
Abstract
Circadian (~24 h) rhythms in physiology and behavior are evolutionarily conserved and found in almost all living organisms. The rhythms are endogenously driven by daily oscillatory activities of so-called “clock genes/proteins”, which are widely distributed throughout the mammalian brain. Mammalian (mechanistic) target of rapamycin (mTOR) signaling is a fundamental intracellular signal transduction cascade that controls important neuronal processes including neurodevelopment, synaptic plasticity, metabolism, and aging. Dysregulation of the mTOR pathway is associated with psychiatric disorders including autism spectrum disorders (ASD) and mood disorders (MD), in which patients often exhibit disrupted daily physiological rhythms and abnormal circadian gene expression in the brain. Recent work has found that the activities of mTOR signaling are temporally controlled by the circadian clock and exhibit robust circadian oscillations in multiple systems. In the meantime, mTOR signaling regulates fundamental properties of the central and peripheral circadian clocks, including period length, entrainment, and synchronization. Whereas the underlying mechanisms remain to be fully elucidated, increasing clinical and preclinical evidence support significant crosstalk between mTOR signaling, the circadian clock, and psychiatric disorders. Here, we review recent progress in understanding the trilateral interactions and propose an “interaction triangle” model between mTOR signaling, the circadian clock, and psychiatric disorders (focusing on ASD and MD).
Similar content being viewed by others

General introduction
Circadian rhythms are the approximately 24-h biological rhythms that are found in almost all living organisms on the planet [1]. These rhythms are endogenously driven by circadian clocks, which can function autonomously but are constantly regulated by environmental signals including light, food availability, etc. [2]. Circadian clocks synchronize numerous internal biological processes with the cyclical changes in the external environment, so that organisms can predict and prepare for upcoming environmental changes and adjust their physiological states accordingly [3]. In cells, the circadian clock is driven by interlocking feedback loops of gene expression. Clock genes are widely expressed in almost all cells of the body. In the brain, a variety of neural processes are orchestrated by circadian clocks throughout life. During neurodevelopment, the circadian clock regulates neurogenesis, migration, and progenitor cell differentiation [4, 5]. In the adult brain, circadian rhythms regulate neuronal excitability, synaptic plasticity, learning and memory, mood, and social behaviors [6,7,8]. Disruption of circadian rhythms can cause mental health problems. In the short term, circadian disruption can cause jet lag-like symptoms, including fatigue, insomnia, difficulty concentrating, etc. [9]. Chronic disruption of circadian rhythms and sleep-wakefulness cycles is often associated with neurological and psychiatric disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), mood disorders(MD), and autism spectrum disorders (ASD), although it is not entirely clear whether circadian dysfunction underlies the pathogenesis of these brain disorders or it is simply a consequence caused by the primary pathophysiological changes in these diseases [10]. There is evidence from neurological and psychiatric disorders where sleep and circadian dysfunction contribute to both the vulnerability to and the development and progress of certain disorders.
The mechanistic/mammalian target of rapamycin (mTOR) signaling is an evolutionarily conserved intracellular signal transduction cascade that regulates fundamental cellular processes including cell growth, metabolism, proliferation, and aging from yeasts to humans [11,12,13]. Dysregulation of mTOR signaling is implicated in a number of common human diseases, including cancer, metabolic syndrome, cardiovascular diseases, and neurological and psychiatric disorders [14]. Pharmacological mTOR inhibitors are clinically applied for immunosuppressing after organ transplantation, cancer chemotherapy, as well as treating some neurological and psychiatric disorders in clinical trials. Recent work has uncovered significant crosstalk between mTOR signaling and the circadian clock in different tissues and systems, suggesting it could be a fundamental mechanism of homeostatic integration between cell metabolism and circadian timekeeping [15]. Along these lines, the activities of mTOR signaling are regulated by the circadian clock and exhibit robust diurnal oscillations in the brain and peripheral systems such as the liver [16, 17]. On the other hand, the fundamental properties of the circadian clock (entrainment, synchronization, speed) are regulated by the mTOR signaling [18,19,20]. Importantly, mTOR activities are often found dysregulated in brain diseases where the daily rhythms in patients are disrupted. Understanding the crosstalk mechanisms between mTOR and the clock may help to gain new insights into the pathogenic mechanisms of psychiatric disorders and develop novel therapeutic strategies to treat these diseases. Increasing evidence support an “interaction triangle” model between the circadian clock, mTOR signaling, and psychiatric diseases (Fig. 1). In this review, we will first introduce the mammalian circadian system and a role for mTOR signaling in the mammalian circadian clock, and then we will discuss recent progress on our understanding of circadian and mTOR dysfunctions in highly prevalent psychiatric disorders, ASD and MD.

1. The circadian clock temporally regulates mTOR activities in different tissues. 2. mTOR signaling regulates the central and peripheral circadian clock functions. 3. Psychiatric disorders can cause disruption of circadian rhythms. 4. Circadian dysfunction can contribute to the pathogenesis of psychiatric disorders. 5. Aberrant brain mTOR activities lead to psychiatric diseases. 6. Psychiatric disorders can lead to dysregulation of mTOR activities in the brain.
The mammalian circadian system
In mammals, the circadian system is organized in a hierarchical manner. The central clock is located in the suprachiasmatic nucleus (SCN) of the hypothalamus [21]. The pair of tear drop-like structures is composed of only ~20,000 neurons, but it is essential for circadian timekeeping in animals [22, 23]. The SCN predominantly determines the speed of activity rhythms in animals and serves as the primary responder to the most important clock resetting signal-ambient light [24]. The important functions of the SCN are enabled by its unique anatomical structure. The SCN is located just above the optic chiasm and its ventral neurons receive photic signal via the monosynaptic input from the melanopsin-expressing intrinsically photosensitive retinal ganglion cells via the retinohypothalamic tract [25,26,27,28]. As part of the hypothalamus, the SCN is embedded in a cluster of hypothalamic nuclei (e.g., paraventricular nucleus) that play pivotal roles in neural and hormonal control of homeostasis. The rhythmic output from the SCN to these nuclei regulates daily rhythms in the endocrine system and the autonomic nervous system [29,30,31].
The SCN neurons are highly heterogeneous in their neuropeptide expression, electrophysiological properties, and photic response [32, 33]. The ventral SCN neurons express vasoactive intestinal peptide (VIP) and gastrin-releasing peptide (GRP) and the dorsal SCN neurons express arginine vasopressin (AVP). Cellular oscillators in the SCN are coupled to form a coherent and stable oscillating network. Intercellular synchronization between SCN neurons renders robustness and accuracy to the SCN-generated rhythms [34]. The unique coupling mechanisms distinguish SCN from peripheral circadian oscillators, where the coupling between cellular oscillators is considered weak [34,35,36]. Vasoactive intestinal peptide (VIP) and GABAergic signaling are important for coupling cellular oscillators in the SCN [37,38,39,40,41,42,43]. AVP receptors are essential for the communications between the ventral and dorsal SCN [44]. AVP positive neurons exhibit stronger circadian rhythms in firing rates and membrane properties compared with AVP negative neurons, indicating their important role in transducing SCN rhythms to other brain areas [45]. Light-induced immediate early gene expression and activation of protein kinases first appear in the “core” SCN, including VIP and GRP expressing cells, indicating that the ventral SCN neurons first respond to light stimulation [46,47,48]. The dorsal SCN neurons in turn received resetting signal from the ventral SCN neurons. Besides SCN neurons, recent studies have uncovered a modulatory role for astrocytes in the SCN [49]. Interestingly. astrocytic clocks alone can drive molecular oscillations in SCN neurons via glutamatergic signals.
Whereas SCN is the master pacemaker, clock genes are widely expressed in almost all cells throughout the body. Clock gene oscillations have been identified in a variety of brain regions [50]. The local brain clocks regulate neuronal properties of individual brain regions and presumably also regulate the brain functions in a time-of-day dependent manner [7]. Phase differences between region-specific clock oscillators are observed, but the coupling mechanisms between these oscillators remain to be fully understood [51]. At the cellular level, the circadian clock is driven by transcriptional/translational genetic feedback loops (TTFL) [52]. Decades of research have identified the framework of feedback mechanisms in different specifies. In mammals, the transcriptional activators BMAL1 and CLOCK (or NPAS2) form a heterodimeric complex to activate E-box-mediated transcription of Period (Per1, 2, 3) and Cryptochrome (Cry1, 2) genes [53, 54]. PER and CRY form protein complexes in the cytosol and translocate to the nucleus upon reaching a level enough to suppress their own gene transcription [2, 55]. In a second negative feedback loop, the transcription of Rev-erb α, β is promoted by CLOCK: BMAL1. In turn REV-ERBs repress Bmal1 transcription via the retinoic acid response element (RRE) [56]. The nuclear hormone receptors RORα, β, γ compete with REV-ERBs to activate Bmal1 transcription [57, 58]. Rhythmic expression of clock output genes (so called “Clock-controlled genes”) is transcriptionally regulated by CLOCK: BMAL1 acting on E-box elements in their regulatory regions [59]. Thus, CLOCK: BMAL1 is not only critical for sustaining the TTFL, but also serves as an important mechanism of clock output. Besides transcriptional mechanisms, the clock gene expression, and levels of clock proteins are also regulated at the levels of mRNA translation and post-translational protein degradation [18, 60,61,62,63,64]. Together, gene expression regulation at different levels ensures the precision and robustness of circadian gene expression (Fig. 2). To be in sync with external and internal environmental changes, the circadian clock must be regulated by extracellular and intracellular signals [3, 65]. As signal transduction process is complex, our understanding of the key signal transduction events that couple extracellular and intracellular signals to clock gene expression is not complete. In particular, the crosstalk mechanisms between cell metabolism and core clock mechanisms remain to be fully understood.

The transcription factors CLOCK (or NPAS2) and BMAL1 form heterodimers, which bind to the cis-acting element E-box and activate the expression of Period1/2 and Crypotochorme1/2. PER and CRY proteins form multiprotein complexes in the cytoplasm. Once accumulating to a certain level, the PER/CRY complexes translocate into the nucleus, interact with the CLOCK: BMAL1 complex, and repress their own gene transcription. The CLOCK: BMAL1 complex also promotes the transcription of Rev-erbα/β. REV-ERBs inhibit the Bmal1 transcription whereas RORs promote Bmal1 transcription. PER protein abundance is controlled at the mRNA translation level via an elF4E-dependent mechanism. CRY is phosphorylated by AMPK and PER by CSNK. The levels of CRY and PER are also regulated by phosphorylation and ubiquitin-medicated protein degradation at the post-translational levels. The CLOCK: BMAL1 complex also regulates numerous clock-controlled genes via the E-box enhancer.
mTOR signaling and the circadian clock
Protein synthesis (mRNA translation) is the most energy-consuming step in gene expression and subject to delicate regulation [66]. mTOR is a highly conserved Ser/Thr protein kinase that forms a complex signaling network and integrates extracellular and intracellular signals to impinge on machineries of mRNA translation and control protein synthesis [11,12,13]. mTOR forms two functionally distinct protein complexes mTORC (mTOR complex) 1 and mTORC2 (Fig. 3). Recent work has found it regulates various neuronal processes including neural progenitor cell growth and differentiation, synaptic plasticity, learning and memory, hormone secretion, food uptake, and sleep [67,68,69,70,71,72].

