
Abstract
Cognitive deficits are a core feature of schizophrenia, account for much of the impaired functioning associated with the disorder and are not responsive to existing treatments. In this review, we first describe the clinical presentation and natural history of these deficits. We then consider aetiological factors, highlighting how a range of similar genetic and environmental factors are associated with both cognitive function and schizophrenia. We then review the pathophysiological mechanisms thought to underlie cognitive symptoms, including the role of dopamine, cholinergic signalling and the balance between GABAergic interneurons and glutamatergic pyramidal cells. Finally, we review the clinical management of cognitive impairments and candidate novel treatments.
Similar content being viewed by others
Introduction
Individuals with schizophrenia show a substantial impairment in overall cognitive performance, which, on average, is around two standard deviations below that in healthy controls [1]. Moreover, this deficit contributes to poor clinical outcomes such as unemployment and an inability to live independently [2]. While cognitive function in schizophrenia is an area of increasing research interest (Fig. 1) [3], this has yet to translate into the development of novel treatments for cognitive problems. All currently approved pharmacological treatments for schizophrenia exert their effects via antagonism of the dopamine D2 receptor [4, 5]. This mechanism of action is efficacious for symptoms that are thought to be driven by excessive striatal dopamine signalling, such as hallucinations and delusions. However, antipsychotic medications have little impact on cognitive impairments in schizophrenia, perhaps because the latter are related to different pathophysiological processes [5]. In the current paper, we outline the clinical nature of cognitive impairment in schizophrenia and consider potential aetiological factors. We then discuss pathophysiology, before concluding with an examination of current and potential future treatment options.

The graph illustrates the proportion of PubMed articles on schizophrenia that include ‘cognitive impairment’ in the title.
The nature of cognitive impairments in schizophrenia
Cognitive deficits appear to be distinct from positive and negative symptoms
Factor analyses of the Positive and Negative Syndrome Scale (PANSS) indicate that a five-factor model (positive, negative, disorganised, excited, and depressed) captures the symptom structure of schizophrenia better than the original a priori grouping of positive, negative, and general symptoms [6, 7]. Of these five factors, the disorganisation factor (which includes difficulty in abstract thinking, poor attention, disorientation, stereotyped thinking and conceptual disorganisation) shows the strongest association with cognitive test scores, but still only accounts for a small proportion of the variance [8, 9]. Network analyses have identified broadly similar symptom groupings, and again find that cognitive scores are distinct from positive and negative symptoms, although linked to disorganisation [10, 11]. Deficits in social cognition are also apparent in individuals with schizophrenia, and these too are separable both from the five PANSS factors and other cognitive domains [12, 13]. Social cognition also has a major impact on functioning [13], and may have distinct pathophysiological underpinnings. This important aspect of cognition in schizophrenia has been reviewed in detail elsewhere [14, 15], and is not within the scope of the present review.
Are impairments global or domain-specific?
For a given cognitive domain, patients with schizophrenia perform around one standard deviation below the level of controls. However, the psychometric properties of composite scores are such that they are typically more extreme than their constituent parts [16]. Thus, patients show an impairment of around 1.5 standard deviations on overall composite scores relative to controls [1, 17,18,19,20,21,22]. An unresolved issue is whether there are distinct domains of cognitive impairment in schizophrenia, or whether the deficits are better summarised as global. This issue maps to a parallel debate over whether the pathophysiology of schizophrenia involves specific loci of brain dysfunction or a systems-level disruption.
Factor analyses of the Measurement and Treatment to Improve Cognition in Schizophrenia test battery identified seven cognitive domains: Processing speed, Attention, Working memory, Verbal learning and memory, Visual learning and memory, Reasoning, and Social cognition [23]. Further dimensionality reduction suggests that these seven domains can be reduced to parent domains of Processing speed, Attention/Working memory and learning [24].
Some reports indicate that processing speed is the domain most affected in schizophrenia, and that processing speed deficits are the strongest predictor of general cognitive performance [24,25,26,27]. However, processing speed is particularly affected by antipsychotic medications [28], and in medication-naïve cohorts, the magnitude of this impairment is no greater than that for verbal or working memory [19]. Because processing speed is a component of many cognitive functions, deficits in this domain may reflect impairments in other higher-order functions. For example, patients with schizophrenia may approach processing speed tests, such as symbol-coding, less strategically than controls [29]. Equally, impairments in even lower-level domains such as reaction time could contribute to deficits in higher-order domains [23]. Given the existence of severe impairments of low-level processes, and the presence of deficits across many domains, it could be argued that cognitive impairments in schizophrenia are best conceptualised as a generalised deficit. However, it is unclear whether the limitations of existing cognitive tests are such that they cannot isolate specific cognitive processes, precluding the capture of more specific impairments [30, 31]. Tests that can examine more specific cognitive and perceptual processes may be more sensitive to the non-generalised components of cognitive impairments in schizophrenia.
Variation in cognitive impairments between and within individuals
Cognitive functioning appears to vary significantly between individual patients with schizophrenia [32]. Cluster analyses of raw test scores point to a heterogeneity of cognitive function similar to that seen in samples of healthy controls. While these findings provide evidence for clusters of cognitive performance similar to those observed in the general population, they do not demonstrate that the population of patients with schizophrenia comprises clusters of individuals with distinct patterns of cognitive impairment. Thus, it is still unclear whether there are different forms of cognitive impairment across patients, or a generic impairment which is differentially expressed due to variations in premorbid cognitive ability. Analysis of the variability of cognitive functioning between people at clinical high risk (CHR) for psychosis indicates that there is significantly greater variability among these individuals than among controls across a wide range of cognitive domains. This result suggests that there is a heterogeneity in the magnitude of the impairment across individuals, rather than a uniform deficit (Fig. 2) [33].

Examining the population distribution of cognitive ability can help to determine whether impairments reflect a generalised deficit or are greater in magnitude in some patients than others. Recent data indicate that the distribution is more like that shown in (B) than (A), with more variability in the patient than the control sample. This suggests in psychosis, rather than there being a constant effect on cognition, in some individuals, there is a large impairment, but in others relatively little. It is also valuable to look at intra-individual variability, unlike (A) and (B) which represent data from many individuals, (C) and (D) represent data from a single individual. From this perspective variability again appears greater in schizophrenia and those at risk with the data resembling the distribution in (D) more than that in (C).
Variability within individuals has also been examined, and it appears that both over time and across cognitive domains individuals with schizophrenia and those at risk of the disorder display greater intra-individual variability than control subjects [34, 35]. These findings suggest different individuals may show impairments in different domains. However, precisely delineating this is more challenging, and may require prospective studies that begin even earlier than the clinical high-risk stage, before the first expression of symptoms.
Time course of cognitive deficits in schizophrenia
In people who later develop schizophrenia, relatively global impairments in cognitive function are detectable in childhood [36], and there is an increase in the severity of nonverbal deficits during adolescence due to the slower development of these abilities (Fig. 3) [37, 38]. This alteration in developmental trajectory over adolescence is a stronger predictor of the subsequent onset of schizophrenia than a cross-sectional impairment in cognitive performance at the age of 18 [39].

Cognitive deficits in individuals who develop schizophrenia are apparent in childhood and do not appear to increase markedly in the initial phase of the illness. While CHR individuals as a group score higher than FEP individuals, longitudinal studies do not provide clear evidence for a decline over the period of transition to psychosis. The decline in cognitive function that occurs in later life in healthy individuals is evident at an earlier age in individuals with schizophrenia, potentially related to neurovascular factors.
Much of the impairment in cognitive functioning that is evident in adults with schizophrenia is thus established before the first expression of symptoms [40] or contact with mental health services. This raises the possibility that even in patients in whom cognitive deficits are not clinically obvious, a degree of deterioration relative to the premorbid state has already occurred. This is supported by the finding that people with schizophrenia, when matched to controls with the same current IQ, have higher scores on cognitive tests that are sensitive to premorbid IQ, but lower scores on working memory (which is not) [32, 41].
The first episode of schizophrenia is often preceded by a CHR state in which cognitive deficits are also observed [42]. Individuals at CHR may or may not progress to develop schizophrenia [43], and cognitive deficits are most severe in the subgroup of the CHR population that subsequently develops the disorder [42]. Whether the transition to frank psychosis in these individuals is associated with a further decline in cognitive function remains an important question. A progression of cognitive impairments (as well as psychotic symptoms) would suggest that neurobiological changes occurring over this period underlie the changes in cognitive function. Alternatively, an absence of progression would indicate that the neurobiological factors critical to cognition are pre-existing and neurodevelopmental. At present, there is no evidence of a decline in raw cognitive performance following transition, although this could reflect practice effects [44]. However, there is some evidence for a relative decline: a recent meta-analysis found that CHR individuals who develop psychosis display a relative worsening in processing speed subsequent to baseline compared to CHR individuals who did not transition [45].
A decline in cognitive function over the course of the illness was originally seen as a defining feature of schizophrenia [46]. Meta-analyses of longitudinal studies, however, have tended to show improvement across multiple cognitive domains. However, many of the studies examined were of limited duration and the observed improvements are likely artefacts of practice effects, with less improvement observed in patients than in controls [47,48,49]. Long-term longitudinal studies of over 10 years duration are rare, and have shown both improvement [50] and decline [51, 52] in cognitive function. It is difficult in these studies, either due to lack of controls, relatively small sample sizes, or poor matching to controls, to disambiguate how much observed changes reflect normal cognitive trajectories, versus how much is due to a changing magnitude of impairment. Large-scale cross-sectional studies have suggested that while cognitive functioning decline with age this follows normal trajectories with no increase in the severity of impairment [53]. There is some evidence for a more rapid decline in cognitive function over the age of 65 in institutionalised individuals with schizophrenia than controls [54]. The basis of this decline is unclear, but could partly be secondary to poor physical health in patients with schizophrenia, which becomes increasingly evident in later life (Fig. 3). Both the lower starting point and this subsequent decline likely contribute to the increased incidence and earlier onset of dementia in individuals with schizophrenia [55].
Are cognitive deficits in schizophrenia distinct from those in other psychiatric disorders?
During the development of the DSM-5, the addition of cognitive impairment as a new diagnostic criterion for schizophrenia was considered. However, this was not implemented, because cognitive deficits in schizophrenia were regarded as not sufficiently distinct from those in other conditions (such as bipolar disorder) to be of diagnostic value [56,57,58,59]. Nevertheless, the pattern of deficits across different psychiatric disorders is not the same. For example, cognitive impairments in schizophrenia are more severe than in bipolar disorder and depression, and are clearly present before the expression of symptoms, which is not the case for bipolar disorder or depression [60, 61] Ironically, subjective cognitive impairment is one of the DSM criteria for the latter disorders. In schizophrenia, however, the use of a subjective criterion is complicated by the lack of insight associated with the disorder, although subjective cognitive difficulties can be used as diagnostic criteria for the CHR state [62].
Lived experience of cognitive impairment in schizophrenia
Some individuals with schizophrenia complain of subjective cognitive impairments, and these appear to be even more common among people at clinically high risk for the disorder [63]. In addition to their potential pathophysiological and diagnostic significance, these self-reported symptoms are important because they are associated with distress and a reduction in quality of life [64]. At the same time, it is clear that a significant proportion of individuals with schizophrenia have severe cognitive impairments but do not report subjective deficits [65].
Among patients with schizophrenia there is only a weak correlation between subjective reports of cognitive impairment and objective measures of cognitive performance, and the reporting of subjective impairments appears to be higher in patients with comorbid depression [66, 67]. Although the correlation between subjective reports and objective measures is stronger when the analysis is restricted to the 50% of patients who report the greatest subjective impairment, it is still only modest [65]. Similarly, there is minimal correlation between objective and subjective measures of cognition in individuals at clinically high risk of psychosis [63, 68]. The absence of a strong correlation across the psychosis spectrum is in keeping with findings that individuals at high risk of psychosis report a greater severity of subjective impairment than people with schizophrenia, despite having a lesser degree of impairment on objective testing [63, 69]. A lack of insight and higher levels of disorganised symptoms appear to be key factors that contribute to a poor ability to self-assess cognitive abilities [70, 71], and individuals with high scores on self-certainty measures (e.g., endorsing statements such as ‘my interpretations of my experiences are definitely right’) tend to be those with greater cognitive problems [72]. This has relevance for the delivery of treatments for cognition, as individuals with subjective impairments appear to be more willing to engage in therapy, whereas those with more severe objective impairments may not see the need [73].
Despite the importance of cognitive deficits in schizophrenia, the formal assessment of cognitive function is rarely part of the routine clinical care of people with schizophrenia [74]. With limited time and the absence of formal training, it is difficult to assess accurately [75], and most established instruments require a trained assessor and a lengthy assessment period, which patients often find demanding. Clinicians have an increasingly limited time to see patients, and clinical teams often lack access to a neuropsychologist to conduct cognitive assessments. Moreover, in the absence of effective interventions for cognitive deficits, clinicians may reason that quantifying their severity is unlikely to be of benefit to their patients.
Functional consequences of cognitive impairments
There is a direct correlation between the level of cognitive performance in schizophrenia and the level of real-world functioning [24, 76]. This relationship is particularly strong when functioning is assessed using performance-based measures such as the UCSD Performance-based Skills Assessment (correlations ranging from r = 0.4 to 0.8), as opposed to an interview-based assessment (correlations ranging from 0.1 to 0.3) [24, 76].
A key driver of the substantial health costs associated with schizophrenia is admission to the hospital. Cognitive impairment is linked to reduced adherence to treatment, a greater likelihood of hospital admission, and to longer lengths of hospital stay [77,78,79]. In addition to health costs, schizophrenia is associated with even greater societal costs, as 80–90% of patients are unemployed, and remain so for most of their adult lives [77, 78, 80]. Cognitive impairments, as well as negative symptoms, are a major factor in this lost productivity [78, 81]. Among people with schizophrenia, a greater severity of cognitive symptoms is associated with lower wages, fewer hours worked in supported employment programs, and fewer benefits gained from employment interventions [82, 83]. Measures of functioning are more strongly correlated with measures of cognition in individuals with schizophrenia than in healthy controls [84], consistent with a non-linear relationship between cognitive performance and function in people with the disorder. The latter raises the possibility that in those with more marked deficits, interventions that result in improvements in cognition could have a disproportionately large benefit on the level of functioning.
The aetiology of cognitive impairment in schizophrenia
Genetic factors
Both cognitive ability in healthy individuals and cognitive impairments in schizophrenia appear to be highly heritable. The relatives of people with schizophrenia also show cognitive deficits [85, 86], and twin studies suggest that a significant proportion of the variance in cognition and schizophrenia risk is due to shared genetic factors [87]. Schizophrenia is regarded as a polygenic disorder in which multiple genes of small effect increase risk of illness when certain alleles are present together. The same architecture applies to cognitive abilities in the general population [88, 89].
