
Abstract
Attention-deficit/hyperactivity disorder (ADHD) is a highly prevalent neurodevelopmental disorder that results from the interaction of both genetic and environmental risk factors. Genome-wide association studies have started to identify multiple genetic risk loci associated with ADHD, however, the exact causal genes and biological mechanisms remain largely unknown. We performed a multi-step analysis to identify and characterize modules of co-expressed genes associated with ADHD using data from peripheral blood mononuclear cells of 270 ADHD cases and 279 controls. We identified seven ADHD-associated modules of co-expressed genes, some of them enriched in both genetic and epigenetic signatures for ADHD and in biological pathways relevant for psychiatric disorders, such as the regulation of gene expression, epigenetics and immune system. In addition, for some of the modules, we found evidence of potential regulatory mechanisms, including microRNAs and common genetic variants. In conclusion, our results point to promising genes and pathways for ADHD, supporting the use of peripheral blood to assess gene expression signatures in psychiatric disorders. Furthermore, they highlight that the combination of multi-omics signals provides deeper and broader insights into the biological mechanisms underlying ADHD.
Similar content being viewed by others


Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a highly prevalent neurodevelopmental disorder that affects around 5–6% of children and adolescents worldwide, and in 40–65% of cases persist into adulthood [1]. It is mainly characterized by inattention and/or hyperactivity and high levels of impulsivity.
ADHD is a complex disorder that results from the interaction of both genetic and environmental risk factors, with an estimated heritability of 70–80% throughout the lifespan [2]. Several studies support the role of both common and rare genetic variants in the development of ADHD, although its etiology and pathogenesis still remain largely unknown [2]. The first genome-wide association study (GWAS) meta-analysis identifying genetic risk variants for ADHD (20,183 cases and 35,191 controls) was published in 2019 [3]. They identified 12 independent ADHD risk loci and estimated that common variants account for 22% of the total ADHD heritability. In addition, very recently, a larger GWAS meta-analysis on ADHD reported 21 new loci and a reduced estimated SNP heritability (h2SNP = 14%) [4]. These data highlight that part of the genetic variance still needs to be explained, which may be accounted, in part, for gene by environment interactions [5]. Epigenetic processes (i.e. histone modifications, DNA methylation and microRNAs) are potential mechanisms by which environmental risk factors lead to changes on gene expression and long-lasting alterations in the neuronal circuits found in psychiatric disorders like ADHD [6]. Recently, the first epigenome-wide association study (EWAS) in peripheral blood mononuclear cells (PBMCs) from adults with ADHD was published, identifying four regions differentially methylated and located in genes previously related to autoimmune disorders, cancer, or neuroticism [7]. Additional EWAS in saliva and whole blood have been performed both in adults and children with ADHD diagnosis or ADHD symptoms, however, results among studies are not consistent and further studies with larger sample sizes are needed [8,9,10,11,12].
Although genetic and epigenetic factors that contribute to the etiology of ADHD have started to be identified through GWAS and EWAS, their biological relevance is difficult to characterize, in part, because genetic risk loci were usually associated with the nearest gene, which may not be necessarily the true causal one. In contrast, the analysis of gene expression profiles provides a closer physiological picture of the disorder that is easier to interpret, and reduces the burden of multiple testing. Transcriptome studies in ADHD, nevertheless, are limited by the inaccessibility of brain samples and have been focused on whole blood or PBMCs. To date, eight transcriptomic studies on ADHD have been performed, highlighting alterations in genes involved in several neuronal functions and the immune system [13,14,15,16,17,18,19,20]. However, the studies performed so far were based on differential gene expression analyses between ADHD cases and controls, which assume that every gene acts as an independent unit in the expression landscape and select genes based on statistical significance. In contrast, gene co-expression network analyses use an unsupervised framework to identify groups of genes with similar expression patterns (co-expressed genes) independently of any phenotype and then correlate these gene modules with a phenotype of interest. This approach has been widely used to characterize patterns of co-expression in normal brain [21, 22] and both in brain and blood samples from several psychiatric disorders [23,24,25,26,27,28].
In the present study, we aimed to perform a multi-step analysis to identify and characterize modules of co-expressed genes associated with ADHD using expression data from PBMCs of ADHD cases and controls. To further understand the biological relevance and provide a more accurate picture of the regulatory mechanisms, we performed a comprehensive characterization of genes in each module and combined genomic and transcriptomic data to identify loci that may regulate the ADHD-associated co-expressed genes.
Materials and methods
Study design
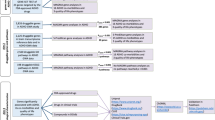
A comprehensive and multi-step approach was applied to identify and characterize modules of co-expressed genes in PBMCs. In the first step we ran a Weighted Gene Correlation Network Analysis (WGCNA) on the processed transcriptomic data from 270 ADHD cases and 279 controls and assessed the association of the resulting co-expression modules with ADHD status. Subsequently, we disentangled the biological relevance of the ADHD-associated co-expression modules by (i) performing enrichment analyses in brain expression, functional pathways, druggable genes and miRNA target genes, (ii) combining results with ADHD genetic, transcriptomic, and epigenetic signatures, and (iii) running a co-expression module eQTL analysis to identify loci regulating the ADHD-associated modules of co-expressed genes (Fig. 1).