Light at night activates ERK MAPK and mTORC1 pathways in the SCN by the neurotransmitter glutamate and PACAP. ERK MAPK in turn activates MSK to phosphorylate CREB and activated Per transcription. ERK MAPK also activated MNK, which in turn phosphorylates the cap-binding protein eIF4E at Ser209 and regulate mRNA translation. The mTORC1 activation also regulates translation through downstream translation pathway effectors such as S6Ks and 4E-BPs, etc. Phosphorylated S6K regulates circadian ribosomal biogenesis. Phosphorylation of 4E-BP activated eIF4E dependent translation. The ERK MAPK and mTOR pathways converge on eIF4E to regulate cap-dependent translation of Per1, Per2, Vip mRNAs, which play important roles in photic entrainment of the circadian clock and SCN cell synchronization. Circadian mTOR activities are controlled by the circadian clock via complex mechanisms, one of which may be through the interaction of Per2 with TSC1.
mTORC1 activity has been extensively studied in the SCN [16, 18, 19, 47, 73]. mTORC1 activity is highly rhythmic in the SCN, high during the day and low at night. It is also highly responsive to light stimulation at night [16]. Transient light exposure can induce rapid (within 15 min) upregulation of the S6K1 and 4E-BP phosphorylation in the ventral SCN [73]. Blocking mTORC1 activation in the SCN before light can significantly affect light-induced behavioral phase shifts in mice [47]. In mice without 4E-BP1, clock re-entrainment by a shifted light/dark cycle is much more quickly, as compared to the wild-type mice. Mechanistic studies found enhanced translation of the precursor protein of VIP, prepro-VIP in the brain of Eif4ebp1KO mice, indicating that enhanced VIP signaling may underlie the circadian behavioral changes in Eif4ebp1 KO mice [18]. Genetic repression of mTOR signaling only in VIP neurons can lead to cellular and behavioral changes similar to those seen in the VIP mutants [19].
Beyond the SCN central clock, mTOR rhythms have been found in numerous cells and tissues and mTOR regulates fundamental functions of the peripheral circadian clocks [15]. It is found that mTOR signaling controls the circadian clock properties in a variety of tissues and organisms, which suggests mTOR is a conserved circadian regulator [15, 20, 74, 75]. Strong circadian rhythms of mTOR activities are found in different tissues and cells in mammals [15]. Pharmacological mTORC1 inhibitors can dampen circadian rhythms of PER2 rhythms in hepatocytes, liver slices and SCN slices in a reversible manner [20]. Genetic mTOR repression can dampen the amplitude and lengthen the circadian period in cellular oscillators, SCN slices, and whole animals [20]. mTOR pharmacological inhibitors can markedly slow down and damp the rhythms in the liver clock. Conversely, mTOR activation can accelerate the speed of cellular clocks [20]. In addition, the mTORC1 target S6K1 was found to phosphorylate the clock protein BMAL1 and regulate rhythmic translation [76]. In TSC mutants where mTOR activities are constitutively elevated, mice demonstrate a shorter wheel-running period and disrupted core body temperature rhythms in constant conditions [77].
How the circadian clock regulates mTOR activities is not completely clear. One study found that the circadian protein Period2 recruits TSC1 to the mTORC1 complex and suppresses mTORC1 activity [78]. Specific functions of the ribosomal protein S6 kinases (S6Ks) and mTORC2 in the circadian clock remain to be identified. There is an urgent need to fill in these knowledge gaps because mTOR inhibitors (rapalogs) are FDA-approved drugs and are being widely used to human patients without regard to the potential effects of these therapeutics on the rhythmicity of gene expression. Knowledge on physiological functions of mTOR would also be important to understand the mTOR-dependent mechanisms whereby circadian dysfunction is involved in the pathogenesis of brain diseases.
Circadian dysfunction, mTOR, and ASD
Introduction of ASD
ASDs are an array of neurodevelopmental disorders that are characterized by core behavioral symptoms, including deficits in social interaction and/or communication, repetitive behaviors, and restricted interests. ASD starts in early childhood around the age of 3 years and persists throughout life [79]. People with ASD commonly experience other comorbidities such as memory and learning deficits, seizures, motor incoordination, changes in sensory perception, anxiety, and sleep disturbances [80,81,82]. According to the data released by the Centers for Disease Control and Prevention (CDC) and Autism and Developmental Disabilities Monitoring network, about 1 in 44 (2.3%) children in the United States have been identified with autism in 2018 [83]. Also, it is four times more common in boys than girls [84]. The etiological factors of ASD remain to be fully understood. The environmental and genetic causes of this disorder are often studied separately, however, ASD may be caused by a combination of both factors. There are no medications that treat the core symptoms of ASD but a combination of multiple treatments (behavioral, social, educational, medical, etc.) are used to reduce symptoms that interfere with daily functioning and quality of life.
Despite the high prevalence of ASD worldwide, the pathogenic mechanisms are not fully understood, although a number of ASD risk genes have been identified. The pathological changes in the brain of ASD patients are not consistent and can comprise alterations in the cell size, synaptic growth as well plasticity, changes in the morphology of the dendritic spines [85, 86]. Abnormalities in the neurotransmission has been implicated in the development of ASD, ranging from an imbalance in glutamatergic and GABAergic (excitatory/inhibitory) neurotransmitters as most explored to dopaminergic, adrenergic, serotonergic, and endo-cannabinoid systems among the less explored pathways [87,88,89]. At the cellular and molecular level, changes include altered neural circuits as well as synaptic plasticity, changes in the morphology of the dendritic spines, abnormal levels of synaptic proteins, and impaired synaptic homeostasis [90]. Due to the complexity of the symptoms, understanding the exact pathophysiology of ASD remains a formidable challenge. This in turn hampers the development of new drug strategies to treat this disorder. Indeed, until today the antipsychotics risperidone and aripiprazole are the only FDA-approved drugs for the management of ASD [91, 92]. Nevertheless, the clinical use of these drugs in ASD is limited as they lack the ability to treat the core ASD symptoms and are accompanied by various side effects [93,94,95]. Therefore, there is an urgent need to elucidate the mechanistic alterations responsible for ASD.
Circadian clock and sleep problems in ASD
Circadian dysfunctions are frequent comorbidities of ASD [96]. Studies suggest that abnormalities in the cortisol, melatonin levels, as well as disrupted sleep wake cycle, have been implicated as underlying features of ASD [97]. The circadian clock controls diurnal oscillations of cortisol and melatonin levels and the timing of sleep onset. The alterations in the circadian timing system may be associated to various neurobehavioral changes including sleep problems, behavioral and cognitive alterations [98]. Literature suggests that ~50–83% ASD individuals show sleep problems, comparing to <30% in the normal population [99, 100]. The most frequently reported circadian and sleep problems in ASD include the symptoms of insomnia indicated by inability to sleep or stay sleep and circadian rhythm sleep wake disorders manifested as a misalignment in the endogenous circadian rhythms and the external environment [101]. In particular, the sleep dysregulations and changes in the biological rhythms lead to a worsening of behaviors, decreased seizure threshold, sensory abnormalities, and an overall affected quality of life in these ASD children [102]. Also, a decrease in the levels of melatonin as well as decreased melatonin synthesis was repeatedly reported in ASD individuals [97, 103, 104]. The therapeutic benefits of melatonin treatment in ASD patients range from improving the sleep latency and sleep quality to improving the behavioral impairements [105,106,107]. A randomized clinical trial found aglomatine, an analogue of melatonin to be effective in the treatment of insomnia and sleep problems in ASD patients [108]. All these data suggest the relevance of an underlying impairment of the circadian timing system to the behavioral ASD symptoms. Therefore, studying the mechanisms associated to circadian dysregulation in ASD may be helpful in identifying the early biomarkers for improving the diagnosis and lifelong prognosis of ASD.
The circadian system is complex, as almost all cells in the body exhibit circadian rhythms of gene expression. As aforementioned, the cellular circadian rhythms are generated by auto regulatory feedback loops of clock genes. Accumulating evidence supports the association between clock gene variants and ASD [109]. Nicholas et al. screened SNPs in eleven clock and clock related genes in over 100 ASD children and their parents and found a significant allelic association of two circadian system related genes (PER1 and NPAS2) with two SNPs in each gene [110]. In addition, a high proportion of all possible haplotypes in NPAS2 were also significant in ASD individuals [110]. Thus, this study supported the hypothesis that the epistatic clock genes may be involved in the ASD etiology. A genome-wide study by Hu et al. reported altered expression of various circadian genes in the lymphoblastic cell lines from ASD individuals as compared to their respective controls [111]. Clock gene sequencing studies identified SNPs in multiple clock genes (TIMELESS, NR1D1, RORA, ARNTL2, PER1, PER2, PER3, BMAL1, CLOCK) in ASD patients [109, 112,113,114,115,116]. The functional mutations of NR1D1 were also detected in ASD subjects with and without sleep disorders [112]. A study by Hoang et al. suggested a possible role of PER2 gene in the sleep dysregulation of ASD individuals [117]. Whole exome sequencing studies identified de novo loss-of-function variants in PER2, RORB and CSNK1E [118,119,120,121]. These studies suggest that the functional deficits of various circadian relevant genes may be related to ASD etiology and pathophysiology.
Next, increasing studies have found circadian clock dysfunctions in animal models of ASD. For example, a recent study by Delorme et al. showed changes in the circadian rhythms indicated by altered locomotor activity rhythms in the mouse model of maternal immune activation of ASD [122]. In particular, the mice whose mothers were injected with Poly I:C in pregnancy developed increased subjective day activities, which suggests a link between circadian rhythm and ASD [122]. SHANK3 mutations are involved in ASD pathogenesis. Interestingly, the protein levels of synaptic SHANK3 exhibit circadian rhythms in the mouse hippocampus [123]. A study shows that the Shank3+/-mouse, a genetic mouse model of ASD, exhibits altered light sensitivity in the SCN clock [124]. Along these lines, another study on mice with a deletion in Shank3 exon 21 reports that these mice have problems falling asleep and exhibit transcriptional down-regulation of clock gene Per3 and Nr1d1 in the prefrontal cortex [125]. These mice also exhibit impaired circadian wheel-running activities in constant darkness [125]. SCN2A is another important ASD risk gene. Ma et al. found that the Scn2a deficient mouse, an ASD genetic animal model, exhibits increased wakefulness and reduced non-rapid-eye-movement sleep. Scn2a deficiency also disrupts the spontaneous firing pattern in SCN neurons [126]. Vijaya Shankara et al. found significant changes in circadian wheel-running behavior in the BTBR T+Itpr3tf/J (BTBR) mouse, an idiopathic model of ASD [127]. The BTBR mice exhibit shorter free running period and higher level of activities as compared to WT C57BL/6J mice. These mice also show increased clock resetting in response to light stimulation at night and accelerated clock resetting to advanced and delayed light cycles. A study by Ferraro et al. demonstrated that in the valproic acid-induced model of ASD, the rhythmic Bmal1 expression in the SCN and diurnal rhythms of corticosteroid are disrupted in a sexually dimorphic manner [128]. Animals exhibit impaired circadian wheel-running behavior under constant dark conditions and reduced clock resetting in response to light at night [128]. Together, these animal studies demonstrate that circadian functions and clock gene expression are often disrupted in ASD animal models.
Emerging evidence supports the hypothesis that circadian dysfunction may contribute to ASD pathogenesis [96, 129, 130]. Biological timing is critically important for neurodevelopment and brain plasticity [131]. Conceivably, the disturbances in the circadian system can lead to sleep disturbance, dysregulation of clock gene expression and diurnal oscillations of many hormones. All these can have detrimental consequences to neurodevelopment and lead to the emergence of a plethora of neurodevelopmental conditions including ASD (Fig. 4). Indeed, impairments of circadian rhythm for a few days can have an impact on the specialization and maturation of brain functions at certain times of development [132]. A study by Kobayashi et al. provides evidence that circadian clock genes Clock and Bmal1 can control the timing of brain plasticity in the development of the neocortex and that the visual cortex of a Clock deficient mice shows delayed maturation of inhibitory PV cells [133]. Another study by Goto et al. reported a role for the clock gene Nr1d1 in mouse brain development and possible implication in ASD [112]. Three NR1D1 mutations are identified in ASD patients. Using mouse models, they further find that Nr1d1 plays a pivotal role in cortical neuron migration, axon extension and dendritic arbor formation. Together these results suggest that functional defects in Nr1d1 may be related to ASD pathophysiology [112]. More experimental evidence supporting a potential role of circadian disruption in the development of ASD is discussed in 4.4. Nevertheless, the mechanistic associations of ASD and circadian dysfunctions remain to be fully understood. Some of the other well-studied pathways in ASD, including the altered excitatory (glutamatergic)/inhibitory (GABAergic) pathways, oxidative stress, changes in the synaptic plasticity, and other metabolic pathways, may also serve as links between circadian dysfunction and ASD pathogenesis [134,135,136,137], as these pathways are directly or indirectly controlled by the circadian clock. Thus, it is possible that disruptions in the circadian clock may be responsible for the alterations of these pathways.