While attempts have been made to characterise the effects of specific alleles [90], most contribute only a minimal degree of risk (median odds ratio 1.05), and are therefore unlikely to have major pathophysiological relevance in isolation [89]. However, functional consequences of increased genetic risk may be detected by assessing polygenic risk scores. Both twin and genome-wide association studies (GWAS) show a strong negative genetic correlation exists between liability for schizophrenia and cognitive ability, indicating that they share common genetic factors [88, 91,92,93,94]. However, this negative genetic correlation is minimal or absent between polygenic risk for bipolar disorder and cognitive function [88, 91, 93, 95]. This is remarkable, given the overlap in genetic risk factors for schizophrenia and bipolar disorder [96]: cognitive function appears to be one phenotypic component that reflects a fundamental difference in their respective genetic architectures. Consistent with this, in healthy cohorts, a high polygenic risk score for schizophrenia is associated with poorer cognition, and low polygenic scores for cognition are associated with an increased risk of schizophrenia [97]. The polygenic risk score for bipolar is also associated with poorer cognition in childhood, primarily as a result of single-nucleotide polymorphisms shared with schizophrenia [98], but bipolar disorder-specific risk alleles are associated with better cognitive performance [99]. Among patients with schizophrenia, cognitive performance is correlated with the polygenic risk scores for IQ and for educational attainment, but not that for schizophrenia [95, 100]. Although this lack of direct correlation suggests that cognitive impairment in schizophrenia is not a consequence of genetic liability for the disorder, cognition may a mediating factor through which genetic risk exerts its effects [101]. As mentioned above, in healthy controls there is an association between higher polygenic risk scores for schizophrenia and lower baseline cognitive performance; however, there is no association with greater cognitive decline, implicating neurodevelopmental rather than neurodegenerative processes [102]. Conclusions drawn from polygenic risk score analyses must be tempered by the fact that these typically account for less than 10% of risk variance, and at present, inferences can only be made in relation to people of European ancestry [103].
Another approach to unpicking GWAS results is to examine associated groups of functionally related genes to determine if genetic pathways are implicated. Counter-intuitively, given the negative correlation at the phenotypic level, these studies have reported a positive correlation between polygenic risk scores for schizophrenia and polygenic risk scores for educational attainment [100]. A recent analysis investigated genes that were positively associated with schizophrenia risk but negatively associated with cognitive ability and looked at how this related to genes associated with both positive and negative educational attainment. For the concordant pathway (i.e., positive educational attainment), gene sets enriched for expression in brain tissue and the CHD8 neurodevelopmental pathway were implicated. When examining the discordant pathway, several synaptic pathways were implicated such as ion channels and synaptic density, and when examining enrichment for drug mechanisms of action, voltage-gated calcium channel genes were over-represented [104].
When considering the genetic underpinning of cognitive abilities, it is important to bear in mind that these do not necessarily imply a direct link. As an example, if individuals of a certain ethnicity have less access to educational opportunities, the alleles associated with this phenotype might show a negative association with cognitive ability. In cases such as this, time will be better spent tracking the environmental mechanism mediating the association than investigating the biological function of the alleles.
Environmental factors
Although the heritability of schizophrenia is substantial at around 80% [105], even identical twins tend to be discordant for the disorder, underlining the importance of environmental influences [106]. Similarly, in the general population, a wide range of environmental factors are associated with cognitive abilities, and these are of greater importance in situations of socioeconomic disadvantage, where they effectively mask genetic heritability [107]. The increased prevalence of negative environmental factors in individuals who develop schizophrenia therefore raises the possibility that similar mechanisms may hold for cognitive impairments in schizophrenia as in the general population, with environmental influences also playing a major role.
In the prenatal period, obstetric complications are a well-established risk factor for schizophrenia [108] and are also associated with lower IQ in both individuals with schizophrenia and in healthy controls [109]. While prenatal infection is associated with risk of schizophrenia, and some prenatal infections are associated with cognitive impairment in the offspring [110], it does not appear that there is a general association with cognitive abilities when evidence of infection is broadly defined [111]. For some infections such as influenza, the deleterious cognitive effects may be more pronounced in individuals who subsequently develop schizophrenia, suggesting that other aetiological factors play a role, or, alternatively, that individuals with schizophrenia represent a group in which a more serious infection is more likely to have occurred [112].
Growing up in an urban environment is linked to a raised incidence of schizophrenia, although the direction of causality remains unclear [113,114,115]. Some of this association may be mediated by higher levels of socioeconomic deprivation. In the general population, socioeconomic deprivation is associated with poorer educational attainment [116], potentially because living in affluent neighbourhoods is associated with more pro-cognitive exposures [117]. However, there appear to be additional factors linking cognition and urbanicity. In children born preterm, living in an urban environment is associated with lower cognitive development scores, even after controlling for socioeconomic factors [118]. In the general population, growing up in an urban environment is associated with poorer spatial navigation abilities [119], and air pollution is associated with both poorer cognitive function and with an increased risk of developing schizophrenia [120,121,122]. Exposure to childhood trauma is associated both with an increased risk of developing schizophrenia, and also with lower performance on cognitive batteries in childhood and adolescence [114, 123, 124].
Acute cannabis use is associated with a clear cognitive impairment [125], and regular users also appear to perform worse on cognitive testing [126]. However, among people with schizophrenia, some studies have reported that cannabis users show better cognitive performance than patients who are non-users [127, 128]. This finding appears counter-intuitive, as cannabis use in healthy volunteers and other studies in schizophrenia has been associated with impairments in cognitive function [125, 129, 130]. The association with better performance in schizophrenia may depend on the pattern of cannabis use, as it is mainly evident in infrequent users rather than in regular or dependent users (Chester et al., in submission). The basis of the association is unclear. One possibility is that occasional cannabis use is a proxy for patients in whom cognition is relatively less impaired: the ability to source illicit cannabis requires motivation, and organisational and social skills. Another is that individuals who develop schizophrenia in the context of cannabis use have a relatively less genetic predisposition and less impairment of cognitive function. However, among people with schizophrenia who use cannabis, those with a family history of schizophrenia have better cognitive performance than those who do not. This has been attributed to cannabidiol (CBD) in cannabis exerting a neuroprotective effect [131]. However, most currently available illicit cannabis contains high levels of THC but minimal amounts of CBD [132].
Ethnic minority status is one of the strongest risk factors for the development of schizophrenia, and ethnically minoritized individuals also tend to have poorer educational attainment [114, 133]. When examining specific ethnic groups, there are examples of correspondence between these outcomes. In England, individuals with Caribbean ancestry show both the greatest increased risk of developing a psychotic disorder and also the poorest educational attainment [114, 133]. Similarly, in individuals of South Asian descent, individuals with Pakistani heritage show both poorer educational attainment and an increased risk of schizophrenia that is not seen in those of Indian heritage [133,134,135]. Among ethnically minoritized individuals, the effect of ethnicity on schizophrenia risk is even greater if they are also a minority in their local neighbourhood [136]. Similarly, an increase in median neighbourhood income is linked to better cognitive test results in black children, but only if they live in an area that has a high proportion of black individuals [137]. Greater exposure to socioeconomic deprivation, childhood trauma, and racism both structural and interpersonal all have the potential to play a role in explaining these associations, and it remains to be determined to what extent the pathways to increased schizophrenia risk and poorer performance on indices of cognitive ability are parallel or intertwined [138].
Individuals with schizophrenia show a further decline in cognitive ability in later life. The latter may reflect an increased prevalence of smoking [139], obesity [140], and hyperglycaemia [141], which can have an adverse effect on cerebrovascular function. Hypertension, diabetes, and metabolic syndrome are all associated with significantly worse cognitive functioning in individuals with schizophrenia [142]. Additional factors include the lack of social and vocational stimulation that can be associated with the disorder [143].
In summary, impaired cognitive function in people with schizophrenia is related to their genetic loading, an increased exposure to environmental factors that are associated with reduced cognitive performance (Fig. 3), and with poor physical health in later life.
The pathophysiology of cognitive impairment in schizophrenia
A wide range of neurotransmitters and brain circuits are implicated in schizophrenia. Many of these appear to converge on a common pathway fundamental to cognitive functioning, namely the balanced interactions between excitatory and inhibitory (E/I) neurons of cortical microcircuits (Fig. 4). Excitation allows neurons to fire in response to stimuli, while inhibition tunes their responses, allowing for precise neural representations. The balance between the two is crucial for the neural computations underlying cognition. We now discuss the role of dopaminergic, cholinergic, glutamatergic, and GABAergic systems. We discuss the evidence for aberrant function in schizophrenia, the role in healthy cognitive function, and their part in modulating E/I balance.

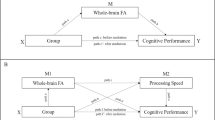
A Muscarinic, dopaminergic, glutamatergic, and GABAergic influences on E/I balance in healthy controls and mechanisms of disruption in schizophrenia. B E/I balance and cognition. In healthy individuals, interactions between excitatory pyramidal cells and inhibitory interneurons generate gamma oscillations, which are associated with functional brain networks observable using fMRI, with all levels showing links to healthy cognitive function. In individuals with schizophrenia, disruptions to muscarinic and dopaminergic signalling, an intrinsic interneuron deficits may underlie a state of cortical disinhibition. This disinhibition would account for the aberrant gamma activity and functional networks observed in the disorder and contribute to cognitive impairments.
Dopamine
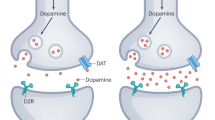
Dopamine occupies a central role in the pathophysiology of schizophrenia [144,145,146]. In preclinical models, increased dopamine signalling is associated with schizophrenia-like behaviours [147]. In patients, post-mortem studies show dopamine receptor upregulation [148], and the potency of antipsychotic medications is tightly linked to their affinity for the D2 receptor [4, 149]. Neuroimaging studies indicate that schizophrenia is associated with presynaptic hyperdopaminergia in the dorsal striatum [150], and experimental stimulation of striatal dopaminergic transmission (e.g., following administration of amphetamine) can induce and exacerbate psychosis [151, 152].
Although dopamine dysfunction has mainly been linked to positive psychotic symptoms, striatal dopamine signalling normally has widespread effects on cortical function [153,154,155,156], and its dysregulation can also cause cognitive symptoms [157]. Measures of striatal dopamine signalling appear to index ‘state’ aspects of psychosis, increasing during acute psychosis and demonstrating an association with the severity of positive psychotic symptoms, positioning striatal dopamine as ‘the wind of the psychotic fire’ [158,159,160]. Cognitive symptoms, however, follow a much more constant trajectory than positive symptoms, and as such, it is less likely that they are directly tied to neurochemical measures that fluctuate over the illness course [161, 162].
In vivo measurement of cortical dopamine signalling is challenging given the relative sparsity of the receptors. However, data from recent studies are consistent with the hypothesis that schizophrenia is associated with a hypodopaminergic state in the cortex [163,164,165]. Moreover, cortical hypodopaminergia may be linked to striatal hyperdopaminergia in schizophrenia: preclinical work has demonstrated that depletion of cortical dopamine increases striatal dopamine levels [166, 167], and there is preliminary in vivo evidence that the same relationship applies in people with schizophrenia [165]. The lack of effect of dopamine antagonists on cognitive symptoms may reflect the fact that correcting the dysregulated striatal signalling in schizophrenia requires not only the blocking of aberrant signals, but also the restoration of the signal-to-noise ratio of adaptive signals [168].
Cortical dopamine signalling plays a role in normal attention, working memory, and executive function, so impaired dopamine function in schizophrenia may therefore affect these processes [169,170,171]. Dopamine promotes the persistent cortical activity required for the maintenance of working memory, while inhibiting unrelated signalling so that relevant information is precisely represented [172]. Studies in people with Parkinson’s disease, and in non-human primates with experimental lesions of prefrontal dopamine function indicate that impaired dopamine signalling leads to working memory and executive functioning deficits [173, 174]. Conversely, The effectiveness of pro-dopaminergic compounds in the treatment of ADHD suggests that augmentation of cortical dopamine signalling has the potential to exert cognitive benefits [175]. Recently the dopaminergic partial agonist cariprazine has demonstrated some advantages on cognitive subscales of the PANSS when compared to antagonists, but more in-depth testing of its cognitive effects is required [176].
However, despite some promising signals in experimental medicine studies [177], pro-dopaminergic treatments have not been approved as treatments for the cognitive deficits of schizophrenia. This may reflect the complexity of the system involved. The effects of dopamine in the cortex are distinct to those in the striatum, and only a fraction of cortical pyramidal neurons express dopamine receptors, with cortical interneurons showing proportionally greater expression [178]. For both pyramidal cells and interneurons, dopamine’s presynaptic effects predominantly reduce neurotransmitter release. Postsynaptically, dopamine enhances the excitability of deep-layer pyramidal neurons, and increases the frequency of interneuron spiking. The net effect of these various mechanisms is that cortical dopamine depresses pyramidal cell firing via its action on interneurons. A link with E/I balance is therefore apparent, in that reduced cortical dopamine signalling is associated with a reduced net inhibitory input, termed disinhibition.
Acetylcholine
Acetylcholine plays a central role in attention and memory. Treatment with anticholinergic drugs can lead to cognitive impairments in these domains as a side effect, and the majority of medications used for treating symptoms of Alzheimer’s disease operate by promoting cholinergic signalling [179, 180]. Central cholinergic innervation is split between two primary networks, the pedunculopontine cholinergic complex which projects to the midbrain, and the forebrain complex which projects to the cortex. The effects of acetylcholine are mediated by two receptor types—ionotropic nicotinic receptors and G protein-coupled muscarinic receptors.
A link between the nicotinic system and schizophrenia is supported by the strikingly high prevalence of tobacco dependence, with around 65% of patients being smokers [181]. This high prevalence has persisted despite clinical initiatives to reduce smoking in patients with schizophrenia, which is a key factor in the 15–20-year reduction in life expectancy associated with the disorder [182]. Most patients are aware of this health risk, but report that they continue to smoke because it improves their concentration and reduces anxiety [183]. This is in keeping with evidence that the acute administration of nicotine ameliorates sensory gating abnormalities and enhances cognitive performance in schizophrenia [184, 185]. However, this enhancement is not seen with chronic use [186], and in the longer term, smoking in schizophrenia is associated with poorer cognitive performance and increases the risk of late-life cognitive decline [187]. Furthermore, smoking cessation appears to improve cognition in schizophrenia [187]. The immediate benefits of tobacco smoking may arise from initial agonism at cholinergic receptors, with the deleterious effects resulting from receptor desensitisation due to chronic exposure. In addition, in people with schizophrenia, the repeated stimulation of nicotinic receptors by smoking leads to less of a reduction in nicotinic receptor expression than in control smokers [188]. Despite this strong association between cholinergic signalling and cognition in schizophrenia, agents that affect nicotine receptor function such as varenicline, or that target cholinergic neurotransmission more broadly, like acetylcholinesterase inhibitors have had disappointing results when used to treat the cognitive deficits of schizophrenia [189, 190].
