 1985;14:1025–32.
Sunderland T, Tariot PN, Weingartner H, Murphy DL, Newhouse PA, Mueller EA, et al. Pharmacologic modelling of Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10:599–610.
Sitaram N, Weingartner H, Gillin JC. Human serial learning: enhancement with arecholine and choline and impairment with scopolamine. Science. 1978;201:274–6.
Bartus RT, Dean IIIRL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–17.
Minzenberg MJ, Poole JH, Benton C, Vinogradov S. Association of anticholinergic load with impairment of complex attention and memory in schizophrenia. Am J Psychiatry. 2004;161:116–24.
Ballesteros A, Sánchez-Torres AM, López-Ilundain JM, Cabrera B, Lobo A, González-Pinto AM, et al. Is cognitive impairment associated with antipsychotic dose and anticholinergic equivalent loads in first-episode psychosis? Psychol Med. 2018;48:2247–56.
Vinogradov S, Fisher M, Warm H, Holland C, Kirshner MA, Pollock BG. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166:1055–62.
Montastruc F, Benevent J, Touafchia A, Chebane L, Araujo M, Guitton‐Bondon E, et al. Atropinic (anticholinergic) burden in antipsychotic‐treated patients. Fund Clin Pharmacol. 2018;32:114–9.
Alt A, Pendri A, Bertekap RL, Li G, Benitex Y, Nophsker M, et al. Evidence for classical cholinergic toxicity associated with selective activation of M1 muscarinic receptorss. J Pharmacol Exp Ther. 2016;356:293–304.
Picciotto MR, Caldarone BJ, King SL, Zachariou V. Nicotinic receptors in the brain: links between molecular biology and behavior. Neuropsychopharmacology. 2000;22:451–65.
Leonard S, Adams C, Breese CR, et al. Nicotinic receptor function in schizophrenia. Schizophr Bull. 1996;22:431–45.
Mexal S, Berger R, Logel J, Ross RG, Freedman R, Leonard S. Differential regulation of alpha7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J Mol Neurosci. 2010;40:185–95. 9233-4
Olincy A, Harris JG, Johnson LL, et al. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–8.
Freedman R, Olincy A, Buchanan RW, et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry. 2008;165:1040–7.
Keefe RSE, Meltzer HA, Dgetluck N, Gawryl M, Koenig G, Moebius HJ, et al. Randomized, double-blind, placebo-controlled study of encenicline, an α7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology. 2015;40:3053–60.
Hilt D, Gawryl M, Koenig G, Dgetluck N, Moebius H. EVP-6124, a selective alpha-7 partial agonist, has positive effects on cognition and clinical function in mild-to-moderate Alzheimer’s disease patients: results of a six-month, double-blind, placebo controlled, dose ranging study. Alzheim Dementia. 2013;8:S746–S746.
Kohlhaas KL, Bitner RS, Gopalakrishnan M, Rueter LE. Effects of alpha 7 nicotinic acetylcholine receptor agonists on antipsychotic efficacy in a preclinical mouse model of psychosis. Psychopharmacology. 2012;220:823–33.
Haig GM, Bain EE, Robieson WZ, Baker JD, Othman AA. A randomized trial to assess the efficacy and safety of ABT-126, a Selective α7 Nicotinic Acetylcholine eceptor Agonist, in the Treatment of Cognitive Impairment in Schizophrenia. Am J Psychiatry. 2016;173:827–35.
Haig GM, Wang D, Zhao J, Othman AA, Bain EE. Efficacy and safety of the α7-nicotinic acetylcholine receptor agonist ABT-126 in the treatment of cognitive impairment associated with schizophrenia: results from a phase 2b randomized controlled study in smokers. J Clin Psychiatry. 2018;79:16m11162.
Felder CC. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. 1995;9:619–25.
Hulme EC, Birdsall NJM, Buckley NJ. Muscarinic receptor subtypes. Ann Rev Pharmacol. 1990;30:633–73.
Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–26.
Bymaster FP, McKinzie DL, Felder CC, Wess J. Use of M1–M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem Res. 2003;28:437–42.
Gomeza J, Zhang L, Kostenis E, Felder C, Bymaster F, Brodkin J, et al. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M4 muscarinic acetylcholine receptor knockout mice. PNAS. 1999;96:10483–8.
Koshimizu H, Leiter LM, Miyakawa T. M4 muscarinic receptor knockout mice display abnormal social behavior and decreased prepulse inhibition. Mol Brain Res. 2012;5:10–10.
Tzavara ET, Bymaster FP, Davis RJ, Wade MR, Perry KW, Wess J, et al. M-4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: relevance to the pathophysiology and treatment of related central nervous system pathologies. FASEB J. 2004;18:1410–2.
Moehle MS, Pancani T, Byun N, Yohn SE, Wilson GH, Dickerson JW. et al. Cholinergic projections to the substantia nigra pars reticulata inhibit dopamine modulation of basal ganglia through the M4 muscarinic receptor. Neuron. 2017;96:1358–72.e1354.
Bubser M, Bridges TM, Dencker D, Gould RW, Grannan M, Noetzel MJ, et al. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem Neurosci. 2014;5:920–42.
Scarr E, Um JY, Cowie TF, Dean B. Cholinergic muscarinic M4 receptor gene polymorphisms: a potential risk factor and pharmacogenomic marker for schizophrenia. Schizophr Res. 2013;146:279–84.
Scarr E, Sundram S, Keriakous D, Dean B. Altered hippocampal muscarinic M4, but Not M1, receptor expression from subjects with schizophrenia. Biol Psychiatry (1969). 2007;61:1161–70.
Pozhidaev I, Boiko AS, Loonen AJM, Paderina DZ, Fedorenko OY, Tenin G, et al. Association of cholinergic muscarinic M4 receptor gene polymorphism with schizophrenia. App Clin Gen. 2020;13:97–105.
Yohn SE, Foster DJ, Covey DP, Moehle MS, Galbraith J, Garcia-Barrantes PM, et al. Activation of the mGlu1 metabotropic glutamate receptor has antipsychotic-like effects and is required for efficacy of M4 muscarinic receptor allosteric modulators. Mol Psychiatry. 2020;25:2786–99.
Cho HP, Garcia-Barrantes PM, Brogan JT, Hopkins CR, Niswender CM, Rodriguez AL, et al. Chemical modulation of mutant mGlu1 receptors derived from deleterious GRM1 mutations found in schizophrenics. ACS Chemical Biology. 2014;9:2334–46.
Ayoub MA, Angelicheva D, Vile D, Chandler D, Morar B, Cavanaugh JA, et al. Deleterious GRM1 mutations in schizophrenia. PloS One. 2012;7:e32849–e32849.
Byun NE, Grannan M, Bubser M, Barry RL, Thompson A, Rosanelli J, et al. Antipsychotic drug-like effects of the selective M4 muscarinic acetylcholine receptor positive allosteric modulator VU0152100.Neuropsychopharmacology. 2014;39:1578–93.
Krystal JH, Kane JM, Correll CU, et al. Emraclidine, a novel positive allosteric modulator of cholinergic M4 receptors, for the treatment of schizophrenia: a two-part, randomised, double-blind, placebo-controlled, phase 1b trial. Lancet. 2023;400:2210–20.
https://clinicaltrials.gov/ct2/show/NCT05227703?term=Emraclidine&draw=2&rank=3, accessed 12/26/2022.
Stanhope KJ, Mirza NR, Bickerdike MJ, Bright JL, Harrington NR, Hesselink MB, et al. The muscarinic receptor agonist xanomeline has an anti psychotic-like profile in the rat. J Pharmacol Exp Ther. 2001;299:782–92.