Modules of co-expressed genes were identified from peripheral blood mononuclear cells (PBMCs) of processed transcriptomic data from 270 ADHD cases and 279 controls by using Weighted Gene Correlation Network Analysis (WGCNA). Then, we assessed the association of the resulting modules with the ADHD status and investigated their biologically relevance by (i) performing enrichment analyses in brain expression (ABAenrichment R package), functional pathways, druggable genes and miRNA target genes using WebGestAlt webtool; (ii) integrating ADHD transcriptomic, genetic and epigenetic data from GWAS meta-analysis [3] and EWAS [7] on ADHD; and (iii) running a co-expression module eQTL analysis to identify loci regulating the ADHD-associated modules of co-expressed genes.
Participants
Analysis of co-expression modules was performed in an in-house sample of 270 ADHD cases (59.3% male, mean age = 34.2 years, s.d = 11.7) and 279 controls (56.9% male, mean age = 36.6 years, s.d = 9.9). All subjects were of European ancestry. Clinical assessment was conducted by structured interviews and self-reported questionnaires as previously described [19]. Detailed information is available in Supplementary Information. The study was approved by the Clinical Research Ethics Committee (CREC) of Hospital Universitari Vall d’Hebron, methods were performed in accordance with the relevant guidelines and regulations and written informed consent was obtained from all subjects before inclusion in the study.
Transcriptome profiling and weighted gene correlation network analysis (WGCNA)
RNA from PBMCs was isolated, hybridized to GeneChip Human Gene 1.1 ST 96-Array plate (Affymetrix) and data were analyzed as previously described [19] (Supplementary Information). Modules of co-expressed genes were identified from processed transcriptomic data by the WGCNA R-package [29]. A soft-thresholding power of 4 was selected (Fig. S1) and one-step network construction and module detection was performed considering an unsigned network type with default values (additional details in Supplementary Information). Gene expression for each module was represented by a module eigengene, derived from its first principal component and treated as a quantitative trait in the downstream analyses. The association between the module eigengenes and ADHD status or potential confounding factors (age, sex, RNA integrity number (RIN), and batch) was tested using regression analyses. Bonferroni correction was applied to correct for multiple testing considering the overall number of co-expression modules constructed (P < 0.05/27 modules <1E-03).
Within each module we examined the correlation between module membership (an indicator of the intramodular connectivity of a gene based on the association between its expression and the module eigengene) and gene significance (effect size of the association between each gene and ADHD) using Pearson correlation (Fig. S2).
Enrichment analyses in the ADHD-associated co-expression modules
We assessed whether genes in each ADHD-associated co-expression module were expressed in specific brain regions at different developmental stages using data from the Allen Human Brain Atlas with ABAEnrichment R-package [30] (additional details in Supplementary Information). Then, enrichment analyses with the webtool WebGestAlt (WEB-based GEne SeT AnaLysis Toolkit, http://www.webgestalt.org/) [31] were performed on: (i) Gene Ontology non-redundant Biological Process (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome, (ii) target genes of pharmacological drugs based on the information from PharmGKB and (iii) miRNA target genes. False discovery rate P-value (PFDR) < 0.05 was set as the significance threshold.
In addition, the correlation between the identified miRNA and its corresponding module eigengene was tested using the non-parametric Spearman rank correlation test in a subset of 310 individuals included in the WGCNA (60% overlap; 150 ADHD cases and 160 controls) from whom miRNA expression profile data from PBMC were available as described in Sanchez-Mora et al. [16]. Expression was available and retrieved from a total of 27 mature miRNAs and Bonferroni correction was used to adjust for multiple testing (P < 0.05/27 tests <1.85E−03).
Integrative analysis of ADHD-associated co-expression modules and ADHD omics data
ADHD transcriptomic signatures
After quality control and sample processing, differential gene expression profiles between the 270 ADHD cases and 279 controls used in the WGCNA analysis were obtained with Limma R-package [32]. Only genes with PFDR < 0.05 and fold change (FC) > |1.15| were considered differentially expressed and were used to test for enrichment in the ADHD-associated co-expression modules using a F-Fisher test and Bonferroni correction across all modules (P < 0.05/7 modules <7.1E−03).
ADHD genetic signatures
The identified ADHD-associated co-expression modules were used as gene sets to test for enrichment in ADHD genetic signatures, considering the European ancestry GWAS summary statistics on ADHD described by Demontis et al. [3]. Gene-based analyses were run in MAGMA_v1.08 [33] using the SNP-wise mean model, and SNPs were assigned to genes based on a positional-approach and eMAGMA [34, 35]. Competitive gene-set analysis was performed using P-values obtained from each gene-based analysis. Bonferroni correction was applied to correct for multiple testing (P < 7.1E-03; Supplementary Information)
ADHD methylation signatures
To test for enrichment in ADHD epigenetic marks in modules of co-expressed genes we used the summary statistics of an EWAS on PBMCs from 103 ADHD patients and 100 controls [7] (90% sample overlap with the in-house sample used in WGCNA), setting the unadjusted P-value < 0.01 to select differentially methylated proves (n = 3967 CpG sites). We considered enrichment in epigenetic signatures in a co-expression module when the three approaches used (methylglm, methylRRA and gsameth [36]) were significant after applying the Bonferroni correction for multiple comparison corrections (P < 7.1E−03; Supplementary Information).