The environmental, genetic and hormonal risk factors cause sleep and circadian disturbances, which may in turn lead to impairments in neurodevelopment and cause neurodevelopmental disorders such as ASD.
The mTOR signaling pathway and ASD
The mTOR signaling cascade regulates numerous neuronal processes from proliferation, differentiation to plasticity and aging [14]. Dysregulation of the mTOR signaling pathway is implicated in the pathogenesis of various neurological disorders including ASD [14]. Hyperactivation of the mTOR pathway have been identified in several syndrome related to ASD [138]. For example, a study by Tang et al. demonstrated the link between ASD and mTOR where an increase in the dendritic spine density with reduced developmental spine pruning in layer V pyramidal neurons was observed in the temporal lobes of postmortem ASD samples [139]. The overactive mTOR signaling may also produce excessive spines thus correlating ASD and mTOR. mTORC1 regulates protein synthesis through downstream effector proteins 4E-BPs and S6Ks [11]. One possible explanation is that mTORC1 activation suppresses 4E-BPs and leads to dysregulated cap-dependent mRNA translation [140]. The complex includes eIF4E (the cap-binding protein), eIF4A (the RNA helicase), and eIF4G (the scaffolding protein bridging RNA to ribosome). The causal role of eIF4E in ASD was first pointed out by the discovery of individuals with ASD whose EIf4e gene had an activating mutation in its promoter region [141]. The development of ASD-like phenotypes via abnormal 4E-BP/eIF4E axis has been demonstrated in mouse models [142]. Dysregulated translation control may lead to abnormal expression of specific ASD risk genes [142, 143]. On the contrary, however, Nicolini et al. found decreased mTOR activities in the postmortem fusiform gyrus samples of patients with idiopathic autism [144]. Interestingly, Rosina et al. observed that there was an increased activities of mTOR and MAPK pathways in the peripheral blood samples of ASD patients [145].
Some of the strongest evidence that mTOR may also be required developmentally comes from genetic mutations associated with neurodevelopmental disorders. Mutations in negative regulators of mTORC1, such as TSC1, TSC2 and PTEN, are found in monogenic ASD [14]. The frequent incidence of autism in the monogenetic mTORopathies has advocated a critical role for mTOR in the pathogenesis of autism [138]. The association of abnormal mTOR activities (particularly hyperactivation) with different syndromic forms of ASDs, such as TSC, Fragile X syndrome (FXS), Angelman syndrome, Hamartoma tumor syndrome, and Rett syndrome, has been documented in various clinical studies. Numerous animal studies reported that the deletion of various genes such as Tsc1/2, Pten, Nf1, Fmr1 resulted in ASD-like phenotypes through the disruption of the mTORC1 mediated signaling. Firstly, studies show that ASD is strongly associated to TSC and the heterozygous deletion of Tsc1 or Tsc2 considerably increase the individual’s risk for ASD development [146]. Patients with mutations of Tsc1 or Tsc2, upstream repressors of mTOR activities, exhibit an array of autistic features that resemble idiopathic autism [147]. The subjects with TSC have reported to show a 100-fold increase in the susceptibility of getting diagnosed with autism in comparison to the normal individuals. Further, the alterations in the TSC-related cell signaling also have a substantial role in ASD pathogenesis [146]. TSC1 or TSC2 normally form an mTOR inhibitor complex and thus their loss causes activation of mTOR signaling which in turn leads to mTORC1-dependent increase in the phosphorylation of S6, S6Ks, and 4E-BPs [146, 148, 149]. This reduced inhibition further causes the mTOR hyperactivity, altered protein synthesis, enhancement of proliferation. Also, a study by Alsaqati et al. demonstrated the TSC disorder which they characterized by hyperactivation of the mTORC1 pathway using the iPSC from ASD patients [150]. Secondly, the mutations in the Fmr1 gene leads to FXS and associated to abnormalities in the mTOR-dependent protein synthesis. Also, the overactivation of mTORC1 has been reported to be associated with FXS [151]. In addition, a study by Hoeffer et al. reported an increase in the phosphorylation of mTOR, S6K1, AKT in their protein lysates in subjects with fragile X syndrome [151]. Dysregulation of mTOR signaling has been found in Fragile X Syndrome. A study by Sharma et al. provides a functional link between elevated mTOR signaling and aberrant synaptic plasticity in fragile X mouse. A study by Qin et al. illustrated that FMRP limits the protein synthesis via activation of metabotropic glutamate protein activation and the functional loss of this FMRP has reported to cause increased protein synthesis [152]. Further, dysfunctions of intracellular signaling pathways including mTOR and PTEN pathways are considered pathogenic in ASD [153]. In addition to this, about 1–5% of ASD cases have demonstrated PTEN gene mutations, a repressor of the phosphatidylinositol 3-kinase/Akt/mTOR signaling pathway. This result suggests that disinhibited mTOR signaling can be a contributing factor for ASD phenotypes in these cases. Moreover, the functional loss of Pten has also been reported to be linked to ASD. The PTEN gene mutations have been found in about 5% of ASD patients with macroencephaly [154]. As the role of PTEN in synaptic functioning is associated with ASD, this link can contribute to better understanding of the ASD pathology and promote potential new therapies. Further, 15% patients with neurofibromatosis type 1, a prototypical disorder caused by heterozygous mutations in the Nf1 gene, meet the criteria for ASD [138, 155]. NF1 is a negative regulator of RAS and thus involved in the mTOR signaling regulation which further shows the link of ASD to mTOR signaling [156]. These findings found that mTOR signaling is dysregulated in different subset of ASD patients may help explain the wide degree of clinical severity and may help provide insight into the treatment strategies for treating individuals with ASD.
In addition to this, dysregulation of the mTOR pathway has also been implicated in animal models of ASD. A study by Nicolini et al. reported decreased mTOR signaling pathway in rats exposed to valproic acid indicated by a decrease in the phosphorylated and total mTOR, Akt, and 4E-BP1 and phosphorylated S6 protein in the VPA exposed rats [144]. Another study by Tang G et al. reported postnatal spine pruning defects, blockade of autophagy, and ASD-like social behaviors in the Tsc2+/− ASD mice model suggesting that mTOR-regulated autophagy is required for developmental spine pruning [139]. Further, specific deletion of Tsc2 in cerebellar Purkinje cells leads to autistic-like changes in mice, indicating cerebellar-specific mTOR signaling regulates mouse social behavior [157]. A study by Kotajima-Murakami et al. demonstrated the implication of over-activation of mTOR in the pathogenesis of syndromic ASD, such as TSC [148]. The results of this study showed that treatment with mTOR inhibitor rapamycin improved social interaction deficits in mouse models of TSC [148]. Enhanced mTORC1/S6K1 activities are found in a mouse model of FXS, the Fmr1 knockout (KO) mice [158]. Moreover, a study by Yan et al. demonstrated that enhanced activity of mTORC1 was related to the reduction in the autophagy and protein degradation in Fmr1 KO mice [159]. A study by Gantois et al. showed that hyperactivation of mTORC1 and ERK MAPK pathways is found in Fmr1 knockout mice and metformin ameliorates core behavioral deficits by normalizing ERK hyperactivation [160]. Targeted Pten deletion in the forebrain leads to aberrant social behaviors in animals [140, 154]. Studies found that the Pten mutant mice showing deficits in the social behaviors resembling ASD individuals was associated with the hyperactivity of mTORC1 and its downstream pathway element S6K1. The studies also showed that rapamycin treatment reversed the ASD phenotypes in the Pten mutant mice further confirming the role of mTORC1 in ASD [161, 162]. Another study showed that treatment with rapamycin ameliorated the social deficits in the BTBR mouse model of ASD [163]. Further studies revealed that the Eif4ebp2 KO and Eif4e overexpression generated an autistic-like behavioral manifestation, impaired social approach, and repetitive behaviors in mice [142, 143]. Thus, this evidence suggests a common biochemical link between dysregulated mTOR signaling and ASD.
The crosstalk between mTOR and circadian clock and its implications in the development of ASD
Emerging evidence suggest that the mTOR pathway may serve as a link between circadian dysfunction and ASD pathogenesis. As mTOR activities in the brain are rhythmically regulated by the circadian clock and exhibit daily oscillations, dysfunction of the circadian clock can lead to aberrant temporal activities of the mTOR signaling and impair neurodevelopment [16]. Indeed, Fang et al. examined the effects of chronic disruption of circadian rhythms on neurodevelopment and animal behaviors in wild-type mice [6]. They find that aberrant light-dark cycle disrupts rhythmic clock gene expression in different brain regions including SCN and the hippocampus and leads to hyperactivation of mTORC1 and MAPK pathways in the brain. Adult WT mice raised in the aberrant light-dark cycle exhibit autistic-like behavioral changes, including impaired social interaction, communication, and repetitive behaviors. Genome-wide changes in gene expression are identified in the hippocampus. A number of ASD risk genes are differentially expressed, including Pon1, Magel2, Ppp1r1b, Slc29a4, Ttc25, Dydc2, Fam92b, Ttn, Tcf7l2, Rorα, Foxp2, Rims3, and Satb2. Aberrant synaptic transmission, immature dendritic spine morphology are also found in the hippocampus of these mice [6]. These results demonstrate that disruption of circadian rhythms during neurodevelopment can lead to aberrant mTOR activities and autistic-like molecular and behavioral changes in adult mice. In the future, it would be important to identify the developmental stages crucial to autism when the crosstalk between mTOR and the clock is most consequential. mTOR activities are dysregulated in monogenic mouse models of ASD, in which circadian dysfunctions are also identified. A study by Sawicka et al. demonstrated circadian rhythm defects in hippocampus dependent memory in the Fmr1 knockout mice [164]. Moreover, Lipton et al. demonstrated that mutation of Tsc1 or 2, the mTOR repressor genes, leads to elevated protein levels of Bmal1, which in turn disrupts the circadian rhythms in mice [77]. It is not clear though in which developmental stage the disruption of clock function emerges and whether the clock dysfunctions contribute to the pathogenesis in these conical ASD models.
Recent evidence demonstrates that disruption of clock gene expression in specific brain regions can deregulate mTORC1 pathway, which in turn may lead to autism-like phenotypes in mice. The essential clock gene Bmal1 is associated with human sociability and its missense mutation has been identified in ASD. A recent study by Liu et al. provides evidence that Bmal1 disruption may contribute to the development of ASD-like traits in mice by mTORC1 overactivation [165]. They find significant social impairments, excessive stereotyped and repetitive behaviors, as well as motor learning disabilities in the Bmal1 KO mice, all of which resemble core behavioral deficits in ASD. Similar autism-like behavioral changes phenotypes are also found in Bmal1+/− mice [166]. Furthermore, pathological, and electrophysiological changes are found in the cerebellar Purkinje cells (PCs) of in the Bmal1 KO mice. By ribosome profiling several signaling pathways of translational control, including mTORC1 signaling, are found to be dysregulated in the cerebellum of Bmal1 KO mice. Interestingly, the antidiabetic drug metformin specifically reversed mTORC1 hyperactivation and alleviated major behavioral and PC deficits in Bmal1 KO mice, suggesting mTORC1 hyperactivation may underlie these changes. Conditional Bmal1 deletion only in cerebellar PCs can recapitulate autistic-like behavioral and cellular changes similar to those identified in Bmal1 KO mice, suggesting that Bmal1 disruption in the cerebellar PCs is responsible for changes in the Bmal1 KO mice. Although it is found that Bmal1 disruption in the cerebellar PCs reduces firing rates in these cells and therefore affects the output signals from the cerebellar cortex, it remains unclear how the Bmal1 disruption in the PCs can affect the activities of the cerebello-thalamo-cortical circuit, which is thought to play a significant role in the pathogenesis of ASD [167]. Together, these results demonstrate a link between molecular clock dysfunction and ASD pathogenesis by mTORC1 hyperactivation.