A separate body of evidence suggests that schizophrenia is associated with aberrant functioning of the muscarinic system. Acute administration of muscarinic antagonists can exacerbate both cognitive deficits and positive symptoms in individuals with schizophrenia, and can induce cognitive deficits and positive symptoms in controls [191]. The G protein-coupled muscarinic receptors either have predominantly excitatory (M1, M3 and M5 subtypes) or inhibitory (M2 and M4) effects. Post-mortem studies in schizophrenia show significant reductions of both M1 and M4 receptors in hippocampus and cortex [191,192,193]. Similarly, PET studies have found reduced muscarinic receptor density in schizophrenia, and have linked this to the presence of cognitive symptoms [194,195,196]. M1 receptors play an important role in learning and memory [197], and M1 agonism may improve both positive symptoms and cognitive performance in people with schizophrenia [198,199,200].
Both nicotinic and muscarinic receptor systems affect GABAergic and glutamatergic signalling and can thereby contribute to the maintenance of E/I balance [197]. Nicotinic receptors are sparsely distributed on pyramidal cells but are more frequently expressed on interneurons, particularly those expressing vasoactive intestinal peptide (VIP) [201,202,203]. VIP interneurons predominantly inhibit other interneurons, and the net effect of nicotinic stimulation upon E/I balance is therefore to increase pyramidal cell firing. This effect may be beneficial if there is reduced pyramidal signalling, but is unlikely to be helpful if there is a state of cortical disinhibition, as may occur in schizophrenia. In contrast to nicotinic receptors, M1 receptors are widely expressed on parvalbumin-positive interneurons and their activation enhances GABA release, playing an important role in learning and memory [197]. M1 receptors are also strongly expressed on pyramidal cells, suggesting that the net effect of M1 agonism might be to increase cortical excitability. Recent work, however, demonstrates that both M1 agonism and antagonism have a net inhibitory effect on cortical pyramidal neurons. The overall effect of modulating activity at M1 receptors is likely to depend on the degree of endogenous tone: at typical levels, this may already be saturated, such that effects are primarily inhibitory via increased activation of interneurons, or a direct effect on pyramidal cells via other pathways [204]. These effects on E/I balance are evident in the modulation of gamma oscillations by muscarinic compounds [205]. Muscarinic agonism is therefore likely to produce cognitive benefits when there is a state of cortical disinhibition, which the evidence presented below suggests occurs in schizophrenia.
In addition to the links with excitation and inhibition, cholinergic signalling also impacts on the dopamine system. At low agonist concentrations, stimulation of nicotinic receptors on GABAergic projections to mesostriatal dopamine neurons inhibits dopamine release, while at higher concentrations this mechanism becomes saturated and stimulation of receptors on glutamatergic projection neurons increases dopamine neuron firing [206]. Stimulation of M4 muscarinic receptors on striatal cholinergic interneurons can reduce acetylcholine release and thereby minimise cholinergic excitation of dopamine neurons projecting to the striatum. M4 agonism can also reduce striatal dopamine release by inhibiting the activity of spiny projection neurons to the midbrain [207, 208]. In contrast, and of particular interest given the reduced cortical dopamine signalling observed in schizophrenia, M1 agonism appears to increase cortical dopamine release [209,210,211].
E/I balance
GWAS have demonstrated that SNPs associated with schizophrenia are concentrated in genes expressed in E/I neurons [89]. Post-mortem studies, in line with a model of cortical disinhibition, have found reductions in the enzymes GAD65 and GAD67, which are required for GABA synthesis, as well as reduced excitatory input to inhibitory neurons, and reduced interneuron numbers [212, 213]. A wide range of animal models that display cognitive deficits analogous to those observed in schizophrenia, demonstrate cortical disinhibition as a common final condition underlying pathological behaviour [214, 215].
E/I balance in humans can be assessed using neuroimaging. Magnetic resonance spectroscopy (MRS) can measure glutamate and GABA concentrations at rest, and functional MRS (fMRS) can examine how these dynamically change in the face of a cognitive challenge [216,217,218]. Studies in individuals with schizophrenia have demonstrated increased Glx (glutamate and glutamine signal combined) [219] and reduced GABA [220], consistent with underlying cortical disinhibition. However, it is not possible to disambiguate intracellular and intrasynaptic signals in MRS data, and there is heterogeneity in the direction of reported findings, which may reflect differences in acquisition methods and medication exposure as much as pathophysiological heterogeneity [219, 221,222,223]. Nevertheless, an increase in patients who do not benefit from standard treatment with dopamine antagonists appears to be a relatively robust finding [220, 224, 225]. fMRS shows that cognitive challenges induce an increase in the ratio of glutamate:GABA concentrations, and there is preliminary evidence that this increase is reduced in schizophrenia [226, 227]. PET ligands can provide a greater level of molecular specificity than MRS. There have been fewer studies, but these have suggested a reduction in both GABA and NMDA receptors [228,229,230]. In addition, reduced availability of the metabotropic glutamate receptors type 5 has been linked to cognitive symptoms in schizophrenia [231].
E/I can also be investigated using electroencephalography (EEG). Interactions between pyramidal cells and interneurons generate neuronal oscillations in the gamma (γ) frequency range (Fig. 4) [232, 233]. In controls, gamma oscillations are associated with working memory, and memory tasks elicit an increase in pyramidal neuron firing which is manifest as an increase in gamma power [234, 235]. Cortical disinhibition is associated with increased resting gamma power, and a reduced ability to mount the normal increase in gamma power in the face of cognitive demands [215, 233, 236,237,238,239]. Several studies suggest that schizophrenia is associated with both increased resting gamma power and reduced task-induced increases in power [240,241,242,243,244,245,246,247], consistent with a state of cortical disinhibition. Furthermore, reduced mismatch negativity (an EEG-detectable response to surprising stimuli) is a marker of cortical disinhibition [248, 249], and is observed in psychosis where it is associated with cognitive symptoms [250,251,252,253,254].
Gamma oscillations are associated with the emergence of whole-brain functional networks observable using fMRI (Fig. 4) [255,256,257]. These fMRI-derived networks are associated with cognition in healthy individuals, and with the cognitive symptoms of schizophrenia [258, 259]. Computational models have demonstrated how a global disruption of cortical disinhibition may be manifest through regionally varying patterns of aberrant resting state fMRI activity that mirror those observed in schizophrenia [260,261,262,263]. These models indicate that despite the ubiquitous nature of glutamatergic and GABAergic signalling, even a globally present microscale deficit may result in non-uniform effects on macroscale neural dynamics across the cortex, as the wiring patterns of the brain are non-random [261]. Regions that are situated centrally in terms of network dynamics may be where functional effects become concentrated and therefore appear to reflect focal alterations in neuroimaging studies, despite having a spatially distributed molecular basis [264]. Nevertheless, however, focal effects at a molecular level are also possible. For example, because parvalbumin interneurons have extremely high energy requirements, oxidative and metabolic stressors can have a disproportionate impact on these neurons, and their effects may be greatest in regions where they are present at high densities [265].
Thus, while the model of E/I imbalance discussed aims to explain systems-level phenomena, it is not inconsistent with findings that certain brain regions, such as the hippocampus, appear to play a central role in both cognitive function and schizophrenia [266]. Similarly, the functional alterations described above are likely to be interlinked with other well-established aspects of schizophrenia pathophysiology. Aberrations of neurotransmitter function will be coupled, both as cause and consequence, to structural alterations in terms of both anatomical connectivity and grey matter loss. The relevance of structural changes to cognition is seen in recent machine learning studies identifying a pattern of grey matter loss linked to subgroups of patients with cognitive deficits [267,268,269].
Finally, at the level of behaviour, computational models predict that cortical disinhibition will be associated with a reduced precision of neural representations, and therefore precision-related deficits on working memory tasks [260]. The pattern of working memory deficits observed in both psychosis and pharmacologically-induced disinhibition are in keeping with these predictions [260, 263]. At a higher level, a ‘cognitive map’ refers to the concept that accumulated knowledge and experiences are linked in an organised structure that allows for subsequent novel inferences. Disruption to the architecture of cognitive maps schizophrenia may potentially provide a framework to account for problems in executive functioning and general reasoning abilities [270]. A key consideration in terms of the inferential reasoning supported by a cognitive map is how easily separate memories can be linked: too high a barrier and no connections can be made; too low and entirely unrelated memories may be inappropriately linked. Impairment in this process of memory linkage has recently been demonstrated in schizophrenia [271]. As inhibitory signalling is crucial for appropriate memory separation [272], disinhibition could promote the aberrant associations and working memory deficits that are evident in people with schizophrenia [270].
There is thus multimodal evidence that E/I balance is disrupted in schizophrenia and is associated with cognitive deficits. While we have highlighted findings consistent with a pattern of cortical disinhibition, the nature of the primary deficit has not been firmly established. One hypothesis is that the loss, and/or aberrant function of parvalbumin-positive interneurons (e.g., secondary to NMDA receptor hypofunction, or reduced dopaminergic stimulation) reflects a direct pathophysiological process, with the implication that augmenting the activity of this cell type could ameliorate cognitive deficits. This would be consistent with the ability of the NMDA antagonist ketamine to induce acute psychotic symptoms in healthy volunteers [273], and the finding that anti-NMDA autoantibodies may cause acute psychosis [274]. Moreover, animal models of interneuron dysfunction result in the expression of schizophrenia-like phenotypes, and various computational models have linked cortical disinhibition to schizophrenia [215, 260, 261]. However, it is also possible that the reduced activity of interneuron populations reflects an adaptive response to a primary reduction in cortical pyramidal cell activity. According to this model, a reduction in inhibitory interneuron activity would restore pyramidal cell firing, so novel treatments designed to increase interneuron activity would therefore be expected to have a deleterious effect. Supporting this interpretation are post-mortem work suggesting that schizophrenia is associated with a reduction in the density of dendritic spines on pyramidal cells, in vivo findings demonstrating reduced synaptic density in the cortex [275,276,277], and data from biophysical network models of fMRI and MEG data pointing to a reduction in synaptic gain on pyramidal cells [278]. It is also consistent with the decline in cognitive function seen in schizophrenia during adolescence, a period when the pruning of dendritic spines is at its peak [279]. A key outstanding question is thus whether reduced inhibitory signalling in schizophrenia represents a primary pathology which treatment should aim to ameliorate, or a compensatory mechanism which, conversely, treatment should try to further reduce.
The treatment of cognitive impairment in schizophrenia
Existing treatments
All medications currently licensed for the treatment of schizophrenia exert their clinical effects via antagonism of the dopamine D2 receptor, and are described as antipsychotics. In the 1990s, the introduction of the so-called ‘atypical’ antipsychotics was accompanied by trials suggesting that these newer types of antipsychotic medication had beneficial effects on cognitive symptoms, in addition to psychotic symptoms. Subsequent research, however, suggested that these purported advantages may have been artefacts of trial design, and the current literature suggests that all antipsychotics have similarly small effects on cognitive function [280].
Other medications used in the management of schizophrenia may also have effects on cognition. The anticholinergic effects of many psychotropic medications have a clear detrimental effect on cognitive function, and reducing the dose of anticholinergic medications used to ameliorate extrapyramidal side effects of antipsychotics can improve cognitive function in schizophrenia [281, 282]. Affective symptoms like depression are often associated with cognitive symptoms [283, 284], and while the non-selective administration of antidepressants in schizophrenia does not appear to have pro-cognitive effects [285], the benefits on cognitive function from their selective use in the subgroup of patients with schizophrenia that have prominent depressive symptoms have yet to be assessed.
In the longer term, treating the high prevalence of obesity, type II diabetes and cardiovascular illness in patients with schizophrenia has the potential to minimise the cognitive decline associated with these comorbidities. However, while there has recently been great progress in the detection and monitoring of physical health problems in schizophrenia, the impact of interventions targeting these is only beginning to be investigated [286]. Sleep disturbance is also common in schizophrenia [287], and given the well-established links between sleep and cognitive function [288], addressing sleep problems may also lead to cognitive benefits in schizophrenia. However, treating sleep dysfunction is difficult, and the effectiveness of this approach in people with schizophrenia is unclear. Cognitive remediation therapy in schizophrenia has been shown to improve cognitive test scores, although as a significant component of the intervention involves the repeated practice of cognitive tests there is a question as to how much this represents practice effects. However, there do appear to be detectable benefits on level of functioning, although of relatively small effect size [289, 290]. Nevertheless, at present, no psychological interventions have been approved for the treatment of cognitive symptoms in schizophrenia [291]. Psychological treatments for impairments in social cognition have a less established evidence base but also show some potential [292]. For a more in-depth discussion of potential mechanisms of psychological interventions for cognitive deficits see recent reviews [293, 294].
Novel treatments
Trace amine-associated receptor 1 (TAAR1) agonists can reduce central dopamine signalling by reducing midbrain dopamine neuron firing [295]. TAAR1 agonism has recently been reported to reduce psychotic symptoms in schizophrenia [296]. Although its effects on cognitive symptoms have yet to be examined, in animal models of schizophrenia it has been found to improve cognitive performance and increase prefrontal cortical activity [297]. The precise mechanisms underlying these latter effects are unclear. Targeting presynaptic neuronal activity may have effects on dopamine signalling that are different from the antagonism of post-synaptic D2 receptors, a hypothesis supported by the absence of extrapyramidal side effects associated with TAAR1 agonism [296]. There is also evidence that TAAR1 agonism affects E/I balance [298].
While the augmentation of cholinergic signalling via drugs that inhibit acetylcholinesterase provides some benefit to cognitive function in Alzheimer’s disease, application of this approach in schizophrenia has not been successful [299, 300]. This suggests a more targeted approach to modulating the cholinergic system may be required. The M1/M4 receptor agonist xanomeline appears to improve both positive and cognitive symptoms in randomised controlled trials in schizophrenia, as well as in Alzheimer’s disease [198, 199, 301]. Initial development of xanomeline was paused due to the high incidence of peripheral pro-cholinergic side effects, but combining it with trospium, a peripheral cholinergic antagonist, markedly reduces its side effects without affecting its efficacy [199]. As discussed above, the mechanism of action here likely involves effects on both dopamine signalling and on E/I balance.