Gene-module eQTL analysis and functional annotation
Genetic information was available from a subset of 231 ADHD subjects and 264 controls included in the WGCNA. A GWAS with each module eigengene as the dependent variable were performed to identify genetic variants associated with each co-expression module. After ascertain normality of module eigengene (Table S1), seven gene-module eQTL analyses were run under an additive linear regression model using PLINK_1.09, adding as covariates the first 10 principal components, sex, age, and the genotyping wave. Lead SNPs were identified in each eQTL analysis considering a P-value < 1E−06 and functionally annotated using the FUMA protocol (Functional Mapping and Annotation of Genome-Wide Association Studies, https://fuma.ctglab.nl/) [37] (Supplementary Information).
Raw data from this article are not publicly available because of limitations in ethical approvals and the summary data will be available upon request.
Results
The WGCNA identified a total of 27 modules of co-expressed genes with size ranging from 33 to 2191 genes (Fig. S1). 42.7% of genes (N = 8114) were not assigned to any module and remained in the module M0. Seven co-expression modules were associated with ADHD after multiple testing correction (modules M1–M7, Table S2). No association between module eigengenes and potential confounders, including age, sex, RIN, or batch, was detected for any module (Table S2). All modules were consistent across samples and have characteristic band structures suggestive of well-defined modules (Fig. S2). Interestingly, modules M1, M3, and M6 showed high module membership—gene significance correlation (r2 > 0.4), suggesting that the higher the connectivity of a gene within the module, the stronger the association with ADHD (Fig. S3).
Different patterns of gene expression in the brain at different developmental stages were found across ADHD-associated co-expression modules. M2 genes are broadly expressed in the whole brain during the lifespan, while genes in M7 are expressed in a specific brain area, the M1C_primary motor cortex, only during the prenatal stage. Besides, genes in modules M1 and M4 show broader expression in different areas from the telencephalon during the prenatal period and are mainly expressed in the cerebellar cortex after birth (Table 1 and S3).
To explore the biological relevance of ADHD-associated co-expression modules further, we performed a functional enrichment analysis in genes in each module and found that several of them were enriched in genes involved in pathways previously related to psychiatric disorders [38], including the posttranscriptional regulation of gene expression and epigenetics (M1 and M7), covalent chromatin remodeling (M4) or immune system and inflammatory response (M5), among others (Table 1 and S4-6). We also performed an enrichment analysis in druggable genes in the ADHD-associated co-expression modules. For six out of the seven modules we identified enrichment in target genes of at least one drug, being Antiinfective for systemic use and Antineoplastic and immunomodulating agents the most common Anatomical Therapeutic Chemical classification categories across all modules (Table 1 and S7-8). Interestingly, module M5, enriched in genes involved in the immune system and inflammatory response, showed enrichment in drugs from all Anatomical Therapeutic Chemical categories, especially those related to the immune response, as expected.
Enrichment in miRNA target genes was identified in modules M1 and M7. Genes in M1 were targeted by 24 families of miRNAs, resulting on 40 mature miRNAs, and genes in M7 were targeted by five mature miRNAs (Table 1 and S9). Consistently, a significant correlation between the eigengene profile of module M1 and the expression of four out of 27 of these miRNAs (hsa-miR-142–5p, hsa-miR-181a-5p, hsa-miR-192–5p, and hsa-miR-215–5p) was found in a subset of 310 individuals (150 ADHD cases and 160 controls) from which miRNA and gene expression from PBMCs was available (Fig. S4 and Table S10).
Then, we further explored the ADHD-associated co-expression modules by integrating transcriptomics with genetic and epigenetic data on ADHD. We explored whether genes differentially expressed between ADHD cases and controls were grouped in any of the identified ADHD-associated modules of co-expressed genes, and found a significant enrichment in module M5 (P < 2.2E−16), which also remained significant when considering only highly connected genes (module membership > 0.8; Table 1). In addition, we found module M4 significantly enriched in genetics (PMAGMA = 1.8E−03; PeMAGMA = 4.2E−03) and epigenetics (Pgsameth = 1.6E−04; PmethylRRA = 1.1E−03; Pmethylglm = 8.1E−05) signatures for ADHD, using data from GWAS meta-analysis [3] and EWAS [7] on ADHD (Table 1 and S11).