Circadian dysfunction, mTOR, and MD
Introduction of MD
MD refer to conditions that severely affect the mood and its associated functions. According to DSM-5, MD broadly include major depressive disorders (MDD), bipolar disorder (BD) I and II, disruptive mood dysregulation disorder, persistent depressive disorder, cyclothymic disorder, and premenstrual psychotic disorder. MDD refers to a heterogeneous and multifactorial psychiatric illness acting at different levels such as psychological, biological, genetic, and social [168]. MDD is one of the most common mental disorders in the United States with a prevalence of about 7.8% of adults having at least one episode of mood disorders (2019; NIMH). The diagnosis of MDD includes consistent depressed/low mood, constant feeling of anhedonia, lack of concentration, poor sleep patterns, a state of worthlessness, changes in appetite, psychomotor impairments, and even suicidal thoughts. The prevalence of MDD is higher in females (9.6%) than in males (6%) [169, 170]. Also, it is maximum in the age group 18–25 (15.2%) has the highest MDD episodes [170]. BD is a serious type of MD identified by recurrent episodes of depression altering with periods of mania that are usually separated by periods of relatively normal mood and functioning [171].
Although a plethora of studies have been conducted to apprehend the causal mechanisms of MD, a clear outlook on the underlying mechanisms involved has not yet been possible. Nevertheless, MD are thought to be a multicausal disorder, the etiology of which is a combination of genetic, neurobiological, and environmental factors. The management of MD includes varied approaches such as pharmacological treatment options, psychotherapeutic interventional as well as lifestyle modifications. The combination of psychotherapy and medication has proved to be more effective than any of these treatments alone. The FDA-approved drugs for mood disorders treatment include SSRIs, SNRIs, Serotonin modulators, atypical antidepressants, and TCAs, MAOIs. Despite the presence of multiple treatment options for this disorder, a considerable population of affected individuals shows very low to no improvement in the symptoms. A major drawback of the present treatment options available is the failure of these drugs to target the complex pathophysiology of mood disorders without causing major side effects. The clinical use of ketamine (esketamine), a nasal spray for mood disorders is also limited due to its accompanying side effects (dissociative effects, change in sensory perception), and its potential abuse liability. Therefore, identifying novel molecular mechanisms and potential drug targets for the treatment of MD are urgently needed. Recent work has found significant associations between the circadian clock, mTOR signaling and MDs in human and animal studies.
Clock dysfunction in MD
MD are often accompanied by disturbances or perturbations of the regular and daily circadian rhythms [172]. One of the most noticed disruptions occurs in the sleep-wake cycles of mood disorders patients [173,174,175]. For instance, patients with depression find difficulties in the initiation and maintaining their sleep. Further, there is shortening of the REM (rapid eye movement) latency and early awakening in the morning [172]. As the endogenous circadian system controls the daily physiological rhythms, this chronometry programming is often compromised in depressed people. In addition, the depressed patients often present abnormal levels and patterns of melatonin secretion, which in turn has an impact on the circadian controls [176, 177]. Also, agomelatine, a melatonergic agonist, was found to have therapeutic benefits in the patients with mood disorders [178, 179]. Therefore, the altered rhythms could be a possible biomarker for the diagnosis of mood disorders, developing therapeutic options, and further targeting the circadian system to improve mood.
A number of studies have identified disrupted clock gene expression in patients with MD. A study by Li et al. established a direct link between disruptions in the circadian patterns and MDD by developing a time-of-death analysis to 24 h sinusoidal gene expression data from postmortem brains of 34 MDD patients compared to controls [180]. This study indicates that the disruption of circadian gene expression (BMAL1, BHLHE, PER1/2/3, BHLHE41, and NR1D1) is linked to the functional regulation of various neuronal processes as well as behaviors (including mood) [180]. A study by Soria et al. found significant associations in CRY1 (rs2287161), NPAS2 (rs11123857), and VIPR2 (rs885861) genes with patients with mood disorders (MDD and BD) [181]. Similarly, Lavebratt et al. studied the relation of genetic variability in the circadian clock linking genes in the predisposition to MDDs and found the association of the clock gene CRY2 with depression [182]. Also, a study by Kovanen et al. demonstrated the association of CRY2 genetic variants in the Finnish population with mood disorders, thus suggesting CRY2 as the diagnostic marker for MDD [183]. Further, a study by Bruney et al. on the postmortem brain tissue of MDD patients showed dysregulated patterns of clock genes in different brain regions with the most robust changes in anterior cingulate (ACC) [184]. A study by Li et al. reported disruptions in the relative expression of clock genes mRNA (PER1, PER2, CRY1, BMAL1, NPAS2, and GSK3β) in patients with MDD as compared to healthy controls [185]. In addition, a study by Saus et al. indicated that abnormal processing of pre-miR-182 in patients carrying the T allele of the rs76481776 polymorphism may be a contributing factor to the disruptions in circadian rhythms in MDD patients with insomnia [186]. A cross-sectional study by Gouin et al. demonstrated the presence of higher mRNA levels of Clock, Bmal1, Per1 in the patients with a history of depression as compared to the control or the non-depressed patients [187]. A genetic connection between circadian gene variation and major depression was again established by Shi et al., where genetic polymorphisms in circadian genes (especially CLOCK and PER3) were found to have an influence risk on developing depression in a sex- and stress-dependent manner [188]. Numerous studies show the association of genetic polymorphism in the circadian genes with MD populations [189,190,191,192,193]. Pirovano et al. found the association of MD and sleep disturbances with two new SNPs which are known downstream for T3111C polymorphism [194]. Some studies linking the disruption of various clock gene expression to human patients and animal models of MD are summarized in the table below (Table 1).
Seasonal affective disorder (SAD) is a special form of MDD that exhibits a seasonal pattern. In SAD, the recurrent major depressive episodes are associated with shortened day length (photoperiod) and commonly occurs during autumn or winter. SAD is more prevalent in the places where the day length changes according to different seasons are extreme such as high latitudes. The circadian phase shifts and melatonin daily rhythms are considered to be a possible justification of this seasonal form of depression [195]. The demonstration of light during the night increases alertness and thus suppresses the release of melatonin in mammals. As per the circadian phase shift theory hypothesis, late sunrise in the winter seasons leads to a significant delay in the circadian rhythmicity [196]. A study by Partonen et al. reported variations in the circadian genes PER2, ARNTLl, NPAS2 in patients with SAD [197]. Similarly, a study by Kim et al. reported that the polymorphisms of CLOCK, NPAS2 and ARNTL are associated with the seasonal variations in behavior and mood. Further, a few studies show that the presentation of bright light early in the morning was helpful in the treatment of SAD possibly due to phase advancing the circadian system putting it back in sync with the sleep/wake cycle [198, 199]. There are other hypotheses such as serotonin hypothesis, genetic factors, and other comorbid conditions such as alcoholism, delayed sleep phase syndrome that can help to understand the etiology of SAD (Fig. 5).

Exposure to stress or other negative stimuli may cause depression and related disorders by inhibition of the brain mTOR activities. On the contrary, anti-depressing agents or lifestyle may work through augmenting mTOR activities directly or indirectly in the brain.
The strong relationship between disrupted circadian rhythms and MD is demonstrated in mouse models. In a series of elegant studies, the McClung group have established an animal model of BD using the ClockΔ19 mutant mice and investigated the underlying neural mechanisms [200]. Mice carrying the ClockΔ19 mutation display behaviors that are similar to human mania, including rapid mood cycling, hyperactivity, decreased sleep, lowered depression-like behavior, lower anxiety, and an increase in reward seeking [201]. Knockdown of Clock in the ventral tegmental area results in a mixed state of mania and depression-like behavior [202]. Mechanistically, the abnormalities in VTA dopamine neuron firing and reduced level of cholecystokinin, and excitatory signaling via Gria1 expression in the NAc appears to underlie the manic-like phenotypes [203,204,205]. Valproate can be used to treat the manic and mixed phases of bipolar disorder. Using the ClockΔ19 mouse model, Logan et al. found that the therapeutic actions of VPA for bipolar mania is partially via the inhibition of histone deacetylase protein 2 in the VTA [206]. Landgraf et al. demonstrated that the disruption of circadian rhythms by knocking down Bmal1 expression in the SCN caused depression and anxiety-like behaviors in mice [207]. A study by Schnell et al. reported that the Cry1 knockout mice showed depressive-like behaviors [208]. Xing et al. reported that sleep deprivation alters circadian oscillations of clock genes and causes depressive-like behavior in rats [209]. Similarly, a study by Christiansen et al. reported that altered expression of clock genes (Per1, Per2, and Bmal1) is associated with the induction of a depression-like state in the chronic mild stress model in rats [210]. A study by Guo et al. showed that Abelson helper integration site 1 (Ahi1) deficient mice exhibit depressive-like behaviors via changes in the circadian clock pathways [211]. A study by LeGates et al. reported that aberrant light directly impairs mood through intrinsically photosensitive retinal ganglion cells. The antidepressant fluoxetine ameliorates the depressive-like behavior in mice under irregular light cycles by modulating the increased corticosterone levels, suggesting that this mechanism is associated with depression caused by jet lag [212].
mTOR and MD
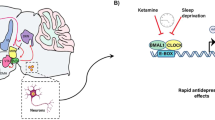
Within the last decade, the glutamatergic system has been implicated in the neurobiology and treatment of depression. Also, the NMDA, N-methyl-D-aspartate receptor antagonists have appeared as the central players in the pathophysiology as well as treatment of depression. Ketamine, an NMDA receptor antagonist have been a successful drug for the different forms of treatment-resistant depression [213]. However, the regular clinical use of ketamine in patients with depression is restricted due to its accompanying harmful effects such as changes in the sensory perception, dissociative properties, and its abuse liability. These limitations have engaged the scientists in exploring the primary targets and thorough mechanism of action underlying the antidepressant effect of ketamine which can result in novel therapeutics for depression. These new treatment options are anticipated to mimic ketamine’s unique antidepressant actions while lacking its undesirable effects. Several reports over the years have linked the mTOR signaling pathway to the antidepressant effect of ketamine. A role of mTOR in the anti-depressive effects of ketamine has been explored in a pioneering study by Li et al., where it was found that ketamine activates the mTOR pathway and causes an increase in the levels of synaptic signaling proteins as well as increased number and functionality of dendritic spines in the prefrontal cortex of rats [214]. Particularly, the administration of ketamine is implicated in increased phosphorylation of mTOR, S6K1, and 4E-BPs. These chemical changes further were correlated with an increase in the levels of synaptic proteins, synapse number in dendritic spines in the PFC. Furthermore, studies have shown the abrogation of the ketamine’s anti-depressive effects on pre-administration with rapamycin which in turn suggests the involvement of mTOR signaling in the mechanism of action of ketamine [213,214,215]. Further, ketamine has also been observed to disinhibit translation of BDNF in a manner dependent on the S6K1 substrate eEF2K [216]. Therefore, mTOR is an emerging signaling pathway of interest in mood disorders pathophysiology and treatment (Fig. 6).

The various hypothesis such as phase shift, melatonin, and serotonin hypothesis, have been proposed for pathogenies of SAD. These factors may convergently influence the brain circadian clock, which may further influence the neural mechanisms underlying SAD and other mood disorders. Like mood disorders, SAD is also characterized by poor mood, anhedonia, loss of energy, weight gain, hypersomnia etc.