CBD has become increasingly recognised as a potentially effective treatment for schizophrenia. The largest trial of adjunctive CBD in schizophrenia to date demonstrated a clear benefit of CBD on positive symptoms. Although this study was not powered to assess effects on cognition, there was a trend for an improvement in the speed of processing [302]. Human imaging studies suggest that the effects of CBD may be mediated by modulating hippocampal and striatal function [303], but CBD acts on a variety of molecular targets, and the precise mechanism underlying its therapeutic effects remains unclear [304]. However, its efficacy in treating seizures, ability to improve cognition in a mouse model of E/I imbalance, and electrophysiological data from rodents suggest that it may modulate E/I balance, and this may involve antagonising the G protein-coupled receptor GPR55 [305,306,307].
A wide range of compounds targeting the glutamate system directly have been tested as potential treatments for cognitive dysfunction in schizophrenia, but the results have been disappointing [308, 309]. Agonism of metabotropic mGlu2/3 receptors was initially seen as promising and led to large-scale trials, but development ceased when these failed to show an effect on positive or negative symptoms [310, 311], and the effects on cognitive impairments were not directly tested. Both riluzole and memantine, drugs approved for the treatment of motor neuron disease and Alzheimer’s disease respectively, have complex effects on the E/I system and have shown promise as treatments for cognitive symptoms in schizophrenia [312, 313]. Although demonstrating efficacy on cognitive symptoms in meta-analyses, memantine did not show cognitive benefits in a large trial, although this was in an entirely unselected patient population [314]. More recently a glycine transport inhibitor has demonstrated efficacy as an augmentation agent for treating cognitive symptoms [315]. There is also preliminary evidence that luvadaxistat, a D-amino acid oxidase inhibitor that augments glutamatergic signalling by elevating serine levels, may have beneficial effects on cognitive symptoms [316].
Future directions
There remain several questions as to the nature of cognitive deficits in schizophrenia. Phenotypically similar cognitive impairments are also evident in other psychiatric disorders, mainly differing from those in schizophrenia in terms of their severity. It is unclear whether the cognitive deficits in these conditions have the same pathophysiological basis as those in schizophrenia. If so, then transdiagnostic approaches to developing interventions may be of benefit. Identifying whether variation in cognitive performance in schizophrenia relates to variation in the severity and nature of the illness, as opposed to variation in premorbid function also has relevance for whether precision psychiatry approaches (which aim to stratify patients according to differing underlying pathophysiologies [280]) are likely to be indicated. In the case of the former, it may be possible to match specific treatments to distinct pathophysiological mechanisms, whereas the latter case represents a uniform insult and so individual differences in treatment effects are less likely.
In terms of work with direct clinical relevance, clinical guidelines recommend the assessment of cognition in standard clinical practice [317], and raising clinician awareness of the need to formally assess cognitive function will become increasingly important as novel interventions become available. It is therefore critical that clinicians are provided with the tools to facilitate the measurement of cognitive impairments so that they can assess the effect of treatment. Traditional cognitive batteries are too time consuming for use in routine clinical care, but the introduction of briefer batteries that can be administered without a trained assessor has major potential here.
In clinical trials, there may be an advantage in evaluating candidate treatments in specific patient subpopulations. Recruiting patients early in the illness course may be of benefit, as although most cognitive deficits will already be present, the higher degree of neuronal plasticity at this stage may increase the chances that intervention will have beneficial effects. Studying individuals early in the disorder also reduces the likelihood that outcomes will be confounded by effects of prior drug treatment. Another subgroup that could be targeted is patients with the greatest degree of cognitive impairment. A recent trial of xanomeline-trospium in schizophrenia found that its impact on cognition was restricted to those with a prominent degree of impairment at baseline [199]. However, it could also be argued that patients with relatively severe cognitive deficits are less likely to improve, due to the severity of neurobiological damage or a high burden of risk factors. To date, most trials of interventions designed to improve cognition in schizophrenia have recruited patients without stratifying samples according to the severity of cognitive impairments. Until it is clear whether enriching samples for patients with marked cognitive deficits will increase or decrease the detection of therapeutic effects, it may be sensible to continue with this approach.
Tying the effects of interventions to real-world functional outcomes is a major challenge but is critical if the aim is to produce outcomes that are meaningful to patients. The use of virtual reality tools may be of benefit here [281]. Non-linear relationships have been observed between symptom dimensions and cognition in schizophrenia [318, 319]. If non-linearities are also seen in the relationship between cognition and function, it is possible that cognitive impairments may only have marked effects on a patient’s level of functioning when they exceed a certain severity threshold. Clarifying the nature of this relationship in schizophrenia could be a goal for future research. If multiple medications are found to be effective in clinical trials precision psychiatry approaches will be important to optimise clinical benefits. Finally, trials aimed at determining whether novel treatments produce transdiagnostic improvements, or whether these are diagnosis specific has major relevance for both clinical practice and development of future treatments.
Conclusion
Over the past three decades, cognitive impairment has emerged as an increasingly important treatment target for schizophrenia. However, the complexity of the neurobiological substrate has made the development of treatments for these deficits particularly challenging. Nevertheless, there are now a number of clinical interventions which have the potential to improve cognitive function in individuals with schizophrenia, and several new treatments with entirely novel mechanisms of action are ready to be rigorously tested. The development of effective treatments for cognitive impairments in schizophrenia would represent an advance in its treatment comparable to the advent of D2 antagonists over 70 years ago.
Change history
02 February 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41380-023-01984-6
References
Keefe RSE, Fox KH, Harvey PD, Cucchiaro J, Siu C, Loebel A. Characteristics of the MATRICS Consensus Cognitive Battery in a 29-site antipsychotic schizophrenia clinical trial. Schizophr Res. 2011;125:161–8.
Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–30.
Sabe M, Pillinger T, Kaiser S, Chen C, Taipale H, Tanskanen A, et al. Half a century of research on antipsychotics and schizophrenia: a scientometric study of hotspots, nodes, bursts, and trends. Neurosci Biobehav Rev. 2022;136:104608.
Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217–9.
Kaar SJ, Natesan S, McCutcheon R, Howes OD. Antipsychotics: mechanisms underlying clinical response and side-effects and novel treatment approaches based on pathophysiology. Neuropharmacology. 2020;172:107704. https://doi.org/10.1016/j.neuropharm.2019.107704.
van der Gaag M, Hoffman T, Remijsen M, Hijman R, de Haan L, van Meijel B, et al. The five-factor model of the Positive and Negative Syndrome Scale II: a ten-fold cross-validation of a revised model. Schizophr Res. 2006;85:280–7.
Wallwork RS, Fortgang R, Hashimoto R, Weinberger DR, Dickinson D. Searching for a consensus five-factor model of the Positive and Negative Syndrome Scale for schizophrenia. Schizophr Res. 2012;137:246–50.
Rodriguez-Jimenez R, Bagney A, Mezquita L, Martinez-Gras I, Sanchez-Morla EM, Mesa N, et al. Cognition and the five-factor model of the Positive and Negative Syndrome Scale in schizophrenia. Schizophr Res. 2013;143:77–83.
Bell MD, Lysaker PH, Milstein RM, Beam-Goulet JL. Concurrent validity of the cognitive component of schizophrenia: relationship of PANSS scores to neuropsychological assessments. Psychiatry Res. 1994;54:51–58.
Moura BM, Van Rooijen G, Schirmbeck F, Wigman H, Madeira L, Van Harten P, et al. A network of psychopathological, cognitive, and motor symptoms in schizophrenia spectrum disorders. Schizophr Bull. 2021;47:915–26.
Chang WC, Wong CSM, Or PCF, Chu AOK, Hui CLM, Chan SKW, et al. Inter-relationships among psychopathology, premorbid adjustment, cognition and psychosocial functioning in first-episode psychosis: a network analysis approach. Psychol Med. 2020;50:2019–27.
Browne J, Penn DL, Raykov T, Pinkham AE, Kelsven S, Buck B, et al. Social cognition in schizophrenia: factor structure of emotion processing and theory of mind. Psychiatry Res. 2016;242:150–6.
Fett AKJ, Viechtbauer W, Dominguez MdeG, Penn DL, van Os J, Krabbendam L. The relationship between neurocognition and social cognition with functional outcomes in schizophrenia: a meta-analysis. Neurosci Biobehav Rev. 2011;35:573–88.
Green MF, Horan WP, Lee J. Social cognition in schizophrenia. Nat Rev Neurosci. 2015;16:620–31.
Green MF, Horan WP, Lee J. Nonsocial and social cognition in schizophrenia: current evidence and future directions. World Psychiatry. 2019;18:146–61.
Schneider WJ. Why are WJ IV cluster scores more extreme than the average of their parts? A gentle explanation of the composite score extremity effect. Woodcock-Johnson IV assessment service bulletin no. 7; 2016.
Fioravanti M, Carlone O, Vitale B, Cinti ME, Clare L. A meta-analysis of cognitive deficits in adults with a diagnosis of schizophrenia. Neuropsychol Rev. 2005;15:73–95.
Mesholam-Gately RI, Giuliano AJ, Goff KP, Faraone SV, Seidman LJ. Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology. 2009;23:315–36.
Fatouros-Bergman H, Cervenka S, Flyckt L, Edman G, Farde L. Meta-analysis of cognitive performance in drug-naïve patients with schizophrenia. Schizophr Res. 2014;158:156–62.
Pietrzak RH, Olver J, Norman T, Piskulic D, Maruff P, Snyder PJ. A comparison of the CogState Schizophrenia Battery and the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) Battery in assessing cognitive impairment in chronic schizophrenia. J Clin Exp Neuropsychol. 2009;31:848–59.
Keefe RSE, Goldberg TE, Harvey PD, Gold JM, Poe MP, Coughenour L. The Brief Assessment of Cognition in Schizophrenia: reliability, sensitivity, and comparison with a standard neurocognitive battery. Schizophr Res. 2004;68:283–97.
August SM, Kiwanuka JN, McMahon RP, Gold JM. The MATRICS Consensus Cognitive Battery (MCCB): clinical and cognitive correlates. Schizophr Res. 2012;134:76–82.
Nuechterlein KH, Barch DM, Gold JM, Goldberg TE, Green MF, Heaton RK. Identification of separable cognitive factors in schizophrenia. Schizophr Res. 2004;72:29–39.
Burton CZ, Vella L, Harvey PD, Patterson TL, Heaton RK, Twamley EW. Factor structure of the MATRICS Consensus Cognitive Battery (MCCB) in schizophrenia. Schizophr Res. 2013;146:244–8.
Dickinson D, Ramsey ME, Gold JM. Overlooking the obvious. Arch Gen Psychiatry. 2007;64:532.
Harvey PD. Domains of cognition and their assessment. Dialogues Clin Neurosci. 2019;21:227–37.
Ojeda N, Peña J, Schretlen DJ, Sánchez P, Aretouli E, Elizagárate E, et al. Hierarchical structure of the cognitive processes in schizophrenia: the fundamental role of processing speed. Schizophrenia Res. 2012;135:72–78.
Knowles EEM, David AS, Reichenberg A. Processing speed deficits in schizophrenia: reexamining the evidence. Am J Psychiatry. 2010;167:828–35.
Knowles EEM, Weiser M, David AS, Glahn DC, Davidson M, Reichenberg A. The puzzle of processing speed, memory, and executive function impairments in schizophrenia: fitting the pieces together. Biol Psychiatry. 2015;78:786–93.
Dickinson D, Ragland JD, Gold JM, Gur RC. General and specific cognitive deficits in schizophrenia: Goliath Defeats David? Biol Psychiatry. 2008;64:823–7.
Keefe RSE, Bilder RM, Harvey PD, Davis SM, Palmer BW, Gold JM, et al. Baseline neurocognitive deficits in the CATIE schizophrenia trial. Neuropsychopharmacology. 2006;31:2033–46.
Joyce EM, Roiser JP. Cognitive heterogeneity in schizophrenia. Curr Opin Psychiatry. 2007;20:268–72.
Catalan A, Radua J, Mccutcheon R, Aymerich C, Pedruzo B, González-torres MÁ, et al. Examining the variability of neurocognitive functioning in individuals at clinical high risk for psychosis: a meta-analysis. Transl Psychiatry. 2022;12:198. https://doi.org/10.1038/s41398-022-01961-7.
Shin YS, Kim SN, Shin NY, Jung WH, Hur J-W, Byun MS, et al. Increased intra-individual variability of cognitive processing in subjects at risk mental state and schizophrenia patients. PLoS One. 2013;8:e78354.
Reichenberg A, Weiser M, Rapp MA, Rabinowitz J, Caspi A, Schmeidler J, et al. Premorbid intra-individual variability in intellectual performance and risk for schizophrenia: a population-based study. Schizophr Res. 2006;85:49–57.
Woodberry KA, Giuliano AJ, Seidman LJ. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165:579–87.
Mollon J, David AS, Zammit S, Lewis G, Reichenberg A. Course of cognitive development from infancy to early adulthood in the psychosis spectrum. JAMA Psychiatry. 2018;75:270–9.
Reichenberg A, Caspi A, Harrington H, Houts R, Keefe RSE, Murray RM, et al. Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry. 2010;167:160–9.
MacCabe JH, Wicks S, Löfving S, David AS, Berndtsson Å, Gustafsson JE, et al. Decline in cognitive performance between ages 13 and 18 years and the risk for psychosis in adulthood: a Swedish longitudinal cohort study in males. JAMA Psychiatry. 2013;70:261–70.
Sheffield JM, Karcher NR, Barch DM. Cognitive deficits in psychotic disorders: a lifespan perspective. Neuropsychol Rev. 2018;28:509–33.
Wilk CM, Gold JM, McMahon RP, Humber K, Iannone VN, Buchanan RW. No, it is not possible to be schizophrenic yet neuropsychologically normal. Neuropsychology. 2005;19:778–86.
Catalan A, Salazar De Pablo G, Aymerich C, Damiani S, Sordi V, Radua J, et al. Neurocognitive functioning in individuals at clinical high risk for psychosis: a systematic review and meta-analysis. JAMA Psychiatry. 2021;78:859–67.
Salazar de Pablo G, Radua J, Pereira J, Bonoldi I, Arienti V, Besana F, et al. Probability of transition to psychosis in individuals at clinical high risk: an updated meta-analysis. JAMA Psychiatry. 2021;78:970–8.