Deutch AY. The regulation of subcortical dopamine systems by the prefrontal cortex: interactions of central dopamine systems and the pathogenesis of schizophrenia. J Neural Transm Suppl. 1992;36:61–89.
Yoon JH, Minzenberg MJ, Raouf S, D’Esposito M, Carter CS. Impaired prefrontal-basal ganglia functional connectivity and substantia nigra hyperactivity in schizophrenia. Biol Psychiatry. 2013;74:122–9.
Dean B, McLeod M, Keriakous D, McKenzie J, Scarr E. Decreased muscarinic(1) receptors in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2002;7:1083–91.
Mancama D, Arranz MJ, Landau S, Kerwin R. Reduced expression of the muscarinic 1 receptor cortical subtype in schizophrenia. Am J Medical Gen Part B. 2003;119B:2–6.
Dean B, Crook JM, Pavey G, Opeskin K, Copolov DL. Muscarinic1 and 2 receptor mRNA in the human caudate-putamen: No change in m1 mRNA in schizophrenia. Mol Psychiatry. 2000;5:203–7.
Haroutunian V, Davidson M, Kanof PD, et al. Cortical cholinergic markers in schizophrenia. Schizophr Res. 1994;12:137–44.
Melancon BJ, Tarr JC, Panarese JD, Wood MR, Lindsley CW. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discov Today. 2013;18:1185–99.
Marino MJ, Conn PJ. Direct and indirect modulation of the N-methyl D-aspartate receptor. Curr Drug Targets CNS Neurol Disord. 2002;1:1–16.
Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U. S. A. 1998;95:11465–70.
Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, et al. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nature Neurosci. 2003;6:51–58.
Davis AA, Fritz JJ, Wess J, Lah JJ, Levey AI. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J Neurosci. 2010;30:4190–6.
Medeiros R, Kitazawa M, Caccamo A, Baglietto-Vargas D, Estrada-Hernandez T, Cribbs DH, et al. Loss of muscarinic M1 receptor exacerbates alzheimer’s disease–like pathology and cognitive decline. Am J Pathology. 2011;179:980–91.
Fisher A. Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: perspectives and challenges in treatment of Alzheimer’s disease. J Neurochem. 2012;120:22–33.
Beach TG, Walker DG, Potter PE, Sue LI, Fisher A. Reduction of cerebrospinal fluid amyloid β after systemic administration of M1 muscarinic agonists. Brain research. 2001;905:220–3.
Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, et al. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–82.
Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exper Neurol. 2000;163:495–529.
Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36:375–99.
Fisher A. Cholinergic treatments with emphasis on M1 muscarinic agonists as potential disease-modifying agents for alzheimer’s disease. Neurotherapeutics. 2008;5:433–42.
Keefe RS, Malhotra AK, Meltzer HY, Kane JM, Buchanan RW, Murthy A, et al. Efficacy and safety of donepezil in patients with schizophrenia or schizoaffective disorder: significant placebo/practice effects in a 12-week, randomized, double-blind, placebo-controlled trial. Neuropsychopharmacology. 2008;33:1217–28.
Buchanan RW, Conley RR, Dickinson D, Ball MP, Feldman S, Gold JM, et al. Galantamine for the treatment of cognitive impairments in people with schizophrenia. Am J Psychiatry. 2008;165:82–89.
Barak S, Weiner I. The M1/M4 preferring agonist xanomeline reverses amphetamine-, MK801- and scopolamine-induced abnormalities of latent inhibition: putative efficacy against positive, negative and cognitive symptoms in schizophrenia. Int J Neuropsychopharmacol. 2011;14:1233–46.
Sauder C, Allen LA, Baker E, Miller AC, Paul SM, Brannan SK. Effectiveness of KarXT (xanomeline-trospium) for cognitive impairment in schizophrenia: post hoc analyses from a randomised, double-blind, placebo-controlled phase 2 study. Transl Psychiatry. 2022;12:491. Published 2022 Nov 21.