We performed a co-expression module eQTL analysis to identify loci regulating ADHD-associated modules in a subset of 495 individuals included in the WGCNA (91.3%) from whom genomic and gene expression profiles were available. After strict quality control criteria, we ran a GWAS on module eigengenes of each of the seven ADHD-associated co-expression modules independently (M1–M7; Fig. S5). QQ plots indicate minimal effects of genomic inflation, and consequently population substructure, on the analyses (Fig. S6). No SNP overcame the genome-wide significance threshold, but 12 independent genomic loci showed suggestive evidence of association (P < 1E−06) with different module eigengenes (Table 2 and Fig. S7). Functional annotations revealed that these loci lay on regions of open chromatin and that most of the signals were intergenic or intronic (Fig. 2). Several SNPs in these genomic risk loci were likely to affect the binding of transcription factors (RBD score = 2b; rs73866266, rs59928606, rs10830974 and rs36098630), had CADD scores > 12.37, suggesting high deleteriousness (rs73170573, rs13408514, rs1508617, rs9565360, and rs10830974; Fig. 2, Table 2 and S12) or were located in regulatory regions of the brain (rs73170578, rs62096513, and rs12583109), according to the information of enhancer and promotor histone marks from the HaploReg webtool [39] (Table 2). In addition, four SNPs, rs62096513, rs6707596, rs66506812 and rs2462337, lay in nearby genes encoding transcription factors (ZSCAN30, SP3, CSRNP3 and CUX1, respectively; Table 2). Of them, rs6707596 nearby SP3 and rs62096513 located in intron 1 of ZSCAN30 were cis-eQTL of these genes in PBMCs in our sample (Fig. 3). Interestingly, the co-expression module M1, which showed suggestive evidence of association with rs62096513 that lies in blood and brain regulatory regions of ZSCAN30 and is cis-eQTL in PBMCs, is enriched in target genes for this specific transcription factor (P = 1.27E-07), which suggest that ZSCAN30 may be upstream regulator of the M1 module of co-expressed genes.

Regulome DB score predicts likelihood of regulatory functionality, lower scores indicate higher likelihood. Further information can be found in Boyle et al. [68]. Minimum Chromatin State across 127 tissue and cell types, lower scores indicate higher accessibility, with states 1–7 referring to open chromatin states.

In the top figure, regional plots for module eigengenes for the (A) rs62096513 and (B) rs6707596 loci and in the bottom figure, boxplots showing the effect of rs62096513 in ZSCAN30 and rs6707596 in SP3 gene expression. Results from the linear regression p-value are shown: *P < 0.5, **P < 0.01, ***P < 0.001.
Discussion
In the present study, we used a network-based approach to identify novel ADHD-associated modules of co-expressed genes in PBMCs. To further investigate the biological significance of the ADHD-associated networks identified, we performed a comprehensive characterization of each module by performing enrichment analysis in biological pathways and drug or miRNA target genes. We also performed an integrative analysis by combining transcriptomic, genetic and epigenetic data on ADHD and run an eQTL analysis to identify genetic variants that could regulate the ADHD-associated modules of co-expressed genes. Our results identified seven ADHD-associated modules of co-expressed genes and support that the study of gene correlation networks may improve our understanding of the complex molecular systems underling ADHD.
Two of the ADHD-associated co-expression modules identified (M1 and M7), were enriched in genes involved in posttranscriptional regulation of gene expression and epigenetic modifications, two relevant pathways in the pathogenesis of ADHD [6]. In the same line, we found enrichment in target genes for several miRNAs in these two modules. In particular, the expression of four of them (hsa-miR-142–5p, hsa-miR-181a-5p, hsa-miR-192–5p and hsa-miR-215–5p) also correlates with the eigengene profile of module M1, pointing them as potential upstream regulatory mechanisms underlying the M1 co-expression network. Some of these miRNAs have been previously related to ADHD, like miR-192–5p upregulated in PBMCs of ADHD patients [16], and comorbid psychiatric disorders, such as miR-192–5p and miR-215–5p that were differentially expressed in the dorsolateral prefrontal cortex of major depression patients [40] or miR-181a-5p extensively related to drug addiction both in mice and human studies [41,42,43,44,45]. Interestingly, these miRNAs share many target genes, suggesting a complex and redundant regulatory system, particularly in the case of miR-291–5p and miR-215–5p which recognize the same seed sequence. Several of these miRNAs may regulate a number of central genes (those with high intramodular connectivity) from module M1, such as CPSF6 encoding a subunit of a cleavage factor required for the RNA cleavage and polyadenylation processing, which was previously related to externalizing behaviors including ADHD [46], and RICTOR, which plays an essential role during the neurodevelopment and has been associated with hyperactivity and reduced anxiety-like behavior in conditional knock-out mice in the dorsal neural progenitor cells [47].
Module M1, as well as M2 and M6, were also enriched in genes that encode proteins involved in the processing of messenger RNA (mRNA), which includes any process related to the conversion of a primary mRNA transcript into one or more mature mRNAs. mRNA processing and alternative splicing are key processes for both the diversification of protein isoforms and the spatio-temporal control of transcripts, essential for the neuronal development, maturation, and synaptic function [48], and genetic variants in genes encoding these proteins have been related to rare neurodevelopmental disorders [49], as well as common psychiatric disorders like schizophrenia [50].
Module M5 was enriched in genes involved in immune system and inflammatory response, pathways known to play an important role in the development of neuropsychiatric disorders [38, 51, 52], particularly in ADHD [53]. Moreover, genes in module M5, and to a less extent in module M2, are targeted by a great variety of known therapeutic drugs, especially by those that target the immune system (including the Anatomical Therapeutic Chemical categories Antiinfective for systemic use and Antineoplastic and immunomodulating agents), pointing to genes in these co-expression networks as potential therapeutic targets. Importantly, a recent study that explored the druggable genome in ADHD also pointed to drugs to treat autoimmune disorders and malignancies as a potential novel path for the treatment of ADHD [54]. Besides, in module M5 we also found an enrichment in genes differentially expressed in ADHD patients compared with controls, suggesting that differentially expressed genes in ADHD cases are co-expressed and participate in the same biologic pathways. Furthermore, this enrichment was also significant when considering only highly connected genes, highlighting that the genes differentially expressed are central nodes highly connected in this network, reinforcing their relevance in the pathophysiology of ADHD.