Bora E, Murray RM. Meta-analysis of cognitive deficits in ultra-high risk to psychosis and first-episode psychosis: Do the cognitive deficits progress over, or after, the onset of psychosis? Schizophr Bull. 2014;40:744–55.
Hedges EP, See C, Si S, McGuire P, Dickson H, Kempton MJ. Meta-analysis of longitudinal neurocognitive performance in people at clinical high-risk for psychosis. Psychol Med. 2022;52:2009–16.
Kraepelin E. Psychiatrie; ein Lehrbuch für Studierende und Ärzte. Leipzig: Barth 1913.
Watson AJ, Harrison L, Preti A, Wykes T, Cella M. Cognitive trajectories following onset of psychosis: a meta-analysis. Br J Psychiatry. 2022;221:714–21.
Szöke A, Trandafir A, Dupont M-E, Méary A, Schürhoff F, Leboyer M. Longitudinal studies of cognition in schizophrenia: meta-analysis. Br J Psychiatry. 2008;192:248–57.
Hedman AM, van Haren NEM, van Baal CGM, Kahn RS, Hulshoff Pol HE. IQ change over time in schizophrenia and healthy individuals: a meta-analysis. Schizophr Res. 2013;146:201–8.
Bonner-Jackson A, Grossman LS, Harrow M, Rosen C. Neurocognition in schizophrenia: a 20-year multi-follow-up of the course of processing speed and stored knowledge. Compr Psychiatry. 2010;51:471–9.
Zanelli J, Mollon J, Sandin S, Morgan C, Dazzan P, Pilecka I, et al. Cognitive change in schizophrenia and other psychoses in the decade following the first episode. Am J Psychiatry. 2019;176:811–9.
Fett AKJ, Velthorst E, Reichenberg A, Ruggero CJ, Callahan JL, Fochtmann LJ, et al. Long-term changes in cognitive functioning in individuals with psychotic disorders: findings from the Suffolk County Mental Health Project. JAMA Psychiatry. 2020;77:387–96.
Velthorst E, Mollon J, Murray RM, de Haan L, Germeys IM, Glahn DC, et al. Cognitive functioning throughout adulthood and illness stages in individuals with psychotic disorders and their unaffected siblings. Mol Psychiatry. 2021;26:4529–43.
Friedman JI, Harvey PD, Coleman T, Moriarty PJ, Bowie C, Parrella M, et al. Six-year follow-up study of cognitive and functional status across the lifespan in schizophrenia: a comparison with Alzheimer’s disease and normal aging. Am J Psychiatry. 2001;158:1441–8.
Richmond-Rakerd LS, D’Souza S, Milne BJ, Caspi A, Moffitt TE. Longitudinal associations of mental disorders with dementia: 30-year analysis of 1.7 million New Zealand citizens. JAMA Psychiatry. 2022;48109:333–40.
Reichenberg A, Harvey PD, Bowie CR, Mojtabai R, Rabinowitz J, Heaton RK, et al. Neuropsychological function and dysfunction in schizophrenia and psychotic affective disorders. Schizophr Bull. 2009;35:1022–9.
Barch DM, Bustillo J, Gaebel W, Gur R, Heckers S, Malaspina D, et al. Logic and justification for dimensional assessment of symptoms and related clinical phenomena in psychosis: relevance to DSM-5. Schizophr Res. 2013;150:15–20.
Li W, Zhou FC, Zhang L, Ng CH, Ungvari GS, Li J, et al. Comparison of cognitive dysfunction between schizophrenia and bipolar disorder patients: a meta-analysis of comparative studies. J Affect Disord. 2020;274:652–61.
Bortolato B, Miskowiak KW, Köhler CA, Vieta E, Carvalho AF. Cognitive dysfunction in bipolar disorder and schizophrenia: a systematic review of meta-analyses. Neuropsychiatr Dis Treat. 2015;11:3111–25.
Mortensen EL, Sørensen HJ, Jensen HH, Reinisch JM, Mednick SA. IQ and mental disorder in young men. Br J Psychiatry. 2005;187:407–15.
Trotta A, Murray RM, Maccabe JH. Do premorbid and post-onset cognitive functioning differ between schizophrenia and bipolar disorder? A systematic review and meta-analysis. Psychol Med. 2015;45:381–94.
Schultze-Lutter F, Ruhrmann S, Fusar-Poli P, Bechdolf A, Schimmelmann BG, Klosterkötter J. Basic symptoms and the prediction of first-episode psychosis. Curr Pharm Des. 2012;18:351–7.
Glenthøj LB, Mariegaard L, Kristensen TD, Wenneberg C, Medalia A, Nordentoft M. Self-perceived cognitive impairments in psychosis ultra-high risk individuals: associations with objective cognitive deficits and functioning. npj Schizophr. 2020;6:1–6.
Paudel S, Coman D, Freudenreich O. Subjective experience of cognitive difficulties as an important attribute of quality of life among individuals with schizophrenia spectrum disorders. Schizophr Res. 2020;215:476–8.
Raffard S, Lebrun C, Bayard S, Macgregor A, Capdevielle D. Self-awareness deficits of cognitive impairment in individuals with schizophrenia. Really? Front Psychiatry. 2020;11:731. https://doi.org/10.3389/fpsyt.2020.00731.
Homayoun S, Nadeau-Marcotte F, Luck D, Stip E. Subjective and objective cognitive dysfunction in schizophrenia is there a link?. Front Psychol. 2011;2:148. https://doi.org/10.3389/fpsyg.2011.00148.
Potvin S, Pelletier J, Stip E. La conscience des déficits neurocognitifs dans la schizophrénie: une méta-analyse. Sante Ment Que. 2014;39:183–200.
Ohmuro N, Katsura M, Obara C, Kikuchi T, Hamaie Y, Sakuma A, et al. The relationship between cognitive insight and cognitive performance among individuals with at-risk mental state for developing psychosis. Schizophr Res. 2018;192:281–6.
Saperstein AM, Thysen J, Medalia A. The Measure of Insight into Cognition: reliability and validity of clinician-rated and self-report scales of neurocognitive insight for schizophrenia. Schizophr Res. 2012;134:54–58.
Santarelli V, Marucci C, Collazzoni A, Rossetti MC, Pizziconi G, Pacitti F, et al. Could the severity of symptoms of schizophrenia affect ability of self-appraisal of cognitive deficits in patients with schizophrenia? Lack of insight as a mediator between the two domains. Eur Arch Psychiatry Clin Neurosci. 2020;270:723–8.
Haugen I, Stubberud J, Ueland T, Haug E, Øie MG. Executive dysfunction in schizophrenia: predictors of the discrepancy between subjective and objective measures. Schizophr Res Cogn. 2021;26:100201. https://doi.org/10.1016/j.scog.2021.100201.
Moritz S, Balzan RP, Bohn F, Veckenstedt R, Kolbeck K, Bierbrodt J, et al. Subjective versus objective cognition: evidence for poor metacognitive monitoring in schizophrenia. Schizophr Res. 2016;178:74–79.
Balzan RP, Neaves A, Denson LA, Liu D, Galletly C. Cognitive deficit awareness in schizophrenia: absent, intact, or somewhere in-between? Cogn Neuropsychiatry. 2014;19:471–84.
Green MF, Barnes TR, Danion J-M, Gallhofer B, Meltzer HY, Pantelis C. The FOCIS international survey on psychiatrists’ opinions on cognition in schizophrenia. Schizophr Res. 2005;74:253–61.
Belgaied W, Samp J, Vimont A, Rémuzat C, Aballéa S, El Hammi E, et al. Routine clinical assessment of cognitive functioning in schizophrenia, major depressive disorder, and bipolar disorder. Eur Neuropsychopharmacol. 2014;24:133–41.
Halverson TF, Orleans-Pobee M, Merritt C, Sheeran P, Fett AK, Penn DL. Pathways to functional outcomes in schizophrenia spectrum disorders: meta-analysis of social cognitive and neurocognitive predictors. Neurosci Biobehav Rev. 2019;105:212–9.
Sevy S, Davidson M. The cost of cognitive impairment in schizophrenia. Schizophr Res. 1995;17:1–3.
Kitchen H, Rofail D, Heron L, Sacco P. Cognitive impairment associated with schizophrenia: a review of the humanistic burden. Adv Ther. 2012;29:148–62.
Kadakia A, Fan Q, Shepherd J, Dembek C, Bailey H, Walker C, et al. Healthcare resource utilization and quality of life by cognitive impairment in patients with schizophrenia. Schizophr Res Cogn. 2022;28:100233.
Knapp M, Mangalore R, Simon J. The global costs of schizophrenia. Schizophr Bull. 2004;30:279–93.
Marwaha S, Johnson S. Schizophrenia and employment: a review. Soc Psychiatry Psychiatr Epidemiol. 2004;39:337–49.
McGurk SR, Mueser KT, Harvey PD, LaPuglia R, Marder J. Cognitive and symptom predictors of work outcomes for clients with schizophrenia in supported employment. Psychiatr Serv. 2003;54:1129–35.
Bell MD, Bryson G. Work rehabilitation in schizophrenia: does cognitive impairment limit improvement? Schizophr Bull. 2001;27:269–79.
Gold JM, Barch DM, Carter CS, Dakin S, Luck SJ, MacDonald AW, et al. Clinical, functional, and intertask correlations of measures developed by the cognitive neuroscience test reliability and clinical applications for schizophrenia consortium. Schizophr Bull. 2012;38:144–52.
Bora E, Lin A, Wood SJ, Yung AR, Mcgorry PD, Pantelis C. Cognitive deficits in youth with familial and clinical high risk to psychosis: a systematic review and meta-analysis. Acta Psychiatr Scand. 2014;130:1–15.
Snitz BE, MacDonald AW, Carter CS. Cognitive deficits in unaffected first-degree relatives of schizophrenia patients: a meta-analytic review of putative endophenotypes. Schizophr Bull. 2006;32:179–94.
Blokland GAM, Mesholam-Gately RI, Toulopoulou T, Del Re EC, Lam M, Delisi LE, et al. Heritability of neuropsychological measures in schizophrenia and nonpsychiatric populations: a systematic review and meta-analysis. Schizophr Bull. 2017;43:788–800.
Davies G, Lam M, Harris SE, Trampush JW, Luciano M, Hill WD, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9:1–16.
Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–8. https://doi.org/10.1038/s41586-022-04434-5.
Sekar A, Bialas AR, De Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83.
Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, De Leeuw CA, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet. 2018;50:912–9.
Toulopoulou T, Picchioni M, Rijsdijk F, Hua-Hall M, Ettinger U, Sham P, et al. Substantial genetic overlap between neurocognition and schizophrenia: genetic modeling in twin samples. Arch Gen Psychiatry. 2007;64:1348–55.
Hagenaars SP, Harris SE, Davies G, Hill WD, Liewald DCM, Ritchie SJ, et al. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N = 112 151) and 24 GWAS consortia. Mol Psychiatry. 2016;21:1624–32.
Lencz T, Knowles E, Davies G, Guha S, Liewald DC, Starr JM, et al. Molecular genetic evidence for overlap between general cognitive ability and risk for schizophrenia: a report from the Cognitive Genomics consorTium (COGENT). Mol Psychiatry. 2014;19:168–74.
Richards AL, Pardiñas AF, Frizzati A, Tansey KE, Lynham AJ, Holmans P, et al. The relationship between polygenic risk scores and cognition in Schizophrenia. Schizophr Bull. 2020;46:336–44.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs Cross-Disorder Group of the Psychiatric Genomics Consortium. Nat Genet. 2014;45:984–94.
Hubbard L, Tansey KE, Rai D, Jones P, Ripke S, Chambert KD, et al. Evidence of common genetic overlap between schizophrenia and cognition. Schizophr Bull. 2016;42:832–42.
Ranlund S, Calafato S, Thygesen JH, Lin K, Cahn W, Crespo-Facorro B, et al. A polygenic risk score analysis of psychosis endophenotypes across brain functional, structural, and cognitive domains. Am J Med Genet B Neuropsychiatr Genet. 2018;177:21–34.
Smeland OB, Bahrami S, Frei O, Shadrin A, O’Connell K, Savage J, et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol Psychiatry. 2020;25:844–53.
Mallet J, Le Strat Y, Dubertret C, Gorwood P. Polygenic risk scores shed light on the relationship between schizophrenia and cognitive functioning: review and meta-analysis. J Clin Med Res. 2020;9:341.
Toulopoulou T, Zhang X, Cherny S, Dickinson D, Berman KF, Straub RE, et al. Polygenic risk score increases schizophrenia liability through cognition-relevant pathways. Brain. 2019;142:471–85.
Kępińska AP, MacCabe JH, Cadar D, Steptoe A, Murray RM, Ajnakina O. Schizophrenia polygenic risk predicts general cognitive deficit but not cognitive decline in healthy older adults. Transl Psychiatry. 2020;10:422. https://doi.org/10.1038/s41398-020-01114-8.
Wray NR, Lin T, Austin J, McGrath JJ, Hickie IB, Murray GK, et al. From basic science to clinical application of polygenic risk scores: a primer. JAMA Psychiatry. 2021;78:101–9.
Lam M, Hill WD, Trampush JW, Yu J, Knowles E, Davies G, et al. Pleiotropic meta-analysis of cognition, education, and schizophrenia differentiates roles of early neurodevelopmental and adult synaptic pathways. Am J Hum Genet. 2019;105:334–50.
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–92.
Hilker R, Helenius D, Fagerlund B, Skytthe A, Christensen K, Werge TM, et al. Heritability of schizophrenia and schizophrenia spectrum based on the Nationwide Danish Twin Register. Biol Psychiatry. 2018;83:492–8.
Tucker-Drob EM, Briley DA, Harden KP. Genetic and environmental influences on cognition across development and context. Curr Dir Psychol Sci. 2013;22:349–55.
Davies C, Segre G, Estradé A, Radua J, De Micheli A, Provenzani U, et al. Prenatal and perinatal risk and protective factors for psychosis: a systematic review and meta-analysis. Lancet Psychiatry. 2020;7:399–410.
Wortinger LA, Engen K, Barth C, Lonning V, Jorgensen KN, Andreassen OA, et al. Obstetric complications and intelligence in patients on the schizophrenia-bipolar spectrum and healthy participants. Psychol Med. 2020;50:1914–22.
Lee YH, Papandonatos GD, Savitz DA, Heindel WC, Buka SL. Effects of prenatal bacterial infection on cognitive performance in early childhood. Paediatr Perinat Epidemiol. 2020;34:70–79.
Dreier JW, Berg-Beckhoff G, Andersen PK, Andersen AMN. Prenatal exposure to fever and infections and academic performance: a multilevel analysis. Am J Epidemiol. 2017;186:29–37.