Bradley SJ, Bourgognon JM, Sanger HE, et al. M1 muscarinic allosteric modulators slow prion neurodegeneration and restore memory loss. J Clin Invest. 2017;127:487–99.
Bradley SJ, Molloy C, Bundgaard C, et al. Bitopic binding mode of an M1 muscarinic acetylcholine receptor agonist associated with adverse clinical trial outcomes. Mol Pharmacol. 2018;93:645–56.
Scarpa M, Hesse S, Bradley SJ. M1 muscarinic acetylcholine receptors: a therapeutic strategy for symptomatic and disease-modifying effects in Alzheimer’s disease? Adv Pharmacol. 2020;88:277–310.
Westfall, TC, Squire, LR. in Encyclopedia of Neuroscience (Academic Press, 2009).
Shannon HE, Bymaster FP, Calligaro DO, Greenwood B, Mitch CH, Sawyer BD, et al. Xanomeline: a novel muscarinic receptor agonist with functional selectivity for M1 receptors. J Pharmacol Exp Ther. 1994;269:271–81.
Bymaster FP, Whitesitt CA, Shannon HE, DeLapp N, Ward JS, Calligaro DO, et al. Xanomeline: a selective muscarinic agonist for the treatment of Alzheimer’s disease. Drug Development Research. 1997;40:158–70.
Ehlert, FJ, Pak, KJ, Griffin, MT. In Muscarinic Receptors (eds. Allison DF, Arthur C, Neil MN) (Springer Berlin Heidelberg, 2012).
Fetscher C, Fleichman M, Schmidt M, Krege S, Michel MC. M(3) muscarinic receptors mediate contraction of human urinary bladder. Br J Pharmacol. 2002;136:641–3.
Staskin D, Kay G, Tannenbaum C, Goldman HB, Bhashi K, Ling J, et al. Trospium chloride has no effect on memory testing and is assay undetectable in the central nervous system of older patients with overactive bladder. Int J CLin Pract. 2010;64:1294–1300.
Haro JM, Kamath SA, Ochoa S, Novick D, Rele K, Fargas A, et al. The clinical global impression–schizophrenia scale: a simple instrument to measure the diversity of symptoms present in schizophrenia. Acta Psychiatrica Scandinavica. 2003;107:16–23.
Joshi YB, Thomas ML, Braff DL, Green MF, Gur RC, Gur RE, et al. Anticholinergic medication burden-associated cognitive impairment in schizophrenia. Am J Psychiatry. 2021;178:838–47.
Greenwood TA, Lazzeroni LC, Maihofer AX, Swerdlow NR, Calkins ME, Freedman R, et al. Genome-wide association of endophenotypes for schizophrenia from the consortium on the genetics of schizophrenia (COGS) study. JAMA Psychiatry. 2019;76:1274–84.
Eum S, Hill SK, Rubin LH, Carnahan RM, Reilly JL, Ivleva EI, et al. Cognitive burden of anticholinergic medications in psychotic disorders. Schizophr Res. 2017;190:129–35.
Eum S, Hill SK, Alliey-Rodriguez N, Stevenson JM, Rubin LH, Lee AM, et al. Genome-wide association study accounting for anticholinergic burden to examine cognitive dysfunction in psychotic disorders. Neuropsychopharmacology. 2021;46:1802–10.
Harvey PD. Domains of cognition and their assessment. Dialogues Clin Neurosci. 2019;21:227–37.
Czaja SJ, Charness N, Fisk AD, Hertzog C, Nair SN, Rogers WA, et al. Factors predicting the use of technology: findings from the center for research and education on aging and technology enhancement (CREATE). Psychol Aging. 2006;21:333–52.
Rabin LA, Smart CM, Amariglio RE. Subjective cognitive decline in preclinical alzheimer’s disease. Ann Rev Clin Psychol. 2017;13:369–96.
Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230–8.