Repressed mTOR activities are found in patients with MDs. A study by Jernigan et al. reported a decrease in the expression of mTOR and its downstream components (S6K, eIF4E, and eIF4B) in the postmortem samples of the prefrontal cortex of 12 patients with MDs as compared to healthy controls [217]. A similar outcome regarding mTOR was also observed in the blood samples from BD patients taken during the depressive episodes where expression of mTor, Akt were reduced in comparison to the healthy participants. These peripheral alterations indirectly correspond to that observed in the brain samples [218]. In addition to this, the administration of subanesthetic ketamine in three patients with depression caused an abrupt decrease in the depressive symptoms which may be associated with an acute increase in the plasma mTOR, GSK-3 expression, and phosphorylation of the eEF2 [219]. In a study by Wang et al., bioinformatics analysis was performed to investigate the mechanism of MDs in males and found that downregulated microRNA-124-3p suppresses the mTOR signaling pathway by targeting DNA damage-inducible transcript 4 (DDIT4) in the male population with mood disorders, thus suggesting miRNA as a gender-specific novel target for MDs [220]. A study by Zhu et al. reported genome-wide profiling of DNA methylation and gene expression in monozygotic twin pairs with mood disorders and found distinct modulation enriched in signaling pathways such as insulin receptor signaling, growth factor receptor signaling, and mTOR signaling [221]. A study by Nowak et al. revealed the subanesthetic dose of ketamine shows an antidepressant effect by stimulating the mTOR-associated gene expression [222]. In addition, an open-label study on treatment-resistant depression patients by Roy et al. reported ketamine infusion improved the brain atrophy as well as increased the insulin/mTOR /GSK3 β in the responders [223].
Inhibition of the mTOR pathway can lead to changes in affective behaviors in animals, whereas enhancing mTOR activity can be therapeutic to some models of mood disorders. A recent study by Koehl M et al. found that inhibition of the mTOR signaling pathway via deletion of S6K1 leads to anxiety-like behaviors in mice [224]. Mice and rats exposed to chronic unpredictable stress (CUS) exhibited depressive-like behaviors associated with a reduction in phosphorylation levels of mTOR and its downstream signaling components, such as S6K, in the prefrontal cortex [225], hippocampus [226], and amygdala [227]. An in vitro and in vivo study by Harraz et al. reported that the antidepressant effect of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation [228]. A study by Szewczyk et al. found that the antidepressant action of zinc is via activation of mTOR-dependent signaling pathway [229]. Further, a study by Gordillo-Salas et al. reported that the antidepressant effect of GluN2A receptor antagonist, NVP-AAM077 occurred via increased GluA1 subunit of AMPA and mTOR signaling [230]. Similarly, other studies have explored the antidepressant effects of various drugs and found activation of the mTOR signaling as a common pathway [231, 232]. Overall, mTOR signaling mediates the behavioral response of many antidepressant drugs [57, 214, 216, 233,234,235,236], which also indicates a critical role of the mTOR signaling in depression.
Little is known, however, regarding the role of mTOR as a potential link between circadian disruption and the pathogenesis of mood disorders. It is well established that photoperiod regulates affective behaviors in diurnal and nocturnal animals [237]. Evans et al. found that the day length profoundly regulates the rhythms of clock gene expression and neuronal network properties in the SCN and presumably also in other brain regions that exhibit circadian rhythms of gene expression [238, 239]. Interestingly, circadian mTOR activities in the SCN are also regulated by photoperiod [16, 240]. As the SCN is the master circadian pacemaker that regulates rhythms of gene expression and neuronal activities in other brain regions, it is expected that photoperiod will also regulate mTOR rhythms in other brain regions that are important to regulate affective behaviors such as the prefrontal cortex (PFC). Along these lines, genetic repression of mTORC1/S6K1 activities in PFC can lead to depressive-like behaviors whereas increase expression of S6K1 can produce antidepressant effects in rats [241]. Together, these results suggest a model in which photoperiod may regulate animal affective behaviors by regulating rhythmic mTOR activities in specific brain regions.
Conclusions
Circadian rhythm is ubiquitous, and rhythmic gene expression is found in a variety of brain regions. Thus, numerous neurophysiological processes exhibit significant daily oscillations in activities and functions. Intact circadian rhythm is critical for mental health, as disruption of daily rhythms by either environmental or genetic factors can lead to or exacerbate neurological and psychiatric problems in laboratory and clinical studies. Indeed, disruption of clock gene expression and circadian rhythm is frequently found in common psychiatric diseases such as ASD and MD. mTOR signaling is a fundamentally important signal transduction pathway, the disruption of which has been implicated in ASD and MD. As mTOR activities are closely regulated by the circadian clock and functionally integrated with the circadian timing process, circadian dysfunction inevitably leads to deregulation of temporal mTOR activities in the brain, which in turn may contribute to pathophysiological changes associated with psychiatric disorders. Although the underlying mechanisms remain to be fully understood, the interactions between the circadian clock, mTOR signaling, and psychiatric disorders appear to be significant in ASD and MD and should be considered in basic research and pharmaceutical developments. As mTOR inhibitors are FDA approved drugs, understanding the interactions between the circadian clock, mTOR, and psychiatric disorders may open new therapeutic avenues to regulate the brain clock function and treat psychiatric diseases.
References
Rosbash M. The implications of multiple circadian clock origins. PLoS Biol. 2009;7:e62.
Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–75.
Herzog ED, Hermanstyne T, Smyllie NJ, Hastings MH. Regulating the suprachiasmatic nucleus (SCN) circadian clockwork: interplay between cell-autonomous and circuit-level mechanisms. Cold Spring Harb Perspect Biol. 2017;9:a027706. https://doi.org/10.1101/cshperspect.a027706.
Malik A, Kondratov RV, Jamasbi RJ, Geusz ME. Circadian clock genes are essential for normal adult neurogenesis, differentiation, and fate determination. PLoS ONE. 2015;10:e0139655.
Bouchard-Cannon P, Mendoza-Viveros L, Yuen A, Kaern M, Cheng HY. The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep. 2013;5:961–73.
Fang K, Liu D, Pathak SS, Yang B, Li J, Karthikeyan R, et al. Disruption of circadian rhythms by ambient light during neurodevelopment leads to autistic-like molecular and behavioral alterations in adult mice. Cells 2021;10:3314. https://doi.org/10.3390/cells10123314.
Paul JR, Davis JA, Goode LK, Becker BK, Fusilier A, Meador-Woodruff A, et al. Circadian regulation of membrane physiology in neural oscillators throughout the brain. Eur J Neurosci. 2020;51:109–38.
Chaudhury D, Colwell CS. Circadian modulation of learning and memory in fear-conditioned mice. Behav Brain Res. 2002;133:95–108.
Lee Y, Field JM, Sehgal A. Circadian rhythms, disease and chronotherapy. J Biol Rhythms. 2021;36:503–31.
Logan RW, McClung CA. Rhythms of life: circadian disruption and brain disorders across the lifespan. Nat Rev Neurosci. 2019;20:49–65.
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell 2006;124:471–84.
Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203.
Lipton JO, Sahin M. The neurology of mTOR. Neuron 2014;84:275–91.
Cao R. mTOR signaling, translational control, and the circadian clock. Front Genet. 2018;9:367.
Cao R, Anderson FE, Jung YJ, Dziema H, Obrietan K. Circadian regulation of mammalian target of rapamycin signaling in the mouse suprachiasmatic nucleus. Neuroscience 2011;181:79–88.
Jouffe C, Cretenet G, Symul L, Martin E, Atger F, Naef F, et al. The circadian clock coordinates ribosome biogenesis. PLoS Biol. 2013;11:e1001455.
Cao R, Robinson B, Xu H, Gkogkas C, Khoutorsky A, Alain T, et al. Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron 2013;79:712–24.
Liu D, Stowie A, de Zavalia N, Leise T, Pathak SS, Drewes LR, et al. mTOR signaling in VIP neurons regulates circadian clock synchrony and olfaction. Proc Natl Acad Sci USA. 2018;115:E3296–304.
Ramanathan C, Kathale ND, Liu D, Lee C, Freeman DA, Hogenesch JB, et al. mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 2018;14:e1007369.
Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature 2002;418:935–41.
Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci USA. 1972;69:1583–6.
Moore RY, Eichler VB. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972;42:201–6.
Moore RY. Retinohypothalamic projection in mammals: a comparative study. Brain Res. 1973;49:403–9.
Hattar S, Liao HW, Takao M, Berson DM, Yau KW. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science 2002;295:1065–70.
Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science 2002;295:1070–3.
Panda S, Sato TK, Castrucci AM, Rollag MD, DeGrip WJ, Hogenesch JB, et al. Melanopsin (Opn4) requirement for normal light-induced circadian phase shifting. Science 2002;298:2213–6.
Bellingham J, Foster RG. Opsins and mammalian photoentrainment. Cell Tissue Res. 2002;309:57–71.
Jones JR, Chaturvedi S, Granados-Fuentes D, Herzog ED. Circadian neurons in the paraventricular nucleus entrain and sustain daily rhythms in glucocorticoids. Nat Commun. 2021;12:5763.
Gizowski C, Zaelzer C, Bourque CW. Clock-driven vasopressin neurotransmission mediates anticipatory thirst prior to sleep. Nature 2016;537:685–8.
Yao Y, Taub AB, LeSauter J, Silver R. Identification of the suprachiasmatic nucleus venous portal system in the mammalian brain. Nat Commun. 2021;12:5643.
Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron 1995;14:697–706.
Herzog ED, Takahashi JS, Block GD. Clock controls circadian period in isolated suprachiasmatic nucleus neurons. Nat Neurosci. 1998;1:708–13.
Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, et al. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell 2007;129:605–16.
Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, Ueda M, et al. Resetting central and peripheral circadian oscillators in transgenic rats. Science 2000;288:682–5.
Yamaguchi S, Isejima H, Matsuo T, Okura R, Yagita K, Kobayashi M, et al. Synchronization of cellular clocks in the suprachiasmatic nucleus. Science 2003;302:1408–12.
Harmar AJ, Marston HM, Shen S, Spratt C, West KM, Sheward WJ, et al. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell 2002;109:497–508.
Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, et al. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R939–49.
Maywood ES, Reddy AB, Wong GK, O’Neill JS, O’Brien JA, McMahon DG, et al. Synchronization and maintenance of timekeeping in suprachiasmatic circadian clock cells by neuropeptidergic signaling. Curr Biol. 2006;16:599–605.
Aton SJ, Colwell CS, Harmar AJ, Waschek J, Herzog ED. Vasoactive intestinal polypeptide mediates circadian rhythmicity and synchrony in mammalian clock neurons. Nat Neurosci. 2005;8:476–83.
Shen S, Spratt C, Sheward WJ, Kallo I, West K, Morrison CF, et al. Overexpression of the human VPAC2 receptor in the suprachiasmatic nucleus alters the circadian phenotype of mice. Proc Natl Acad Sci USA. 2000;97:11575–80.
Liu C, Reppert SM. GABA synchronizes clock cells within the suprachiasmatic circadian clock. Neuron 2000;25:123–8.
Freeman GM Jr., Krock RM, Aton SJ, Thaben P, Herzog ED. GABA networks destabilize genetic oscillations in the circadian pacemaker. Neuron 2013;78:799–806.
Yamaguchi Y, Suzuki T, Mizoro Y, Kori H, Okada K, Chen Y, et al. Mice genetically deficient in vasopressin V1a and V1b receptors are resistant to jet lag. Science 2013;342:85–90.
Schaap J, Pennartz CM, Meijer JH. Electrophysiology of the circadian pacemaker in mammals. Chronobiol Int. 2003;20:171–88.
Butcher GQ, Lee B, Obrietan K. Temporal regulation of light-induced extracellular signal-regulated kinase activation in the suprachiasmatic nucleus. J Neurophysiol. 2003;90:3854–63.
Cao R, Li A, Cho HY, Lee B, Obrietan K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J Neurosci. 2010;30:6302–14.
Dziema H, Oatis B, Butcher GQ, Yates R, Hoyt KR, Obrietan K. The ERK/MAP kinase pathway couples light to immediate-early gene expression in the suprachiasmatic nucleus. Eur J Neurosci. 2003;17:1617–27.
Brancaccio M, Edwards MD, Patton AP, Smyllie NJ, Chesham JE, Maywood ES, et al. Cell-autonomous clock of astrocytes drives circadian behavior in mammals. Science 2019;363:187–92.