Ellman LM, Yolken RH, Buka SL, Torrey EF, Cannon TD. Cognitive functioning prior to the onset of psychosis: the role of fetal exposure to serologically determined influenza infection. Biol Psychiatry. 2009;65:1040–7.
Sariaslan A, Fazel S, D’Onofrio BM, Långström N, Larsson H, Bergen SE, et al. Schizophrenia and subsequent neighborhood deprivation: revisiting the social drift hypothesis using population, twin and molecular genetic data. Transl Psychiatry. 2016;6:1–7.
Radua J, Ramella-Cravaro V, Ioannidis JPA, Reichenberg A, Phiphopthatsanee N, Amir T, et al. What causes psychosis? An umbrella review of risk and protective factors. World Psychiatry. 2018;17:49–66.
Paksarian D, Trabjerg BB, Merikangas KR, Mors O, Borglum AD, Hougaard DM, et al. The role of genetic liability in the association of urbanicity at birth and during upbringing with schizophrenia in Denmark. Psychol Med. 2018;48:305–14.
Garner CL, Raudenbush SW. Neighborhood effects on educational attainment: a multilevel analysis. Socio Educ. 1991;64:251.
Chen J, Brooks‐Gunn J. Neighborhoods and Cognitive Development. Emerging Trends in the Social and Behavioral Sciences: An Interdisciplinary, Searchable, and Linkable Resource. In: Scoutt R, Kosslyn K, editors. Hoboken, NJ: Wiley 2015. p. 1–15.
Gouin M, Flamant C, Gascoin G, Rouger V, Florin A, Guimard P, et al. The association of urbanicity with cognitive development at five years of age in preterm children. PLoS One. 2015;10:1–15.
Coutrot A, Manley E, Goodroe S, Gahnstrom C, Filomena G, Yesiltepe D, et al. Entropy of city street networks linked to future spatial navigation ability. Nature. 2022;604:104–10.
Newbury JB, Arseneault L, Beevers S, Kitwiroon N, Roberts S, Pariante CM, et al. Association of air pollution exposure with psychotic experiences during adolescence. JAMA Psychiatry. 2019;76:614–23.
Antonsen S, Mok PLH, Webb RT, Mortensen PB, McGrath JJ, Agerbo E, et al. Exposure to air pollution during childhood and risk of developing schizophrenia: a national cohort study. Lancet Planet Health. 2020;4:e64–e73.
Clifford A, Lang L, Chen R, Anstey KJ, Seaton A. Exposure to air pollution and cognitive functioning across the life course – a systematic literature review. Environ Res. 2016;147:383–98.
Majer M, Nater UM, Lin JMS, Capuron L, Reeves WC. Association of childhood trauma with cognitive function in healthy adults: a pilot study. BMC Neurol. 2010;10:61. https://doi.org/10.1186/1471-2377-10-61.
Bücker J, Kapczinski F, Post R, Ceresér KM, Szobot C, Yatham LN, et al. Cognitive impairment in school-aged children with early trauma. Compr Psychiatry. 2012;53:758–64.
Crean RD, Crane NA, Mason BJ. An evidence-based review of acute and long-term effects of cannabis use on executive cognitive functions. J Addiction Med. 2011;5:1–8.
Figueiredo PR, Tolomeo S, Steele JD, Baldacchino A. Neurocognitive consequences of chronic cannabis use: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2020;108:358–69.
Yücel M, Bora E, Lubman DI, Solowij N, Brewer WJ, Cotton SM, et al. The impact of cannabis use on cognitive functioning in patients with schizophrenia: a meta-analysis of existing findings and new data in a first-episode sample. Schizophr Bull. 2012;38:316–30.
Rabin RA, Zakzanis KK, George TP. The effects of cannabis use on neurocognition in schizophrenia: a meta-analysis. Schizophr Res. 2011;128:111–6.
Ferraro L, Quattrone D, La Barbera D, La Cascia C, Morgan C, Kirkbride JB, et al. First-episode psychosis patients who deteriorated in the premorbid period do not have higher polygenic risk scores than others: a cluster analysis of EU-GEI data. Schizophr Bull. 2023;49:218–27. https://doi.org/10.1093/schbul/sbac100.
Sánchez-Gutiérrez T, Fernandez-Castilla B, Barbeito S, González-Pinto A, Becerra-García JA, Calvo A. Cannabis use and nonuse in patients with first-episode psychosis: a systematic review and meta-analysis of studies comparing neurocognitive functioning. Eur Psychiatry. 2020;63:e6. https://doi.org/10.1192/j.eurpsy.2019.9.
González-Pinto A, González-Ortega I, Alberich S, De Azúa SR, Bernardo M, Bioque M, et al. Opposite cannabis-cognition associations in psychotic patients depending on family history. PLoS One. 2016;11:1–16.
Potter DJ, Hammond K, Tuffnell S, Walker C, Di Forti M. Potency of Δ9 -tetrahydrocannabinol and other cannabinoids in cannabis in England in 2016: implications for public health and pharmacology. Drug Test Anal. 2018;10:628–35.
Strand S. The limits of social class in explaining ethnic gaps in educational attainment. Br Educ Res J. 2011;37:197–229.
Weich S, Nazroo J, Sporston K, McManus S, Blanchard M, Erens B, et al. Common mental disorders and ethnicity in England: the EMPIRIC study. Psychol Med. 2004;34:1543–51.
Kirkbride JB, Hameed Y, Ioannidis K, Ankireddypalli G, Crane CM, Nasir M, et al. Ethnic minority status, age-at-immigration and psychosis risk in rural environments: evidence from the SEPEA study. Schizophr Bull. 2017;43:1251–61.
Bosqui TJ, Hoy K, Shannon C. A systematic review and meta-analysis of the ethnic density effect in psychotic disorders. Soc Psychiatry Psychiatr Epidemiol. 2014;49:519–29.
López Turley RN. When do neighborhoods matter? The role of race and neighborhood peers. Soc Sci Res. 2003;32:61–79.
Nazroo JY, Bhui KS, Rhodes J. Where next for understanding race/ethnic inequalities in severe mental illness? Structural, interpersonal and institutional racism. Socio Health Illn. 2020;42:262–76.
Sabia S, Elbaz A, Dugravot A, Head J, Shipley M, Hagger-Johnson G, et al. Impact of smoking on cognitive decline in early old age: the Whitehall II cohort study. Arch Gen Psychiatry. 2012;69:627–35.
Smith E, Hay P, Campbell L, Trollor JN. A review of the association between obesity and cognitive function across the lifespan: implications for novel approaches to prevention and treatment. Obes Rev. 2011;12:740–55.
Strachan MWJ, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7:108–14.
Hagi K, Nosaka T, Dickinson D, Lindenmayer JP, Lee J, Friedman J, et al. Association between cardiovascular risk factors and cognitive impairment in people with schizophrenia: a systematic review and meta-analysis. JAMA Psychiatry. 2021;78:510–8.
Vélez-Coto M, Rute-Pérez S, Pérez-García M, Caracuel A. Unemployment and general cognitive ability: a review and meta-analysis. J Econ Psychol. 2021;87:102430. https://doi.org/10.1016/j.joep.2021.102430.
McCutcheon RA, Abi-Dargham A, Howes OD. Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci. 2019;42:205–20. https://doi.org/10.1016/j.tins.2018.12.004.
Meltzer HY, Stahl SM. The dopamine hypothesis of schizophrenia: a review. Schizophr Bull. 1976;2:19–76.
Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–86.
Simpson EH, Kellendonk C. Insights about striatal circuit function and schizophrenia from a mouse model of dopamine D2 receptor upregulation. Biol Psychiatry. 2017;81:21–30.
McCutcheon RA, Krystal JH, Howes OD. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry. 2020;19:15–33. https://doi.org/10.1002/wps.20693.
Creese I, Burt D, Snyder S. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–3.
McCutcheon R, Beck K, Jauhar S, Howes OD. Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophr Bull. 2018;44:1301–11.
Bell DS. The experimental reproduction of amphetamine psychosis. Arch Gen Psychiatry. 1973;29:35–40.
Wallis GG, Mcharg JF, Scott OCA. Acute psychosis caused by dextro-amphetamine. Br Med J. 1949;2:1394.
Lohani S, Poplawsky AJ, Kim S-G, Moghaddam B. Unexpected global impact of VTA dopamine neuron activation as measured by opto-fMRI. Mol Psychiatry. 2017;22:585–94.
McCutcheon RA, Brown K, Nour MM, Smith SM, Veronese M, Zelaya F, et al. Dopaminergic organization of striatum is linked to cortical activity and brain expression of genes associated with psychiatric illness. Sci Adv. 2021;7:1–13.
McCutcheon RA, Nour MM, Dahoun T, Jauhar S, Pepper F, Expert P, et al. Mesolimbic dopamine function is related to salience network connectivity: an integrative positron emission tomography and magnetic resonance study. Biol Psychiatry. 2019;85:368–78.
Li N, Jasanoff A. Local and global consequences of reward-evoked striatal dopamine release. Nature. 2020;580:239–44.
Simpson EH, Kellendonk C, Kandel E. A possible role for the striatum in the pathogenesis of the cognitive symptoms of schizophrenia. Neuron. 2010;65:585–96.
Howes OD, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry. 2011;16:885–6.
Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J Psychopharmacol. 1999;13:358–71.
Laruelle M, Abi-dargham A, Van Dyck CH, Gil R, Souza CDD, Erdos J, et al. Single photon emission computerized tomography imaging of schizophrenic subjects. Proc Natl Acad Sci USA. 1996;93:9235–40.
Anda L, Brønnick KS, Johnsen E, Kroken RA, Jørgensen H, Løberg E-M. The course of neurocognitive changes in acute psychosis: relation to symptomatic improvement. PLoS One. 2016;11:e0167390.
Islam MA, Habtewold TD, van Es FD, Quee PJ, van den Heuvel ER, Alizadeh BZ, et al. Long-term cognitive trajectories and heterogeneity in patients with schizophrenia and their unaffected siblings. Acta Psychiatr Scand. 2018;138:591–604.
Slifstein M, Van De Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry. 2015;72:316–24.
Rao N, Northoff G, Tagore A, Rusjan P, Kenk M, Wilson A, et al. Impaired prefrontal cortical dopamine release in schizophrenia during a cognitive task: a [11 C]FLB 457 positron emission tomography study. Schizophr Bull. 2019;45:670–9.
Frankle WG, Himes M, Mason NS, Mathis CA, Narendran R. Prefrontal and striatal dopamine release are inversely correlated in schizophrenia. Biol Psychiatry. 2022;92:791–9. https://doi.org/10.1016/j.biopsych.2022.05.009.
Pycock CJ, Kerwin RW, Carter CJ. Effect of lesion of cortical dopamine terminals on subcortical dopamine receptors in rats. Nature. 1980;286:74–77.
Clarke HF, Cardinal RN, Rygula R, Hong YT, Fryer TD, Sawiak SJ, et al. Orbitofrontal dopamine depletion upregulates caudate dopamine and alters behavior via changes in reinforcement sensitivity. J Neurosci. 2014;34:7663–76.
Maia TV, Frank MJ. An integrative perspective on the role of dopamine in schizophrenia. Biol Psychiatry. 2017;81:52–66.
Cools R, D’Esposito M. Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol Psychiatry. 2011;69:e113–e125.
Floresco SB, Magyar O. Mesocortical dopamine modulation of executive functions: beyond working memory. Psychopharmacology. 2006;188:567–85.
Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67:53–83.
Krystal JH, Anticevic A, Yang GJ, Dragoi G, Driesen NR, Wang XJ, et al. Impaired tuning of neural ensembles and the pathophysiology of schizophrenia: a translational and computational neuroscience perspective. Biol Psychiatry. 2017;81:874–85.
Owen AM, Iddon JL, Hodges JR, Summers BA, Robbins TW. Spatial and non-spatial working memory at different stages of Parkinson’s disease. Neuropsychologia. 1997;35:519–32.
Castner SA, Goldman-Rakic PS, Williams GV. Animal models of working memory: Insights for targeting cognitive dysfunction in schizophrenia. Psychopharmacology. 2004;174:111–25.
De Crescenzo F, Cortese S, Adamo N, Janiri L. Pharmacological and non-pharmacological treatment of adults with ADHD: a meta-review. Evid Based Ment Health. 2017;20:4–11.
Fleischhacker W, Galderisi S, Laszlovszky I, Szatmári B, Barabássy Á, Acsai K, et al. The efficacy of cariprazine in negative symptoms of schizophrenia: post hoc analyses of PANSS individual items and PANSS-derived factors. Eur Psychiatry. 2019;58:1–9.
Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophr Res. 2005;77:43–58.
Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50.
Marucci G, Buccioni M, Ben DD, Lambertucci C, Volpini R, Amenta F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology. 2021;190:108352.
Hasselmo ME. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 2006;16:710–5.
Fornaro M, Carvalho AF, De Prisco M, Mondin AM, Billeci M, Selby P, et al. The prevalence, odds, predictors, and management of tobacco use disorder or nicotine dependence among people with severe mental illness: systematic review and meta-analysis. Neurosci Biobehav Rev. 2022;132:289–303.
Chesney E, Robson D, Patel R, Shetty H, Richardson S, Chang CK, et al. The impact of cigarette smoking on life expectancy in schizophrenia, schizoaffective disorder and bipolar affective disorder: an electronic case register cohort study. Schizophr Res. 2021;238:29–35.
Gurpegui M, Martínez-Ortega JM, Jurado D, Aguilar MC, Diaz FJ, de Leon J. Subjective effects and the main reason for smoking in outpatients with schizophrenia: a case-control study. Compr Psychiatry. 2007;48:186–91.
Adler E, Hoffer LD, Wiser A. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150:1856–61.
Dondé C, Brunelin J, Mondino M, Cellard C, Rolland B, Haesebaert F. The effects of acute nicotine administration on cognitive and early sensory processes in schizophrenia: a systematic review. Neurosci Biobehav Rev. 2020;118:121–33.
Hickling LM, Perez-Iglesias R, Ortiz-García de la Foz V, Balanzá-Martínez V, McGuire P, Crespo-Facorro B, et al. Tobacco smoking and its association with cognition in first episode psychosis patients. Schizophr Res. 2018;192:269–73.
Vermeulen JM, Schirmbeck F, Blankers M, Van Tricht M, Bruggeman R, Van Den Brink W, et al. Association between smoking behavior and cognitive functioning in patients with psychosis, siblings, and healthy control subjects: Results from a prospective 6-year follow-up study. Am J Psychiatry. 2018;175:1121–8.
Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23:351–64.
Tanzer T, Shah S, Benson C, De Monte V, Gore-Jones V, Rossell SL, et al. Varenicline for cognitive impairment in people with schizophrenia: systematic review and meta-analysis. Psychopharmacology. 2020;237:11–19.
Choi KH, Til W, Kurtz MM. Adjunctive pharmacotherapy for cognitive deficits in schizophrenia: meta-analytical investigation of efficacy. Br J Psychiatry. 2013;203:172–8.
Carruthers SP, Gurvich CT, Rossell SL. The muscarinic system, cognition and schizophrenia. Neurosci Biobehav Rev. 2015;55:393–402.
Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B. Decreased muscarinic receptor binding in subjects with schizophrenia: a study of the human hippocampal formation. Biol Psychiatry. 2000;48:381–8.
Dean B, Soulby A, Evin GM, Scarr E. Levels of [3H]pirenzepine binding in Brodmann’s area 6 from subjects with schizophrenia is not associated with changes in the transcription factor SP1 or BACE1. Schizophr Res. 2008;106:229–36.
Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, et al. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am J Psychiatry. 2003;160:118–27.
Bakker G, Vingerhoets C, Boucherie D, Caan M, Bloemen O, Eersels J, et al. Relationship between muscarinic M1 receptor binding and cognition in medication-free subjects with psychosis. Neuroimage Clin. 2018;18:713–9.
Scarr E, Cowie TF, Kanellakis S, Sundram S, Pantelis C, Dean B. Decreased cortical muscarinic receptors define a subgroup of subjects with schizophrenia. Mol Psychiatry. 2009;14:1017–23.
Yi F, Ball J, Stoll KE, Satpute VC, Mitchell SM, Pauli JL, et al. Direct excitation of parvalbumin-positive interneurons by M1 muscarinic acetylcholine receptors: roles in cellular excitability, inhibitory transmission and cognition. J Physiol. 2014;592:3463–94.
Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, et al. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–9.
Brannan SK, Sawchak S, Miller AC, Lieberman JA, Paul SM, Breier A. Muscarinic cholinergic receptor agonist and peripheral antagonist for schizophrenia. N Engl J Med. 2021;384:717–26.
Yohn SE, Weiden PW, Felder CC, Stahl SM. Muscarinic acetylcholine receptors for psychotic disorders: bench-side to clinic. Trends Pharmacol Sci. 2022;43:1098–112. https://doi.org/10.1016/j.tips.2022.09.006.
Poorthuis RB, Bloem B, Schak B, Wester J, De Kock CPJ, Mansvelder HD. Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb Cortex. 2013;23:148–61.
Askew CE, Lopez AJ, Wood MA, Metherate R. Nicotine excites VIP interneurons to disinhibit pyramidal neurons in auditory cortex. Synapse. 2019;73:1–12.
Porter JT, Cauli B, Tsuzuki K, Lambolez B, Rossier J, Audinat E. Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci. 1999;19:5228–35.
Vijayraghavan S, Major AJ, Everling S. Muscarinic M1 receptor overstimulation disrupts working memory activity for rules in primate prefrontal cortex. Neuron. 2018;98:1256–68.e4.
Betterton RT, Broad LM, Tsaneva-Atanasova K, Mellor JR. Acetylcholine modulates gamma frequency oscillations in the hippocampus by activation of muscarinic M1 receptors. Eur J Neurosci. 2017;45:1570–85.
Mackowick KM, Barr MS, Wing VC, Rabin RA, Ouellet-Plamondon C, George TP. Neurocognitive endophenotypes in schizophrenia: modulation by nicotinic receptor systems. Prog Neuropsychopharmacol Biol Psychiatry. 2014;52:79–85.
Foster DJ, Bryant ZK, Conn PJ. Targeting muscarinic receptors to treat schizophrenia. Behav Brain Res. 2021;405:113201. https://doi.org/10.1016/j.bbr.2021.113201.
Foster DJ, Wilson JM, Remke DH, Mahmood MS, Uddin MJ, Wess J, et al. Antipsychotic-like effects of M4 positive allosteric modulators are mediated by CB2 receptor-dependent inhibition of dopamine release. Neuron. 2016;91:1244–52.
Stanhope KJ, Mirza NR, Bickerdike MJ, Bright JL, Harrington NR, Hesselink MB, et al. The muscarinic receptor agonist xanomeline has an antipsychotic-like profile in the rat. J Pharm Exp Ther. 2001;299:782–92.
Li Z, Snigdha S, Roseman AS, Dai J, Meltzer HY. Effect of muscarinic receptor agonists xanomeline and sabcomeline on acetylcholine and dopamine efflux in the rat brain; comparison with effects of 4-[3-(4-butylpiperidin-1-yl)-propyl]-7-fluoro-4H-benzo[1,4]oxazin-3-one (AC260584) and N-desmethylclozapine. Eur J Pharmacol. 2008;596:89–97.
Perry KW, Nisenbaum LK, George CA, Shannon HE, Felder CC, Bymaster FP. The muscarinic agonist xanomeline increases monoamine release and immediate early gene expression in the rat prefrontal cortex. Biol Psychiatry. 2001;49:716–25.
Kaar SJ, Angelescu I, Marques TR, Howes OD. Pre-frontal parvalbumin interneurons in schizophrenia: a meta-analysis of post-mortem studies. J Neural Transm. 2019;126:1637–51.
Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67.
O’Donnell P. Adolescent onset of cortical disinhibition in schizophrenia: insights from animal models. Schizophr Bull. 2011;37:484–92.
Del Pino I, García-Frigola C, Dehorter N, Brotons-Mas JR, Alvarez-Salvado E, Martínez de Lagrán M, et al. Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron. 2013;79:1152–68.
Jelen LA, King S, Mullins PG, Stone JM. Beyond static measures: a review of functional magnetic resonance spectroscopy and its potential to investigate dynamic glutamatergic abnormalities in schizophrenia. J Psychopharmacol. 2018;32:497–508.
Barron HC, Vogels TP, Emir UE, Makin TR, O’Shea J, Clare S, et al. Unmasking latent inhibitory connections in human cortex to reveal dormant cortical memories. Neuron. 2016;90:191–203.
Woodcock EA, Anand C, Khatib D, Diwadkar VA, Stanley JA. Working memory modulates glutamate levels in the dorsolateral prefrontal cortex during 1H fMRS. Front Psychiatry. 2018;9:66.
Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of glutamate alterations in schizophrenia a meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry. 2016;73:665–74. https://doi.org/10.1001/jamapsychiatry.2016.0442.
Nakahara T, Tsugawa S, Noda Y, Ueno F, Honda S, Kinjo M, et al. Glutamatergic and GABAergic metabolite levels in schizophrenia-spectrum disorders: a meta-analysis of 1H-magnetic resonance spectroscopy studies. Mol Psychiatry. 2022;27:744–57. https://doi.org/10.1038/s41380-021-01297-6.
Kaminski J, Mascarell-Maricic L, Fukuda Y, Katthagen T, Heinz A, Schlagenhauf F. Glutamate in the dorsolateral prefrontal cortex in patients with schizophrenia: a meta-analysis of 1H-magnetic resonance spectroscopy studies. Biol Psychiatry. 2021;89:270–7.
Merritt K, McGuire PK, Egerton A, Aleman A, Block W, Bloemen OJN, et al. Association of age, antipsychotic medication, and symptom severity in schizophrenia with proton magnetic resonance spectroscopy brain glutamate level: a mega-analysis of individual participant-level data. JAMA Psychiatry. 2021;78:667–81.
Smucny J, Carter CS, Maddock RJ. Medial prefrontal cortex glutamate is reduced in schizophrenia and moderated by measurement quality: a meta-analysis of proton magnetic resonance spectroscopy studies. Biol Psychiatry. 2021;90:643–51.
Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, et al. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry. 2014;75:e11–e13.
Egerton A, Broberg BV, Van Haren N, Merritt K, Barker GJ, Lythgoe DJ, et al. Response to initial antipsychotic treatment in first episode psychosis is related to anterior cingulate glutamate levels: a multicentre 1 H-MRS study (OPTiMiSE). Mol Psychiatry. 2018;23:2145–55.
Jelen LA, King S, Horne CM, Lythgoe DJ, Young AH, Stone JM. Functional magnetic resonance spectroscopy in patients with schizophrenia and bipolar affective disorder: glutamate dynamics in the anterior cingulate cortex during a working memory task. Eur Neuropsychopharmacol. 2019;29:222–34.
Taylor R, Neufeld RWJ, Schaefer B, Densmore M, Rajakumar N, Osuch EA, et al. Functional magnetic resonance spectroscopy of glutamate in schizophrenia and major depressive disorder: anterior cingulate activity during a color-word Stroop task. NPJ Schizophr. 2015;1:15028.
Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry. 2006;11:118–9.
Beck K, Arumuham A, Veronese M, Santangelo B, McGinnity CJ, Dunn J, et al. N-methyl-D-aspartate receptor availability in first-episode psychosis: a PET-MR brain imaging study. Transl Psychiatry. 2021;11:1–8.
Marques TR, Ashok AH, Angelescu I, Borgan F, Myers J, Lingford-Hughes A, et al. GABA-A receptor differences in schizophrenia: a positron emission tomography study using [11C]Ro154513. Mol Psychiatry. 2021;26:2616–25.
Régio Brambilla C, Veselinović T, Rajkumar R, Mauler J, Orth L, Ruch A, et al. mGluR5 receptor availability is associated with lower levels of negative symptoms and better cognition in male patients with chronic schizophrenia. Hum Brain Mapp. 2020;41:2762–81.
Buzsáki G, Wang X-J. Mechanisms of gamma oscillations. Annu Rev Neurosci. 2012;35:203–25.
Gonzalez-Burgos G, Lewis DA. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull. 2012;38:950–7.
Miller EK, Lundqvist M, Bastos AM. Working memory 2.0. Neuron. 2018;100:463–75.
Alekseichuk I, Turi Z, Amador de Lara G, Antal A, Paulus W. Spatial working memory in humans depends on theta and high gamma synchronization in the prefrontal cortex. Curr Biol. 2016;26:1513–21.
Pinault D. N-methyl D-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry. 2008;63:730–5.
Hong LE, Summerfelt A, Buchanan RW, O’Donnell P, Thaker GK, Weiler MA, et al. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology. 2010;35:632–40.
Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–69.
Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, McPartland JC, et al. Searching for cross-diagnostic convergence: neural mechanisms governing excitation and inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry. 2017;81:848–61.
Winterer G, Coppola R, Goldberg TE, Egan MF, Jones DW, Sanchez CE, et al. Prefrontal broadband noise, working memory, and genetic risk for schizophrenia. Am J Psychiatry. 2004;161:490–500.
Tikka SK, Yadav S, Nizamie SH, Das B, Tikka DL, Goyal N. Schneiderian first rank symptoms and gamma oscillatory activity in neuroleptic naïve first episode schizophrenia: a 192 channel EEG study. Psychiatry Investig. 2014;11:467–75.
Hong LE, Summerfelt A, Mitchell BD, McMahon RP, Wonodi I, Buchanan RW, et al. Sensory gating endophenotype based on its neural oscillatory pattern and heritability estimate. Arch Gen Psychiatry. 2008;65:1008–16.
Spencer KM. Baseline gamma power during auditory steady-state stimulation in schizophrenia. Front Hum Neurosci. 2011;5:190.
Arikan MK, Metin B, Metin SZ, Tülay EE, Tarhan N. High frequencies in QEEG are related to the level of insight in patients with schizophrenia. Clin EEG Neurosci. 2018;49:316–20.
Baradits M, Kakuszi B, Bálint S, Fullajtár M, Mód L, Bitter I, et al. Alterations in resting-state gamma activity in patients with schizophrenia: a high-density EEG study. Eur Arch Psychiatry Clin Neurosci. 2019;269:429–37.
Tanaka-Koshiyama K, Koshiyama D, Miyakoshi M, Joshi YB, Molina JL, Sprock J, et al. Abnormal spontaneous gamma power is associated with verbal learning and memory dysfunction in schizophrenia. Front Psychiatry. 2020;11:832.
Ramyead A, Kometer M, Studerus E, Koranyi S, Ittig S, Gschwandtner U, et al. Aberrant current source-density and lagged phase synchronization of neural oscillations as markers for emerging psychosis. Schizophr Bull. 2015;41:919–29.
Catts VS, Lai YL, Weickert CS, Weickert TW, Catts SV. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: how can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol. 2016;116:57–67.
Todd J, Harms L, Schall U, Michie PT. Mismatch negativity: translating the potential. Front Psychiatry. 2013;4:171.
Baldeweg T, Klugman A, Gruzelier J, Hirsch SR. Mismatch negativity potentials and cognitive impairment in schizophrenia. Schizophr Res. 2004;69:203–17.
Kiang M, Light GA, Prugh J, Coulson S, Braff DL, Kutas M. Cognitive, neurophysiological, and functional correlates of proverb interpretation abnormalities in schizophrenia. J Int Neuropsychol Soc. 2007;13:653–63.
Näätänen R, Kujala T, Kreegipuu K, Carlson S, Escera C, Baldeweg T, et al. The mismatch negativity: an index of cognitive decline in neuropsychiatric and neurological diseases and in ageing. Brain. 2011;134:3435–53.
Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005;76:1–23.
Erickson MA, Ruffle A, Gold JM. A meta-analysis of mismatch negativity in schizophrenia: from clinical risk to disease specificity and progression. Biol Psychiatry. 2016;79:980–7.
Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–7.
Niessing J, Ebisch B, Schmidt KE, Niessing M, Singer W, Galuske RAW. Neuroscience: hemodynamic signals correlate tightly with synchronized gamma oscillations. Science. 2005;309:948–51.
Schirner M, McIntosh AR, Jirsa V, Deco G, Ritter P. Inferring multi-scale neural mechanisms with brain network modelling. Elife. 2018;7:1–30.
van den Heuvel MP, Hulshoff Pol HE. Exploring the brain network: a review on resting-state fMRI functional connectivity. Eur Neuropsychopharmacol. 2010;20:519–34.
Sheffield JM, Barch DM. Cognition and resting-state functional connectivity in schizophrenia. Neurosci Biobehav Rev. 2016;61:108–20.
Murray JD, Anticevic A, Gancsos M, Ichinose M, Corlett PR, Krystal JH, et al. Linking microcircuit dysfunction to cognitive impairment: effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb Cortex. 2014;24:859–72.
Yang GJ, Murray JD, Wang XJ, Glahn DC, Pearlson GD, Repovs G, et al. Functional hierarchy underlies preferential connectivity disturbances in schizophrenia. Proc Natl Acad Sci USA. 2016;113:E219–E228.