Nitsch RM, Wurtman RJ, Growdon JH. Regulation of APP processing potential for the therapeutical reduction of brain amyloid burden. PANAS. 1996;777:175–82.
Geerlings MI, Jonker C, Bouter LM, Adèr HJ, Schmand B. Association between memory complaints and incident alzheimer’s disease in elderly people with normal baseline cognition. Am J Psychiatry. 1999;156:531–7.
Atkins AS, Khan A, Ulshen D, Vaughan A, Balentin D, Dickerson H, et al. Assessment of instrumental activities of daily living in older adults with subjective cognitive decline using the virtual reality functional capacity assessment tool (VRFCAT). J Prev Alzheimer’s Dis. 2018;5:216–24.
Lenze EJ, Stevens A, Waring JD, Pham VT, Haddad R, Shimony J, et al. Augmenting computerized cognitive training with vortioxetine for age-related cognitive decline: a randomized controlled trial. Am J Psychiatry. 2020;177:548–55.
Harvey PD, Sand M. Pharmacological augmentation of psychosocial and remediation training efforts in schizophrenia. Front Psychiatry. 2017;8:177–177.
Howes OD, McCutcheon R, Agid O, de Bartolomeis A, van Beveren NJM, Birnbaum ML, et al. Treatment-resistant schizophrenia: treatment response and resistance in psychosis (TRRIP) working group consensus guidelines on diagnosis and terminology. Am J Psychiatry. 2017;174:216–29.
Nucifora FC, Woznica E, Lee BJ, Cascella N, Sawa A. Treatment resistant schizophrenia: clinical, biological, and therapeutic perspectives. Neurobiol Dis. 2019;131:104257–104257.
Giraud-Baro E, Dassa D, De Vathaire F, Garay RP, Obeid J. Schizophrenia-spectrum patients treated with long-acting injectable risperidone in real-life clinical settings: Functional recovery in remitted versus stable, non-remitted patients (the EVeREST prospective observational cohort study). BMC Psychiatry. 2016;16:8–8.
Rubio JM, Schoretsanitis G, John M, Tiihonen J, Taipale H, Guinart D, et al. Psychosis relapse during treatment with long-acting injectable antipsychotics in individuals with schizophrenia-spectrum disorders: an individual participant data meta-analysis. Lancet. Psychiatry. 2020;7:749–61.
Koblan KS, Kent J, Hopkins SC, Krystal JH, Cheng H, Goldman R, et al. A Non–D2-Receptor-Binding Drug for the Treatment of Schizophrenia. NEJM. 2020;382:1497–506.
Paul SM, Correll CU, Angelov AS, Kaul I., Brannan SK. Safety and efficacy of KarXT in patients with schizophrenia: results from a phase 3, randomized double-blind placebo controlled trial (Emergent-2). 2022. Ann Meeting Am Coll Neuropsychopharmacolog, M178.
Author information
Authors and Affiliations
Contributions
Both authors contributed equally to this paper. Tasks included conceptualizing the important issues, literature reviews, consolidating information, writing drafts of the paper, and responding to reviews.
Corresponding author
Ethics declarations
Competing interests
Last Year. Dr. Harvey has received consulting fees or travel reimbursements from Alkermes, Bio Excel, Boehringer Ingelheim, Karuna Therapeutics, Merck Pharma, Minerva Pharma, Roche Pharma, and Sunovion (DSP) Pharma in the past year. He receives royalties from the Brief Assessment of Cognition in Schizophrenia (Owned by WCG Verasci, Inc. and contained in the MCCB). He is chief scientific officer of i-Function, Inc. Dr. Jones has no Bomedical Conflicts of Interest to report.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jones, S.E., Harvey, P.D. Cross-diagnostic determinants of cognitive functioning: the muscarinic cholinergic receptor as a model system. Transl Psychiatry 13, 100 (2023). https://doi.org/10.1038/s41398-023-02400-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-023-02400-x