The integrative analysis of transcriptomics, genomics, and epigenomics data on ADHD revealed that genes in module M4, also involved in the regulation of gene expression and epigenetic mechanisms, were enriched in both genetic and epigenetic signatures previously described for ADHD [3, 7]. We used two complementary approaches to assign ADHD-associated SNPs to genes, based on position or eQTL results, and found consistent results. PNPLA2 and IQSEC1 were the central genes in the module more significantly associated with ADHD using both methods. PNPLA2 encodes an enzyme involved in the hydrolysis of triglycerides in adipose tissue, and has been related to obesity [55], a highly comorbid disorder in ADHD [56]. In addition, a recent study pointed PNPLA2 as one of the most high-confidence causal genes for ADHD, after combining GWAS, eQTL and gene expression data [57]. IQSEC1 encodes a guanine nucleotide exchange factor, essential for the maintenance of glutamatergic synapses [58], one of the key neurotransmitter systems involved in the pathophysiology of ADHD in combination with dopamine [59, 60].
The eQTL analysis did not reveal any genetic variant that overcame the genome-wide significance threshold, but we found 12 independent genomic loci that showed suggestive evidence of association (P < 1e−06) with the different module eigengenes. We identified a genetic variant associated with the co-expression module M1, rs62096513, which is located in a blood and brain regulatory region of a transcription factor, ZSCAN30, and regulates its expression in PBMCs. Interestingly, module M1 was enriched in target genes for ZSCAN30 that is also included in the same module, suggesting that this transcription factor is an upstream regulator of the co-expressed genes in the module. Besides, we identified another genetic variant associated with the M1 module eigengene, rs73170578, located in CNTNAP2, which encodes a neuronal transmembrane protein member of the neurexin superfamily that function as cell adhesion molecules and receptors. Both rare and common genetic variants in CNTNAP2 have been associated with neurodevelopmental disorders [61, 62], with a special relevance in ADHD and autism [63, 64]. In addition, module M4 was associated with rs6707596, that is an eQTL of the SP3 gene in PBMCs, a transcription factor involved in synaptic plasticity [65]. Finally, we identified four genetic variants associated with M6 module, among them, rs2462337 is located in a blood regulatory region upstream the CUX1 gene, a transcription factor involved in the control of neuronal differentiation and the regulation of dendritic branching, spine development, and synapse formation in cortical neurons [66].
Gene networks analyses reduce the dimensionality of genome-wide gene expression data without losing important biological information and alleviate the multiple testing burden associated with the traditional gene-based methods. Similar network-based studies have been performed using gene expression data in both brain and blood in several psychiatric disorders like autism, schizophrenia and bipolar disorder [23,24,25,26,27,28]. These studies were usually performed in small sample sizes (n < 100 individuals), limiting their statistical power. In contrast, we improved the resolution and robustness of gene networks by considering more than 500 subjects, which allowed the identification of seven ADHD-associated modules enriched in relevant and highly significant biological pathways. However, although our transcriptomic analyses were performed mainly in medication-naive ADHD patients without comorbid disorders (93.7% of all ADHD cases), we cannot discard that these conditions may have influenced the results of the present study. So, further studies in the same cell type are required to confirm our results. Additionally, the identified modules were based on expression data from PBMCs, a non-invasive peripheral tissue whose expression profile has been proposed as a surrogate for expression profiling in the central nervous system [67], and further evidence in the brain is required to confirm their role in the pathophysiology of the disorder.
In summary, we conducted a multi-step analysis to identify and characterize modules of co-expressed genes associated with ADHD using expression data from PBMCs in ADHD cases and controls. We identified seven ADHD-associated modules of co-expressed genes, some of them being enriched in both genetic and epigenetic signatures for ADHD and on biological pathways relevant for psychiatric disorders, such as the regulation of gene expression, epigenetic mechanisms and immune signaling. We also found preliminary evidence for some potential regulatory mechanisms, including microRNAs and genetic variants, for some of the ADHD-associated modules of co-expressed genes identified. These results pinpoint promising genes and pathways for ADHD, support the use of peripheral blood to assess gene expression signatures for the disorder and highlight that the combination of multi-omics signals provides deeper and broader insights into the biological mechanisms underlying the disorder.
References
Sayal K, Prasad V, Daley D, Ford T, Coghill D. ADHD in children and young people: prevalence, care pathways, and service provision. Lancet Psychiatry. 2018;5:175–86.
Faraone SV, Larsson H. Genetics of attention deficit hyperactivity disorder. Mol Psychiatry. 2019;24:562–75.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75.
Demontis D, Walters GB, Athanasiadis G, Walters R, Therrien K, Farajzadeh L, et al. Genome-wide analyses of ADHD identify 27 risk loci, refine the genetic architecture and implicate several cognitive domains. medRxiv. 2022; https://doi.org/10.1101/2022.02.14.22270780.