Anticevic A, Lisman J. How can global alteration of excitation/inhibition balance lead to the local dysfunctions that underlie schizophrenia? Biol Psychiatry. 2017;81:818–20.
Starc M, Murray JD, Santamauro N, Savic A, Diehl C, Cho YT, et al. Schizophrenia is associated with a pattern of spatial working memory deficits consistent with cortical disinhibition. Schizophr Res. 2017;181:107–16.
Crossley NA, Mechelli A, Scott J, Carletti F, Fox PT, Mcguire P, et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain. 2014;137:2382–95.
Ruden JB, Dugan LL, Konradi C. Parvalbumin interneuron vulnerability and brain disorders. Neuropsychopharmacology. 2021;46:279–87.
Wegrzyn D, Juckel G, Faissner A. Structural and functional deviations of the hippocampus in schizophrenia and schizophrenia animal models. Int J Mol Sci. 2022;23:5482. https://doi.org/10.3390/ijms23105482.
Chand GB, Dwyer DB, Erus G, Sotiras A, Varol E, Srinivasan D, et al. Two distinct neuroanatomical subtypes of schizophrenia revealed using machine learning. Brain. 2020;143:1027–38.
Xiao Y, Liao W, Long Z, Tao B, Zhao Q, Luo C, et al. Subtyping schizophrenia patients based on patterns of structural brain alterations. Schizophr Bull. 2022;48:241–50.
Chand GB, Singhal P, Dwyer DB, Wen J, Erus G, Doshi J, et al. Schizophrenia imaging signatures and their associations with cognition, psychopathology, and genetics in the general population. Am J Psychiatry. 2022;179:650–60.
Musa A, Khan S, Mujahid M, El-Gaby M. The shallow cognitive map hypothesis: a hippocampal framework for thought disorder in schizophrenia. Schizophrenia. 2022;8:34. https://doi.org/10.1038/s41537-022-00247-7.
Nour MM, Liu Y, Arumuham A, Kurth-Nelson Z, Dolan RJ. Impaired neural replay of inferred relationships in schizophrenia. Cell. 2021;184:4315–28.e17.
Rolls ET. The mechanisms for pattern completion and pattern separation in the hippocampus. Front Syst Neurosci. 2013;7:1–21.
Beck K, Hindley G, Borgan F, Ginestet C, McCutcheon R, Brugger S, et al. Association of ketamine with psychiatric symptoms and implications for its therapeutic use and for understanding schizophrenia: a systematic review and meta-analysis. JAMA Netw Open. 2020;3:1–20.
Pollak TA, McCormack R, Peakman M, Nicholson TR, David AS. Prevalence of anti-N-methyl-d-aspartate (NMDA) receptor antibodies in patients with schizophrenia and related psychoses: a systematic review and meta-analysis. Psychol Med. 2014;44:2475–87.
Radhakrishnan R, Skosnik PD, Ranganathan M, Naganawa M, Toyonaga T, Finnema S, et al. In vivo evidence of lower synaptic vesicle density in schizophrenia. Mol Psychiatry. 2021;26:7690–8. https://doi.org/10.1038/s41380-021-01184-0.
Onwordi EC, Halff EF, Whitehurst T, Mansur A, Cotel MC, Wells L, et al. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat Commun. 2020;11:246. https://doi.org/10.1038/s41467-019-14122-0.
Van Berlekom AB, Muflihah CH, Snijders GJLJ, MacGillavry HD, Middeldorp J, Hol EM, et al. Synapse pathology in schizophrenia: a meta-analysis of postsynaptic elements in postmortem brain studies. Schizophr Bull. 2020;46:374–86.
Adams RA, Pinotsis D, Tsirlis K, Unruh L, Mahajan A, Horas AM, et al. Computational modeling of electroencephalography and functional magnetic resonance imaging paradigms indicates a consistent loss of pyramidal cell synaptic gain in schizophrenia. Biol Psychiatry. 2022;91:202–15.
Howes OD, McCutcheon R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Transl Psychiatry. 2017;7:e1024. https://doi.org/10.1038/tp.2016.278.
Keefe RSE. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64:633.
Joshi YB, Thomas ML, Braff DL, Green MF, Gur RC, Gur RE, et al. Anticholinergic medication burden-associated cognitive impairment in schizophrenia. Am J Psychiatry. 2021;178:838–47.
Georgiou R, Lamnisos D, Giannakou K. Anticholinergic burden and cognitive performance in patients with schizophrenia: a systematic literature review. Front Psychiatry. 2021;12:779607. https://doi.org/10.3389/fpsyt.2021.779607.
Kanchanatawan B, Thika S, Anderson G, Galecki P, Maes M. Affective symptoms in schizophrenia are strongly associated with neurocognitive deficits indicating disorders in executive functions, visual memory, attention and social cognition. Prog Neuropsychopharmacol Biol Psychiatry. 2018;80:168–76.
Möser C, Krieg JC, Zihl J, Lautenbacher S. Attention and memory deficits in schizophrenia: the role of symptoms of depression. Cogn Behav Neurol. 2006;19:150–6.
Vernon JA, Grudnikoff E, Seidman AJ, Frazier TW, Vemulapalli MS, Pareek P, et al. Antidepressants for cognitive impairment in schizophrenia—a systematic review and meta-analysis. Schizophr Res. 2014;159:385–94.
Firth J, Siddiqi N, Koyanagi A, Siskind D, Rosenbaum S, Galletly C, et al. The Lancet Psychiatry Commission: a blueprint for protecting physical health in people with mental illness. Lancet Psychiatry. 2019;6:675–712.
Meyer N, Faulkner SM, McCutcheon RA, Pillinger T, Dijk DJ, MacCabe JH. Sleep and circadian rhythm disturbance in remitted schizophrenia and bipolar disorder: a systematic review and meta-analysis. Schizophr Bull. 2020;46:1126–43.
Ma Y, Liang L, Zheng F, Shi L, Zhong B, Xie W. Association between sleep duration and cognitive decline. JAMA Netw Open. 2020;3:e2013573.
Vita A, Barlati S, Ceraso A, Nibbio G, Ariu C, Deste G, et al. Effectiveness, core elements, and moderators of response of cognitive remediation for schizophrenia: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry. 2021;78:848–58.
Lejeune JA, Northrop A, Kurtz MM. A meta-analysis of cognitive remediation for schizophrenia: efficacy and the role of participant and treatment factors. Schizophr Bull. 2021;47:997–1006.
National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management. Nice. Feb 54 Clinical Guidelines n° 178; 2014.
Horan WP, Green MF. Treatment of social cognition in schizophrenia: current status and future directions. Schizophr Res. 2019;203:3–11.
Tripathi A, Kar SK, Shukla R. Cognitive deficits in schizophrenia: understanding the biological correlates and remediation strategies. Clin Psychopharmacol Neurosci. 2018;16:7–17.
Billeke P, Aboitiz F. Social cognition in schizophrenia: from social stimuli processing to social engagement. Front Psychiatry. 2013;4:4.
Rutigliano G, Accorroni A, Zucchi R. The case for TAAR1 as a modulator of central nervous system function. Front Pharm. 2018;8:1–18.
Koblan KS, Kent J, Hopkins SC, Krystal JH, Cheng H, Goldman R, et al. A non–D2-receptor-binding drug for the treatment of schizophrenia. N Engl J Med. 2020;382:1497–506.
Revel FG, Moreau JL, Pouzet B, Mory R, Bradaia A, Buchy D, et al. A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol Psychiatry. 2013;18:543–56.
Zhang Y, Li JT, Wang H, Niu WP, Zhang CC, Zhang Y, et al. Role of trace amine‑associated receptor 1 in the medial prefrontal cortex in chronic social stress-induced cognitive deficits in mice. Pharm Res. 2021;167:105571.
Dou KX, Tan MS, Tan CC, Cao XP, Hou XH, Guo QH, et al. Comparative safety and effectiveness of cholinesterase inhibitors and memantine for Alzheimer’s disease: a network meta-analysis of 41 randomized controlled trials. Alzheimers Res Ther. 2018;10:1–10.
Santos B, González-Fraile E, Zabala A, Guillén V, Rueda JR, Ballesteros J. Cognitive improvement of acetylcholinesterase inhibitors in schizophrenia. J Psychopharmacol. 2018;32:1155–66.
Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–73.
McGuire P, Robson P, Cubala WJ, Vasile D, Morrison PD, Barron R, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicenter randomized controlled trial. Am J Psychiatry. 2018;175:225–31.
Bhattacharyya S, Wilson R, Appiah-Kusi E, O’Neill A, Brammer M, Perez J, et al. Effect of cannabidiol on medial temporal, midbrain, and striatal dysfunction in people at clinical high risk of psychosis: a randomized clinical trial. JAMA Psychiatry. 2018;75:1107–17.
Chesney E, Oliver D, McGuire P. Cannabidiol (CBD) as a novel treatment in the early phases of psychosis. Psychopharmacology. 2022;239:1179–90.
Kaplan JS, Stella N, Catterall WA, Westenbroek RE. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2017;114:11229–34.
Khan AA, Shekh-Ahmad T, Khalil A, Walker MC, Ali AB. Cannabidiol exerts antiepileptic effects by restoring hippocampal interneuron functions in a temporal lobe epilepsy model. Br J Pharm. 2018;175:2097–115.
Gomes FV, Llorente R, Del Bel EA, Viveros MP, López-Gallardo M, Guimarães FS. Decreased glial reactivity could be involved in the antipsychotic-like effect of cannabidiol. Schizophr Res. 2015;164:155–63.
Sinkeviciute I, Begemann M, Prikken M, Oranje B, Johnsen E, Lei WU, et al. Efficacy of different types of cognitive enhancers for patients with schizophrenia: a meta-analysis. npj Schizophr. 2018;4:22. https://doi.org/10.1038/s41537-018-0064-6.
Iwata Y, Nakajima S, Suzuki T, Keefe RSE, Plitman E, Chung JK, et al. Effects of glutamate positive modulators on cognitive deficits in schizophrenia: a systematic review and meta-analysis of double-blind randomized controlled trials. Mol Psychiatry. 2015;20:1151–60.
Downing ACM, Kinon BJ, Millen BA, Zhang L, Liu L, Morozova MA, et al. A double-blind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry. 2014;14:351. https://doi.org/10.1186/s12888-014-0351-3.
Stauffer VL, Millen BA, Andersen S, Kinon BJ, LaGrandeur L, Lindenmayer JP, et al. Pomaglumetad methionil: no significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophrenia Res. 2013;150:434–41.
Zheng W, Li XH, Yang XH, Cai DB, Ungvari GS, Ng CH, et al. Adjunctive memantine for schizophrenia: a meta-analysis of randomized, double-blind, placebo-controlled trials. Psychol Med. 2018;48:72–81.
Farokhnia M, Sabzabadi M, Pourmahmoud H, Khodaie-Ardakani MR, Hosseini SMR, Yekehtaz H, et al. A double-blind, placebo controlled, randomized trial of riluzole as an adjunct to risperidone for treatment of negative symptoms in patients with chronic schizophrenia. Psychopharmacology. 2014;231:533–42.
Lieberman JA, Papadakis K, Csernansky J, Litman R, Volavka J, Jia XD, et al. A randomized, placebo-controlled study of memantine as adjunctive treatment in patients with schizophrenia. Neuropsychopharmacology. 2009;34:1322–9.
Fleischhacker WW, Podhorna J, Gröschl M, Hake S, Zhao Y, Huang S, et al. Efficacy and safety of the novel glycine transporter inhibitor BI 425809 once daily in patients with schizophrenia: a double-blind, randomised, placebo-controlled phase 2 study. Lancet Psychiatry. 2021;8:191–201.
ACNP 60th Annual Meeting: Poster Abstracts P551–P830. https://europepmc.org/article/pmc/8637058.
Vita A, Gaebel W, Mucci A, Sachs G, Erfurth A, Barlati S, et al. European Psychiatric Association guidance on assessment of cognitive impairment in schizophrenia. Eur Psychiatry. 2022;65:e58.
Van Der Does AJ, Dingemans PM, Linszen DH, Nugter MA, Scholte WF. Symptom dimensions and cognitive and social functioning in recent-onset schizophrenia. Psychological Med. 1993;23:745–53.
Kravariti E, Russo M, Vassos E, Morgan K, Fearon P, Zanelli JW, et al. Linear and non-linear associations of symptom dimensions and cognitive function in first-onset psychosis. Schizophrenia Res. 2012;140:221–31.
Acknowledgements
RAM is supported by an NIHR clinical lectureship and Wellcome Clinical Research Career Development Fellowship (224625/Z/21/Z).
Author information
Authors and Affiliations
Contributions
RAM wrote the initial draft. PKM and RSEK reviewed and commented on drafts. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
RSEK has received consulting fees in the past 2 years from Karuna, Biogen, Merck, BoehringerIngelheim, WCG and received royalties from the BACS and VRFCAT. RAM has received speaking fees/served on advisory boards of Otsuka, Karuna and Janssen. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: In this article the affiliation details for Prof McGuire were incorrectly given as 1. Department of Psychiatry, University of Oxford, Oxford, UK. 2. Department of Psychosis Studies, Institute of Psychiatry, Psychology & Neuroscience, London, UK. 3. Oxford health NHS Foundation Trust, Oxford health NHS Foundation Trust, Oxford, UK. The correct affiliations are 1. Department of Psychiatry, University of Oxford. 2. NIHR Oxford Health Biomedical Research Centre. 3. Oxford Health NHS Foundation Trust.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McCutcheon, R.A., Keefe, R.S.E. & McGuire, P.K. Cognitive impairment in schizophrenia: aetiology, pathophysiology, and treatment. Mol Psychiatry 28, 1902–1918 (2023). https://doi.org/10.1038/s41380-023-01949-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-023-01949-9
This article is cited by
-
Schizophrenia: from neurochemistry to circuits, symptoms and treatments
Nature Reviews Neurology (2024)
-
A concerted neuron–astrocyte program declines in ageing and schizophrenia
Nature (2024)
-
The magnitude and variability of neurocognitive performance in first-episode psychosis: a systematic review and meta-analysis of longitudinal studies
Translational Psychiatry (2024)
-
Kognitive Beeinträchtigung in Zusammenhang mit Schizophrenie (CIAS): Diagnostik und Therapie
psychopraxis. neuropraxis (2024)
-
Geographical variation in treated psychotic and other mental disorders in Finland by region and urbanicity
Social Psychiatry and Psychiatric Epidemiology (2024)