Harbour VL, Weigl Y, Robinson B, Amir S. Comprehensive mapping of regional expression of the clock protein PERIOD2 in rat forebrain across the 24-h day. PLoS ONE. 2013;8:e76391.
Harbour VL, Weigl Y, Robinson B, Amir S. Phase differences in expression of circadian clock genes in the central nucleus of the amygdala, dentate gyrus, and suprachiasmatic nucleus in the rat. PLoS ONE. 2014;9:e103309.
Rosbash M, Bradley S, Kadener S, Li Y, Luo W, Menet JS, et al. Transcriptional feedback and definition of the circadian pacemaker in Drosophila and animals. Cold Spring Harb Symp Quant Biol. 2007;72:75–83.
Darlington TK, Wager-Smith K, Ceriani MF, Staknis D, Gekakis N, Steeves TD, et al. Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science 1998;280:1599–603.
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998;280:1564–9.
Sangoram AM, Saez L, Antoch MP, Gekakis N, Staknis D, Whiteley A, et al. Mammalian circadian autoregulatory loop: a timeless ortholog and mPer1 interact and negatively regulate CLOCK-BMAL1-induced transcription. Neuron 1998;21:1101–13.
Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002;110:251–60.
Sato S, Bunney B, Mendoza-Viveros L, Bunney W, Borrelli E, Sassone-Corsi P, et al. Rapid-acting antidepressants and the circadian clock. Neuropsychopharmacol. 2022;47:805–816.
Guillaumond F, Dardente H, Giguere V, Cermakian N. Differential control of Bmal1 circadian transcription by REV-ERB and ROR nuclear receptors. J Biol Rhythms. 2005;20:391–403.
Jin X, Shearman LP, Weaver DR, Zylka MJ, de Vries GJ, Reppert SM. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell 1999;96:57–68.
Cao R, Gkogkas CG, de Zavalia N, Blum ID, Yanagiya A, Tsukumo Y, et al. Light-regulated translational control of circadian behavior by eIF4E phosphorylation. Nat Neurosci 2015;18:855–62.
Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009;326:437–40.
Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001;107:855–67.
Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 2013;152:1091–105.
Hirano A, Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, et al. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell 2013;152:1106–18.
Gillette MU, Mitchell JW. Signaling in the suprachiasmatic nucleus: selectively responsive and integrative. Cell Tissue Res. 2002;309:99–107.
Lane N, Martin W. The energetics of genome complexity. Nature 2010;467:929–34.
Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, et al. Hypothalamic mTOR signaling regulates food intake. Science 2006;312:927–30.
Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, et al. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron 2008;60:832–45.
Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, et al. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci 2013;16:441–8.
Vecsey CG, Peixoto L, Choi JH, Wimmer M, Jaganath D, Hernandez PJ, et al. Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol Genomics. 2012;44:981–91.
Tudor JC, Davis EJ, Peixoto L, Wimmer ME, van Tilborg E, Park AJ, et al. Sleep deprivation impairs memory by attenuating mTORC1-dependent protein synthesis. Sci Signal. 2016;9:ra41.
Areal CC, Cao R, Sonenberg N, Mongrain V. Wakefulness/sleep architecture and electroencephalographic activity in mice lacking the translational repressor 4E-BP1 or 4E-BP2. Sleep 2020;43:zsz210. https://doi.org/10.1093/sleep/zsz210.
Cao R, Lee B, Cho HY, Saklayen S, Obrietan K. Photic regulation of the mTOR signaling pathway in the suprachiasmatic circadian clock. Mol Cell Neurosci. 2008;38:312–24.
Zheng X, Sehgal AAKT. and TOR signaling set the pace of the circadian pacemaker. Curr Biol. 2010;20:1203–8.
Ratnayake L, Adhvaryu KK, Kafes E, Motavaze K, Lakin-Thomas P. A component of the TOR (Target Of Rapamycin) nutrient-sensing pathway plays a role in circadian rhythmicity in Neurospora crassa. PLoS Genet. 2018;14:e1007457.
Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, et al. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 2015;161:1138–51.
Lipton JO, Boyle LM, Yuan ED, Hochstrasser KJ, Chifamba FF, Nathan A, et al. Aberrant proteostasis of BMAL1 underlies circadian abnormalities in a paradigmatic mTOR-opathy. Cell Rep. 2017;20:868–80.
Wu R, Dang F, Li P, Wang P, Xu Q, Liu Z, et al. The circadian protein Period2 suppresses mTORC1 activity via recruiting Tsc1 to mTORC1 complex. Cell Metab. 2019;29:653–67.e656.
McNally Keehn R, Ciccarelli M, Szczepaniak D, Tomlin A, Lock T, Swigonski N. A statewide tiered system for screening and diagnosis of autism spectrum disorder. Pediatrics 2020;146.
Arnett AB, Cairney BE, Wallace AS, Gerdts J, Turner TN, Eichler EE, et al. Comorbid symptoms of inattention, autism, and executive cognition in youth with putative genetic risk. J Child Psychol Psychiatry. 2018;59:268–76.
Hunter JE, McLay LK, France KG, Blampied NM. Sleep and stereotypy in children with autism: effectiveness of function-based behavioral treatment. Sleep Med. 2021;80:301–4.
Simonoff E, Pickles A, Charman T, Chandler S, Loucas T, Baird G. Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J Am Acad Child Adolesc Psychiatry. 2008;47:921–9.
Maenner MJ, Shaw KA, Bakian AV, Bilder DA, Durkin MS, Esler A, et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill Summ. 2021;70:1–16.
Tan C, Frewer V, Cox G, Williams K, Ure A. Prevalence and age of onset of regression in children with autism spectrum disorder: a systematic review and meta-analytical update. Autism Res. 2021;14:582–98.
Hutsler JJ, Zhang H. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 2010;1309:83–94.
Irwin SA, Patel B, Idupulapati M, Harris JB, Crisostomo RA, Larsen BP, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am J Med Genet. 2001;98:161–7.
Horder J, Petrinovic MM, Mendez MA, Bruns A, Takumi T, Spooren W, et al. Glutamate and GABA in autism spectrum disorder-a translational magnetic resonance spectroscopy study in man and rodent models. Transl Psychiatry. 2018;8:106.
Papariello A, Taylor D, Soderstrom K, Litwa K. CB(1) antagonism increases excitatory synaptogenesis in a cortical spheroid model of fetal brain development. Sci Rep. 2021;11:9356.
DiCarlo GE, Aguilar JI, Matthies HJ, Harrison FE, Bundschuh KE, West A, et al. Autism-linked dopamine transporter mutation alters striatal dopamine neurotransmission and dopamine-dependent behaviors. J Clin Invest. 2019;129:3407–19.
Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–93.
Ghanizadeh A, Sahraeizadeh A, Berk M. A head-to-head comparison of aripiprazole and risperidone for safety and treating autistic disorders, a randomized double blind clinical trial. Child Psychiatry Hum Dev. 2014;45:185–92.
Blankenship K, Erickson CA, Stigler KA, Posey DJ, McDougle CJ. Aripiprazole for irritability associated with autistic disorder in children and adolescents aged 6-17 years. Ped Health. 2010;4:375–81.
Roke Y, Buitelaar JK, Boot AM, Tenback D, van Harten PN. Risk of hyperprolactinemia and sexual side effects in males 10-20 years old diagnosed with autism spectrum disorders or disruptive behavior disorder and treated with risperidone. J Child Adolesc Psychopharmacol. 2012;22:432–9.
Houghton R, van den Bergh J, Law K, Liu Y, de Vries F. Risperidone versus aripiprazole fracture risk in children and adolescents with autism spectrum disorders. Autism Res. 2021;14:1800–14.
Yoon Y, Wink LK, Pedapati EV, Horn PS, Erickson CA. Weight gain effects of second-generation antipsychotic treatment in autism spectrum disorder. J Child Adolesc Psychopharmacol. 2016;26:822–7.
Lorsung E, Karthikeyan R, Cao R. Biological timing and neurodevelopmental disorders: a role for circadian dysfunction in autism spectrum disorders. Front Neurosci. 2021;15:642745.
Maruani A, Dumas G, Beggiato A, Traut N, Peyre H, Cohen-Freoua A, et al. Morning plasma melatonin differences in autism: beyond the impact of pineal gland volume. Front Psychiatry. 2019;10:11.
Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–88.
Ballester P, Richdale AL, Baker EK, Peiró AM. Sleep in autism: a biomolecular approach to aetiology and treatment. Sleep Med Rev. 2020;54:101357.
Yavuz-Kodat E, Reynaud E, Geoffray MM, Limousin N, Franco P, Bonnet-Brilhault F, et al. Disturbances of continuous sleep and circadian rhythms account for behavioral difficulties in children with autism spectrum disorder. J Clin Med. 2020;9.
Carmassi C, Palagini L, Caruso D, Masci I, Nobili L, Vita A, et al. Systematic review of sleep disturbances and circadian sleep desynchronization in autism spectrum disorder: toward an integrative model of a self-reinforcing loop. Front Psychiatry. 2019;10:366.
Missig G, McDougle CJ, Carlezon WA Jr. Sleep as a translationally-relevant endpoint in studies of autism spectrum disorder (ASD). Neuropsychopharmacology 2020;45:90–103.
Braam W, Ehrhart F, Maas A, Smits MG, Curfs L. Low maternal melatonin level increases autism spectrum disorder risk in children. Res Dev Disabil. 2018;82:79–89.
Melke J, Goubran Botros H, Chaste P, Betancur C, Nygren G, Anckarsäter H, et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry. 2008;13:90–8.
Hayashi M, Mishima K, Fukumizu M, Takahashi H, Ishikawa Y, Hamada I, et al. Melatonin treatment and adequate sleep hygiene interventions in children with autism spectrum disorder: a randomized controlled trial. J Autism Dev Disord. 2022;52:2784–2793.
Gringras P, Nir T, Breddy J, Frydman-Marom A, Findling RL. Efficacy and safety of pediatric prolonged-release melatonin for insomnia in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2017;56:948–57.e944.
Maras A, Schroder CM, Malow BA, Findling RL, Breddy J, Nir T, et al. Long-term efficacy and safety of pediatric prolonged-release melatonin for insomnia in children with autism spectrum disorder. J Child Adolesc Psychopharmacol. 2018;28:699–710.
Ballester P, Martínez MJ, Inda MD, Javaloyes A, Richdale AL, Muriel J, et al. Evaluation of agomelatine for the treatment of sleep problems in adults with autism spectrum disorder and co-morbid intellectual disability. J Psychopharmacol. 2019;33:1395–406.
Yang Z, Matsumoto A, Nakayama K, Jimbo EF, Kojima K, Nagata K, et al. Circadian-relevant genes are highly polymorphic in autism spectrum disorder patients. Brain Dev. 2016;38:91–9.
Nicholas B, Rudrasingham V, Nash S, Kirov G, Owen MJ, Wimpory DC. Association of Per1 and Npas2 with autistic disorder: support for the clock genes/social timing hypothesis. Mol Psychiatry. 2007;12:581–92.
Hu VW, Sarachana T, Kim KS, Nguyen A, Kulkarni S, Steinberg ME, et al. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2009;2:78–97.
Goto M, Mizuno M, Matsumoto A, Yang Z, Jimbo EF, Tabata H, et al. Role of a circadian-relevant gene NR1D1 in brain development: possible involvement in the pathophysiology of autism spectrum disorders. Sci Rep. 2017;7:43945.
Sayad A, Noroozi R, Omrani MD, Taheri M, Ghafouri-Fard S. Retinoic acid-related orphan receptor alpha (RORA) variants are associated with autism spectrum disorder. Metab Brain Dis. 2017;32:1595–601.
Nguyen A, Rauch TA, Pfeifer GP, Hu VW. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. Faseb J. 2010;24:3036–51.
Rudolf G, Lesca G, Mehrjouy MM, Labalme A, Salmi M, Bache I, et al. Loss of function of the retinoid-related nuclear receptor (RORB) gene and epilepsy. Eur J Hum Genet. 2016;24:1761–70.