Palladino VS, McNeill R, Reif A, Kittel-Schneider S. Genetic risk factors and gene-environment interactions in adult and childhood attention-deficit/hyperactivity disorder. Psychiatr Genet. 2019;29:63–78.
Mirkovic B, Chagraoui A, Gerardin P, Cohen D. Epigenetics and attention-deficit/hyperactivity disorder: new perspectives? Front Psychiatry. 2020;11. https://doi.org/10.3389/fpsyt.2020.00579.
Rovira P, Sánchez-Mora C, Pagerols M, Richarte V, Corrales M, Fadeuilhe C, et al. Epigenome-wide association study of attention-deficit/hyperactivity disorder in adults. Transl Psychiatry. 2020;10:1–12.
Wilmot B, Fry R, Smeester L, Musser ED, Mill J, Nigg JT. Methylomic analysis of salivary DNA in childhood ADHD identifies altered DNA methylation in VIPR2. J Child Psychol Psychiatry Allied Discip. 2016;57:152–60.
Walton E, Pingault JB, Cecil CAM, Gaunt TR, Relton CL, Mill J, et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol Psychiatry. 2017;22:250–6.
Mooney MA, Ryabinin P, Wilmot B, Bhatt P, Mill J, Nigg JT. Large epigenome-wide association study of childhood ADHD identifies peripheral DNA methylation associated with disease and polygenic risk burden. Transl Psychiatry. 2020;10:1–12.
Meijer M, Klein M, Hannon E, van der Meer D, Hartman C, Oosterlaan J, et al. Genome-Wide DNA methylation patterns in persistent attention-deficit/hyperactivity disorder and in association with impulsive and callous traits. Front Genet. 2020;11:16.
van Dongen J, Zilhão NR, Sugden K, Heijmans BT, ’t Hoen PAC, van Meurs J, et al. Epigenome-wide Association Study of attention-deficit/hyperactivity disorder symptoms in adults. Biol Psychiatry. 2019;86:599–607.
Marín-Méndez JJ, Patiño-García A, Segura V, Ortuño F, Gálvez MD, Soutullo CA. Differential expression of prostaglandin D2 synthase (PTGDS) in patients with attention deficit-hyperactivity disorder and bipolar disorder. J Affect Disord. 2012;138:479–84.
De Jong S, Newhouse SJ, Patel H, Lee S, Dempster D, Curtis C, et al. Immune signatures and disorder-specific patterns in a cross-disorder gene expression analysis. Br J Psychiatry. 2016;209:202–8.
Garcia-Martínez I, Sánchez-Mora C, Soler Artigas M, Rovira P, Pagerols M, Corrales M, et al. Gene-wide association study reveals RNF122 ubiquitin ligase as a novel susceptibility gene for attention deficit hyperactivity disorder. Sci Rep. 2017;7:1–13.
Sánchez-Mora C, Soler Artigas M, Garcia-Martínez I, Pagerols M, Rovira P, Richarte V, et al. Epigenetic signature for attention-deficit/hyperactivity disorder: identification of miR-26b-5p, miR-185-5p, and miR-191-5p as potential biomarkers in peripheral blood mononuclear cells. Neuropsychopharmacology. 2019;44:890–7.
Lorenzo G, Braun J, Muñoz G, Casarejos MJ, Bazán E, Jimenez-Escrig A. RNA-Seq blood transcriptome profiling in familial attention deficit and hyperactivity disorder (ADHD). Psychiatry Res. 2018;270:544–6.
Nuzziello N, Craig F, Simone M, Consiglio A, Licciulli F, Margari L, et al. Integrated analysis of microRNA and mRNA expression profiles: an attempt to disentangle the complex interaction network in attention deficit hyperactivity disorder. Brain Sci. 2019;9:288.
Mortimer N, Sánchez-Mora C, Rovira P, Vilar-Ribó L, Richarte V, Corrales M, et al. Transcriptome profiling in adult attention-deficit hyperactivity disorder. Eur Neuropsychopharmacol. 2020;41:160–6.
McCaffrey TA, St. Laurent G, Shtokalo D, Antonets D, Vyatkin Y, Jones D, et al. Biomarker discovery in attention deficit hyperactivity disorder: RNA sequencing of whole blood in discordant twin and case-controlled cohorts. BMC Med Genomics. 2020;13. https://doi.org/10.1186/s12920-020-00808-8.
Hu Y, Pan J, Xin Y, Mi X, Wang J, Gao Q, et al. Gene expression analysis reveals novel gene signatures between young and old adults in human prefrontal cortex. Front Aging Neurosci. 2018;10:259.
Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, et al. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–82.
Li X, Zhang Y, Wang L, Lin Y, Gao Z, Zhan X, et al. Integrated analysis of brain transcriptome reveals convergent molecular pathways in autism spectrum disorder. Front Psychiatry. 2019;10:706.
Maschietto M, Tahira AC, Puga R, Lima L, Mariani D, Da Silveira Paulsen B, et al. Co-expression network of neural-differentiation genes shows specific pattern in schizophrenia Bioinformatic and algorithmical studies. BMC Med Genomics. 2015;8:1–15.
Gerring ZF, Gamazon ER, Derks EM. Consortium for the MDDWG of the PG. A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLOS Genet. 2019;15:e1008245.
Radulescu E, Jaffe AE, Straub RE, Chen Q, Shin JH, Hyde TM, et al. Identification and prioritization of gene sets associated with schizophrenia risk by co-expression network analysis in human brain. Mol Psychiatry. 2018;25:791–804.
Jalbrzikowski M, Lazaro MT, Gao F, Huang A, Chow C, Geschwind DH, et al. Transcriptome profiling of peripheral blood in 22q11.2 deletion syndrome reveals functional pathways related to psychosis and autism spectrum disorder. PLoS ONE 2015;10. https://doi.org/10.1371/journal.pone.0132542.
Hess JL, Tylee DS, Barve R, de Jong S, Ophoff RA, Kumarasinghe N, et al. Transcriptomic abnormalities in peripheral blood in bipolar disorder, and discrimination of the major psychoses. Schizophr Res. 2020;217:124.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:1–13.
Grote S, Prüfer K, Kelso J, Dannemann M. ABAEnrichment: an R package to test for gene set expression enrichment in the adult and developing human brain. Bioinformatics. 2016;32:3201–3.
Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019;47:W199–W205.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
Gerring ZF, Mina-Vargas A, Gamazon ER, Derks EM. E-MAGMA: an eQTL-informed method to identify risk genes using genome-wide association study summary statistics. Bioinformatics. 2021;37:2245–9.
Gerring ZF, Gamazon ER, Derks EM. A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet. 2019;15:e1008245.
Maksimovic J, Oshlack A, Phipson B. Gene set enrichment analysis for genome-wide DNA methylation data. Genome Biol. 2021;22:1–26.
Watanabe K, Taskesen E, Van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826.
O’dushlaine C, Rossin L, Lee PH, Duncan L, Parikshak NN, Newhouse S, et al. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18:199.
Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877–D881.
Yoshino Y, Roy B, Dwivedi Y. Differential and unique patterns of synaptic miRNA expression in dorsolateral prefrontal cortex of depressed subjects. Neuropsychopharmacology. 2021;46:900–10.
Sim MS, Soga T, Pandy V, Wu YS, Parhar IS, Mohamed Z. MicroRNA expression signature of methamphetamine use and addiction in the rat nucleus accumbens. Metab Brain Dis. 2017;32:1767–83.
Zhao Y, Zhang K, Jiang H, Du J, Na Z, Hao W, et al. Decreased expression of plasma MicroRNA in patients with methamphetamine (MA) use disorder. J Neuroimmune Pharm. 2016;11:542–8.
Xu W, Zhao M, Lin Z, Liu H, Ma H, Hong Q, et al. Increased expression of plasma hsa-miR-181a in male patients with heroin addiction use disorder. J Clin Lab Anal. 2020;34. https://doi.org/10.1002/jcla.23486.
Chandrasekar V, Dreyer JL. microRNAs miR-124, let-7d and miR-181a regulate Cocaine-induced Plasticity. Mol Cell Neurosci. 2009;42:350–62.
Chandrasekar V, Dreyer JL. Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology. 2011;36:1149–64.
Karlsson Linnér R, Mallard TT, Barr PB, Sanchez-Roige S, Madole JW, Driver MN, et al. Multivariate analysis of 1.5 million people identifies genetic associations with traits related to self-regulation and addiction. Nat Neurosci. 2021;24:1367–76.
Carson RP, Fu C, Winzenburger P, Ess KC. Deletion of rictor in neural progenitor cells reveals contributions of mTORC2 signaling to tuberous sclerosis complex. Hum Mol Genet. 2013;22:140–52.
Furlanis E, Scheiffele P. Regulation of neuronal differentiation, function, and plasticity by alternative splicing. Annu Rev Cell Dev Biol. 2018;34:451–69.
Fliedner A, Kirchner P, Wiesener A, van de Beek I, Waisfisz Q, van Haelst M, et al. Variants in SCAF4 cause a neurodevelopmental disorder and are associated with impaired mRNA processing. Am J Hum Genet. 2020;107:544–54.
Chapman RM, Tinsley CL, Hill MJ, Forrest MP, Tansey KE, Pardiñas AF, et al. Convergent evidence that ZNF804A Is a regulator of pre-messenger RNA processing and gene expression. Schizophr Bull. 2019;45:1267–78.
Jiang NM, Cowan M, Moonah SN, Petri WA. The impact of systemic inflammation on neurodevelopment. Trends Mol Med. 2018;24:794–804.
Han VX, Patel S, Jones HF, Dale RC. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol. 2021;17:564–79.
Dunn GA, Nigg JT, Sullivan EL. Neuroinflammation as a risk factor for attention deficit hyperactivity disorder. Pharmacol Biochem Behav. 2019;182:22–34.
Hegvik TA, Waløen K, Pandey SK, Faraone SV, Haavik J, Zayats T. Druggable genome in attention deficit/hyperactivity disorder and its co-morbid conditions. New avenues for treatment. Mol Psychiatry. 2021;26:4004–15.