Boudry-Labis E, Demeer B, Le Caignec C, Isidor B, Mathieu-Dramard M, Plessis G, et al. A novel microdeletion syndrome at 9q21.13 characterised by mental retardation, speech delay, epilepsy and characteristic facial features. Eur J Med Genet. 2013;56:163–70.
Hoang N, Yuen RKC, Howe J, Drmic I, Ambrozewicz P, Russell C, et al. Sleep phenotype of individuals with autism spectrum disorder bearing mutations in the PER2 circadian rhythm gene. Am J Med Genet A. 2021;185:1120–30.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014;515:216–21.
Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20:602–11.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014;515:209–15.
Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, et al. Integrative analyses of de novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep. 2018;22:734–47.
Delorme TC, Srivastava LK, Cermakian N. Altered circadian rhythms in a mouse model of neurodevelopmental disorders based on prenatal maternal immune activation. Brain Behav Immun. 2021;93:119–31.
Sarowar T, Chhabra R, Vilella A, Boeckers TM, Zoli M, Grabrucker AM. Activity and circadian rhythm influence synaptic Shank3 protein levels in mice. J Neurochem. 2016;138:887–95.
Alamilla J, Ramiro-Cortés Y, Mejía-López A, Chavez JL, Rivera DO, Felipe V, et al. Altered light sensitivity of circadian clock in shank3(+/-) mouse. Front Neurosci. 2021;15:604165.
Ingiosi AM, Schoch H, Wintler T, Singletary KG, Righelli D, Roser LG, et al. Shank3 modulates sleep and expression of circadian transcription factors. Elife 2019;8:e42819. https://doi.org/10.7554/eLife.42819.
Ma Z, Eaton M, Liu Y, Zhang J, Chen X, Tu X, et al. Deficiency of autism-related Scn2a gene in mice disrupts sleep patterns and circadian rhythms. Neurobiol Dis. 2022;168:105690.
Vijaya Shankara J, Horsley KG, Cheng N, Rho JM, Antle MC. Circadian responses to light in the BTBR Mouse. J Biol Rhythms. 2022: 7487304221102279.
Ferraro S, de Zavalia N, Belforte N, Amir S. In utero exposure to valproic-acid alters circadian organisation and clock-gene expression: implications for autism spectrum disorders. Front Behav Neurosci. 2021;15:711549.
Bourgeron T. The possible interplay of synaptic and clock genes in autism spectrum disorders. Cold Spring Harb Symp Quant Biol. 2007;72:645–54.
Wimpory D, Nicholas B, Nash S. Social timing, clock genes and autism: a new hypothesis. J Intellect Disabil Res. 2002;46:352–8.
Reh RK, Dias BG, Nelson CA 3rd, Kaufer D, Werker JF, Kolb B, et al. Critical period regulation across multiple timescales. Proc Natl Acad Sci USA. 2020;117:23242–51.
Fagiolini M, Fritschy JM, Löw K, Möhler H, Rudolph U, Hensch TK. Specific GABAA circuits for visual cortical plasticity. Science 2004;303:1681–3.
Kobayashi Y, Ye Z, Hensch TK. Clock genes control cortical critical period timing. Neuron 2015;86:264–75.
Zhang L, Huang CC, Dai Y, Luo Q, Ji Y, Wang K, et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl Psychiatry. 2020;10:9.
Luhach K, Kulkarni GT, Singh VP, Sharma B. Vinpocetine amended prenatal valproic acid induced features of ASD possibly by altering markers of neuronal function, inflammation, and oxidative stress. Autism Res. 2021;14:2270–86.
Cusmano DM, Mong JA. In utero exposure to valproic acid changes sleep in juvenile rats: a model for sleep disturbances in autism. Sleep 2014;37:1489–99.
Brancaccio M, Patton AP, Chesham JE, Maywood ES, Hastings MH. Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron 2017;93:1420–35.e1425.
Winden KD, Ebrahimi-Fakhari D, Sahin M. Abnormal mTOR activation in autism. Annu Rev Neurosci. 2018;41:1–23.
Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014;83:1131–43.
Kelleher RJ 3rd, Bear MF. The autistic neuron: troubled translation? Cell 2008;135:401–6.
Neves-Pereira M, Muller B, Massie D, Williams JH, O’Brien PC, Hughes A, et al. Deregulation of EIF4E: a novel mechanism for autism. J Med Genet. 2009;46:759–65.
Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013;493:371–7.
Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013;493:411–5.
Nicolini C, Ahn Y, Michalski B, Rho JM, Fahnestock M. Decreased mTOR signaling pathway in human idiopathic autism and in rats exposed to valproic acid. Acta Neuropathol Commun. 2015;3:3.
Rosina E, Battan B, Siracusano M, Di Criscio L, Hollis F, Pacini L, et al. Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl Psychiatry. 2019;9:50.
Martin P, Wagh V, Reis SA, Erdin S, Beauchamp RL, Shaikh G, et al. TSC patient-derived isogenic neural progenitor cells reveal altered early neurodevelopmental phenotypes and rapamycin-induced MNK-eIF4E signaling. Mol Autism. 2020;11:2.
Petrasek T, Vojtechova I, Klovrza O, Tuckova K, Vejmola C, Rak J, et al. mTOR inhibitor improves autistic-like behaviors related to Tsc2 haploinsufficiency but not following developmental status epilepticus. J Neurodev Disord. 2021;13:14.
Kotajima-Murakami H, Kobayashi T, Kashii H, Sato A, Hagino Y, Tanaka M, et al. Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero. Mol Brain. 2019;12:3.
Nobukini T, Thomas G. The mTOR/S6K signalling pathway: the role of the TSC1/2 tumour suppressor complex and the proto-oncogene Rheb. Novartis Found Symp. 2004;262:148–54 .
Alsaqati M, Heine VM, Harwood AJ. Pharmacological intervention to restore connectivity deficits of neuronal networks derived from ASD patient iPSC with a TSC2 mutation. Mol Autism. 2020;11:80.
Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11:332–41.
Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005;25:5087–95.
Chen CJ, Sgritta M, Mays J, Zhou H, Lucero R, Park J, et al. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat Med. 2019;25:1684–90.
DeSpenza T Jr., Carlson M, Panchagnula S, Robert S, Duy PQ, Mermin-Bunnell N, et al. PTEN mutations in autism spectrum disorder and congenital hydrocephalus: developmental pleiotropy and therapeutic targets. Trends Neurosci. 2021;44:961–76.
Garg S, Plasschaert E, Descheemaeker MJ, Huson S, Borghgraef M, Vogels A, et al. Autism spectrum disorder profile in neurofibromatosis type I. J Autism Dev Disord. 2015;45:1649–57.
Walsh KS, Vélez JI, Kardel PG, Imas DM, Muenke M, Packer RJ, et al. Symptomatology of autism spectrum disorder in a population with neurofibromatosis type 1. Dev Med Child Neurol. 2013;55:131–8.
Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012;488:647–51.
Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 2012;76:325–37.
Yan J, Porch MW, Court-Vazquez B, Bennett MVL, Zukin RS. Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc Natl Acad Sci USA. 2018;115:E9707–16.
Gantois I, Khoutorsky A, Popic J, Aguilar-Valles A, Freemantle E, Cao R, et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat Med. 2017;23:674–7.
Mighell TL, Evans-Dutson S, O’Roak BJ. A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am J Hum Genet. 2018;102:943–55.
Lugo JN, Smith GD, Arbuckle EP, White J, Holley AJ, Floruta CM, et al. Deletion of PTEN produces autism-like behavioral deficits and alterations in synaptic proteins. Front Mol Neurosci. 2014;7:27.
Burket JA, Benson AD, Tang AH, Deutsch SI. Rapamycin improves sociability in the BTBR T(+)Itpr3(tf)/J mouse model of autism spectrum disorders. Brain Res Bull. 2014;100:70–5.
Sawicka K, Hale CR, Park CY, Fak JJ, Gresack JE, Van Driesche, SJ et al. FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. Elife 2019;8:e46919. https://doi.org/10.7554/eLife.46919.
Liu D, Nanclares C, Simbriger K, Fang K, Lorsung E, Le N, et al. Autistic-like behavior and cerebellar dysfunction in Bmal1 mutant mice ameliorated by mTORC1 inhibition. Mol Psychiatry. 2022. https://doi.org/10.1038/s41380-022-01499-6.
Singla R, Mishra A, Lin H, Lorsung E, Le N, Tin S, et al. Haploinsufficiency of a circadian clock gene Bmal1 (Arntl or Mop3) causes brain-wide mTOR hyperactivation and autism-like behavioral phenotypes in mice. Int J Mol Sci. 2022;23:6317. https://doi.org/10.3390/ijms23116317.
D’Mello AM, Stoodley CJ. Cerebro-cerebellar circuits in autism spectrum disorder. Front Neurosci. 2015;9:408.
Kim YK. Molecular neurobiology of major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:275–6.
Zhdanava M, Pilon D, Ghelerter I, Chow W, Joshi K, Lefebvre P, et al. The prevalence and national burden of treatment-resistant depression and major depressive disorder in the United States. J Clin Psychiatry. 2021;82:20m13699. https://doi.org/10.4088/JCP.20m13699.
Lu J, Xu X, Huang Y, Li T, Ma C, Xu G, et al. Prevalence of depressive disorders and treatment in China: a cross-sectional epidemiological study. Lancet Psychiatry. 2021;8:981–90.
Smith DJ, Whitham EA, Ghaemi SN. Bipolar disorder. Handb Clin Neurol. 2012;106:251–63.
Crouse JJ, Carpenter JS, Song YJC, Hockey SJ, Naismith SL, Grunstein RR, et al. Circadian rhythm sleep-wake disturbances and depression in young people: implications for prevention and early intervention. Lancet Psychiatry. 2021;8:813–23.
Souêtre E, Salvati E, Belugou JL, Pringuey D, Candito M, Krebs B, et al. Circadian rhythms in depression and recovery: evidence for blunted amplitude as the main chronobiological abnormality. Psychiatry Res. 1989;28:263–78.
Raoux N, Benoit O, Dantchev N, Denise P, Franc B, Allilaire JF, et al. Circadian pattern of motor activity in major depressed patients undergoing antidepressant therapy: relationship between actigraphic measures and clinical course. Psychiatry Res. 1994;52:85–98.
Mendoza J. Circadian insights into the biology of depression: Symptoms, treatments and animal models. Behav Brain Res. 2019;376:112186.
Caumo W, Hidalgo MP, Souza A, Torres ILS, Antunes LC. Melatonin is a biomarker of circadian dysregulation and is correlated with major depression and fibromyalgia symptom severity. J Pain Res. 2019;12:545–56.
Wang XQ, Wang DQ, Bao YP, Liu JJ, Chen J, Wu SW, et al. Preliminary study on changes of sleep EEG power and plasma melatonin in male patients with major depressive disorder after 8 weeks treatment. Front Psychiatry. 2021;12:736318.
Arioz BI, Tastan B, Tarakcioglu E, Tufekci KU, Olcum M, Ersoy N, et al. Melatonin attenuates LPS-induced acute depressive-like behaviors and microglial NLRP3 inflammasome activation through the SIRT1/Nrf2 pathway. Front Immunol. 2019;10:1511.
Gahr M. Agomelatine in the treatment of major depressive disorder: an assessment of benefits and risks. Curr Neuropharmacol. 2014;12:287–398.
Li JZ, Bunney BG, Meng F, Hagenauer MH, Walsh DM, Vawter MP, et al. Circadian patterns of gene expression in the human brain and disruption in major depressive disorder. Proc Natl Acad Sci USA. 2013;110:9950–5.
Soria V, Martínez-Amorós E, Escaramís G, Valero J, Pérez-Egea R, García C, et al. Differential association of circadian genes with mood disorders: CRY1 and NPAS2 are associated with unipolar major depression and CLOCK and VIP with bipolar disorder. Neuropsychopharmacology 2010;35:1279–89.