La Cruz-Color LD, Hernández-Nazará ZH, Maldonado-González M, Navarro-Muñíz E, Domínguez-Rosales JA, Torres-Baranda JR, et al. Association of the PNPLA2, SCD1 and leptin expression with fat distribution in liver and adipose tissue from obese subjects. Exp Clin Endocrinol Diabetes. 2020;128:715–22.
Cortese S. The association between ADHD and obesity: Intriguing, progressively more investigated, but still puzzling. Brain Sci. 2019;9. https://doi.org/10.3390/brainsci9100256.
Fahira A, Li Z, Liu N, Shi Y. Prediction of causal genes and gene expression analysis of attention-deficit hyperactivity disorder in the different brain region, a comprehensive integrative analysis of ADHD. Behav Brain Res. 2019;364:183–92.
Scholz R, Berberich S, Rathgeber L, Kolleker A, Köhr G, Kornau HC. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66:768–80.
Huang X, Wang M, Zhang Q, Chen X, Wu J. The role of glutamate receptors in attention-deficit/hyperactivity disorder: From physiology to disease. Am J Med Genet Part B Neuropsychiatr Genet. 2019;180:272–86.
Cai Y, Xing L, Yang T, Chai R, Wang J, Bao J, et al. The neurodevelopmental role of dopaminergic signaling in neurological disorders. Neurosci Lett. 2021;741. https://doi.org/10.1016/j.neulet.2020.135540.
Poot M. Connecting the CNTNAP2 networks with neurodevelopmental disorders. Mol Syndromol. 2015;6:7–22.
Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet. 2014;22:171.
Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry. 2010;15:637.
Vecchia ED, Mortimer N, Palladino VS, Kittel-Schneider S, Lesch KP, Reif A, et al. Cross-species models of attention-deficit/hyperactivity disorder and autism spectrum disorder: Lessons from CNTNAP2, ADGRL3, and PARK2. Psychiatr Genet. 2019;29:1–17.
Yamakawa H, Cheng J, Penney J, Gao F, Rueda R, Wang J, et al. The transcription factor Sp3 cooperates with HDAC2 to regulate synaptic function and plasticity in neurons. Cell Rep. 2017;20:1319–34.
Cubelos B, Sebastián-Serrano A, Beccari L, Calcagnotto ME, Cisneros E, Kim S, et al. Cux1 and Cux2 regulate dendritic branching, spine morphology, and synapses of the upper layer neurons of the cortex. Neuron. 2010;66:523–35.
Sullivan PF, Fan C, Perou CM. Evaluating the comparability of gene expression in blood and brain. Am J Med Genet - Neuropsychiatr Genet. 2006;141 B:261–8.
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7.
Acknowledgements
The authors are grateful to patients and controls who kindly participated in this research. The microarrays service was carried out at High Technology Unit (UAT) at Vall d’Hebron Research Institute (VHIR), Barcelona (Spain).
Funding
This work was supported by the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR, 2017SGR-1461), the Instituto de Salud Carlos III (PI18/01788, P19/01224, PI20/00041 and FI18/00285 to L.V.R), the Ministry of Science, Innovation and Universities (IJC2018-035346-I to M.S.A), the Network Center for Biomedical Research (CIBER) to J.C.D.; the European Regional Development Fund (ERDF), the European Union H2020 Programme (H2020/2014-2020) under grant agreements no. 728018 (Eat2beNICE), no. 848228 (DISCOvERIE) and no. 2020604 (TIMESPAN) and the ECNP Network ‘ADHD across the Lifespan’.
Author information
Authors and Affiliations
Contributions
JCD, MR, and MSA conceived the project. CF, MC, VR, JARQ participated in the clinical assessment and in the recruitment of patients. JCD, LA, and LVR participated in the RNA isolation and preparation of samples. JCD undertook the statistical analyses. JCD, LVR, NL, SA, MR, and MSA participated in the study design and the discussion of results. JCD, MR, and MSA participated in the manuscript preparation. All authors contributed to the interpretation of the findings and revised and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
J.A.R.Q. was on the speakers bureau and/or acted as consultant for Janssen-Cilag, Novartis, Shire, Takeda, Bial, Shionogi, Sincrolab, Novartis, BMS, Medice, Technofarma, Rubió and Raffo in the last 3 years. He also received travel awards (air tickets + hotel) for taking part in psychiatric meetings from Janssen-Cilag, Rubió, Shire, Takeda, Shionogi, Bial, and Medice. The Department of Mental Health chaired by him received unrestricted educational and research support from the following companies in the last 3 years: Janssen-Cilag, Shire, Oryzon, Roche, Psious, and Rubió. C.F. and V.R. have received fees to give talks for Shire/Takeda and Rubió. All other authors declare no biomedical financial interests or conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cabana-Domínguez, J., Soler Artigas, M., Arribas, L. et al. Comprehensive analysis of omics data identifies relevant gene networks for Attention-Deficit/Hyperactivity Disorder (ADHD). Transl Psychiatry 12, 409 (2022). https://doi.org/10.1038/s41398-022-02182-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02182-8
This article is cited by
-
Children with a history of both maternal immune activation and prematurity are not at increased risk of ADHD symptoms
European Child & Adolescent Psychiatry (2023)