Lavebratt C, Sjöholm LK, Soronen P, Paunio T, Vawter MP, Bunney WE, et al. CRY2 is associated with depression. PLoS ONE 2010;5:e9407.
Kovanen L, Donner K, Kaunisto M, Partonen T. PRKCDBP (CAVIN3) and CRY2 associate with major depressive disorder. J Affect Disord. 2017;207:136–40.
Bunney BG, Li JZ, Walsh DM, Stein R, Vawter MP, Cartagena P, et al. Circadian dysregulation of clock genes: clues to rapid treatments in major depressive disorder. Mol Psychiatry. 2015;20:48–55.
Li SX, Liu LJ, Xu LZ, Gao L, Wang XF, Zhang JT, et al. Diurnal alterations in circadian genes and peptides in major depressive disorder before and after escitalopram treatment. Psychoneuroendocrinology 2013;38:2789–99.
Saus E, Soria V, Escaramís G, Vivarelli F, Crespo JM, Kagerbauer B, et al. Genetic variants and abnormal processing of pre-miR-182, a circadian clock modulator, in major depression patients with late insomnia. Hum Mol Genet. 2010;19:4017–25.
Gouin JP, Connors J, Kiecolt-Glaser JK, Glaser R, Malarkey WB, Atkinson C, et al. Altered expression of circadian rhythm genes among individuals with a history of depression. J Affect Disord. 2010;126:161–6.
Shi SQ, White MJ, Borsetti HM, Pendergast JS, Hida A, Ciarleglio CM, et al. Molecular analyses of circadian gene variants reveal sex-dependent links between depression and clocks. Transl Psychiatry. 2016;6:e748.
Ma HY, Liu ZF, Xu YF, Hu XD, Sun N, Li XR, et al. The association study of CLOCK gene polymorphisms with antidepressant effect in Chinese with major depressive disorder. Per Med. 2019;16:115–22.
Hua P, Liu W, Chen D, Zhao Y, Chen L, Zhang N, et al. Cry1 and Tef gene polymorphisms are associated with major depressive disorder in the Chinese population. J Affect Disord. 2014;157:100–3.
Kishi T, Yoshimura R, Kitajima T, Okochi T, Okumura T, Tsunoka T, et al. SIRT1 gene is associated with major depressive disorder in the Japanese population. J Affect Disord. 2010;126:167–73.
Li Y, Cao Z, Wu S, Wang C, Dong Y, Zhao NO, et al. Association between the CLOCK gene polymorphism and depressive symptom mediated by sleep quality among non-clinical Chinese Han population. J Affect Disord. 2022;298:217–23.
Kishi T, Kitajima T, Ikeda M, Yamanouchi Y, Kinoshita Y, Kawashima K, et al. Association study of clock gene (CLOCK) and schizophrenia and mood disorders in the Japanese population. Eur Arch Psychiatry Clin Neurosci. 2009;259:293–7.
Pirovano A, Lorenzi C, Serretti A, Ploia C, Landoni S, Catalano M, et al. Two new rare variants in the circadian “clock” gene may influence sleep pattern. Genet Med. 2005;7:455–7.
Terman JS, Terman M, Lo ES, Cooper TB. Circadian time of morning light administration and therapeutic response in winter depression. Arch Gen Psychiatry. 2001;58:69–75.
Menculini G, Verdolini N, Murru A, Pacchiarotti I, Volpe U, Cervino A, et al. Depressive mood and circadian rhythms disturbances as outcomes of seasonal affective disorder treatment: a systematic review. J Affect Disord. 2018;241:608–26.
Partonen T, Treutlein J, Alpman A, Frank J, Johansson C, Depner M, et al. Three circadian clock genes Per2, Arntl, and Npas2 contribute to winter depression. Ann Med. 2007;39:229–38.
Albrecht U. Circadian clocks and mood-related behaviors. Handb Exp Pharmacol. 2013;217:227–39.
Terman M, Terman JS. Light therapy for seasonal and nonseasonal depression: efficacy, protocol, safety, and side effects. CNS Spectr. 2005;10:647–63.
Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA. 2007;104:6406–11.
Sidor MM, Spencer SM, Dzirasa K, Parekh PK, Tye KM, Warden MR, et al. Daytime spikes in dopaminergic activity drive rapid mood-cycling in mice. Mol Psychiatry. 2015;20:1406–19.
Mukherjee S, Coque L, Cao JL, Kumar J, Chakravarty S, Asaithamby A, et al. Knockdown of Clock in the ventral tegmental area through RNA interference results in a mixed state of mania and depression-like behavior. Biol Psychiatry. 2010;68:503–11.
Coque L, Mukherjee S, Cao JL, Spencer S, Marvin M, Falcon E, et al. Specific role of VTA dopamine neuronal firing rates and morphology in the reversal of anxiety-related, but not depression-related behavior in the ClockDelta19 mouse model of mania. Neuropsychopharmacology 2011;36:1478–88.
Arey RN, Enwright JF 3rd, Spencer SM, Falcon E, Ozburn AR, Ghose S, et al. An important role for cholecystokinin, a CLOCK target gene, in the development and treatment of manic-like behaviors. Mol Psychiatry. 2014;19:342–50.
Parekh PK, Becker-Krail D, Sundaravelu P, Ishigaki S, Okado H, Sobue G, et al. Altered GluA1 (Gria1) function and accumbal synaptic plasticity in the ClockDelta19 model of bipolar mania. Biol Psychiatry. 2018;84:817–26.
Logan RW, Ozburn AR, Arey RN, Ketchesin KD, Winquist A, Crain A, et al. Valproate reverses mania-like behaviors in mice via preferential targeting of HDAC2. Mol Psychiatry. 2021;26:4066–84.
Landgraf D, Long JE, Proulx CD, Barandas R, Malinow R, Welsh DK. Genetic disruption of circadian rhythms in the suprachiasmatic nucleus causes helplessness, behavioral despair, and anxiety-like behavior in mice. Biol Psychiatry. 2016;80:827–35.
Schnell A, Sandrelli F, Ranc V, Ripperger JA, Brai E, Alberi L, et al. Mice lacking circadian clock components display different mood-related behaviors and do not respond uniformly to chronic lithium treatment. Chronobiol Int. 2015;32:1075–89.
Xing C, Zhou Y, Xu H, Ding M, Zhang Y, Zhang M, et al. Sleep disturbance induces depressive behaviors and neuroinflammation by altering the circadian oscillations of clock genes in rats. Neurosci Res. 2021;171:124–32.
Christiansen SL, Bouzinova EV, Fahrenkrug J, Wiborg O. Altered expression pattern of clock genes in a rat model of depression. Int J Neuropsychopharmacol 2016;19:pyw061. https://doi.org/10.1093/ijnp/pyw061.
Guo D, Zhang S, Sun H, Xu X, Hao Z, Mu C, et al. Tyrosine hydroxylase down-regulation after loss of Abelson helper integration site 1 (AHI1) promotes depression via the circadian clock pathway in mice. J Biol Chem. 2018;293:5090–101.
LeGates TA, Altimus CM, Wang H, Lee HK, Yang S, Zhao H, et al. Aberrant light directly impairs mood and learning through melanopsin-expressing neurons. Nature 2012;491:594–8.
Zanos P, Gould TD. Mechanisms of ketamine action as an antidepressant. Mol Psychiatry. 2018;23:801–11.
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010;329:959–64.
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–61.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011;475:91–5.
Jernigan CS, Goswami DB, Austin MC, Iyo AH, Chandran A, Stockmeier CA, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1774–9.
Machado-Vieira R, Zanetti MV, Teixeira AL, Uno M, Valiengo LL, Soeiro-de-Souza MG, et al. Decreased AKT1/mTOR pathway mRNA expression in short-term bipolar disorder. Eur Neuropsychopharmacol. 2015;25:468–73.
Yang C, Zhou ZQ, Gao ZQ, Shi JY, Yang JJ. Acute increases in plasma mammalian target of rapamycin, glycogen synthase kinase-3β, and eukaryotic elongation factor 2 phosphorylation after ketamine treatment in three depressed patients. Biol Psychiatry. 2013;73:e35–6.
Wang Q, Zhao G, Yang Z, Liu X, Xie P. Downregulation of microRNA‑124‑3p suppresses the mTOR signaling pathway by targeting DDIT4 in males with major depressive disorder. Int J Mol Med. 2018;41:493–500.
Zhu Y, Strachan E, Fowler E, Bacus T, Roy-Byrne P, Zhao J. Genome-wide profiling of DNA methylome and transcriptome in peripheral blood monocytes for major depression: a monozygotic discordant twin study. Transl Psychiatry. 2019;9:215.
Nowak W, Grendas LN, Sanmarco LM, Estecho IG, Arena ÁR, Eberhardt N, et al. Pro-inflammatory monocyte profile in patients with major depressive disorder and suicide behaviour and how ketamine induces anti-inflammatory M2 macrophages by NMDAR and mTOR. EBioMedicine. 2019;50:290–305.
Roy AV, Thai M, Klimes-Dougan B, Westlund Schreiner M, Mueller BA, Albott CS, et al. Brain entropy and neurotrophic molecular markers accompanying clinical improvement after ketamine: preliminary evidence in adolescents with treatment-resistant depression. J Psychopharmacol. 2021;35:168–77.
Koehl M, Ladevèze E, Catania C, Cota D, Abrous DN. Inhibition of mTOR signaling by genetic removal of p70 S6 kinase 1 increases anxiety-like behavior in mice. Transl Psychiatry. 2021;11:165.
Zhu WL, Wang SJ, Liu MM, Shi HS, Zhang RX, Liu JF, et al. Glycine site N-methyl-D-aspartate receptor antagonist 7-CTKA produces rapid antidepressant-like effects in male rats. J Psychiatry Neurosci. 2013;38:306–16.
Zhong P, Wang W, Pan B, Liu X, Zhang Z, Long JZ, et al. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 2014;39:1763–76.
Chandran A, Iyo AH, Jernigan CS, Legutko B, Austin MC, Karolewicz B. Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Prog Neuropsychopharmacol Biol Psychiatry. 2013;40:240–5.
Harraz MM, Tyagi R, Cortés P, Snyder SH. Antidepressant action of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation. Mol Psychiatry. 2016;21:313–9.
Szewczyk B, Pochwat B, Rafało A, Palucha-Poniewiera A, Domin H, Nowak G. Activation of mTOR dependent signaling pathway is a necessary mechanism of antidepressant-like activity of zinc. Neuropharmacology 2015;99:517–26.
Gordillo-Salas M, Pilar-Cuéllar F, Auberson YP, Adell A. Signaling pathways responsible for the rapid antidepressant-like effects of a GluN2A-preferring NMDA receptor antagonist. Transl Psychiatry. 2018;8:84.
Pothula S, Liu RJ, Wu M, Sliby AN, Picciotto MR, Banerjee P, et al. Positive modulation of NMDA receptors by AGN-241751 exerts rapid antidepressant-like effects via excitatory neurons. Neuropsychopharmacology 2021;46:799–808.
Neis VB, Moretti M, Rosa PB, Dalsenter YO, Werle I, Platt N, et al. The involvement of PI3K/Akt/mTOR/GSK3β signaling pathways in the antidepressant-like effect of AZD6765. Pharm Biochem Behav. 2020;198:173020.
Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–16.
Moretti M, Budni J, Freitas AE, Rosa PB, Rodrigues AL. Antidepressant-like effect of ascorbic acid is associated with the modulation of mammalian target of rapamycin pathway. J Psychiatr Res. 2014;48:16–24.
Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74:742–9.
Pałucha-Poniewiera A, Szewczyk B, Pilc A. Activation of the mTOR signaling pathway in the antidepressant-like activity of the mGlu5 antagonist MTEP and the mGlu7 agonist AMN082 in the FST in rats. Neuropharmacology 2014;82:59–68.
Green NH, Jackson CR, Iwamoto H, Tackenberg MC, McMahon DG. Photoperiod programs dorsal raphe serotonergic neurons and affective behaviors. Curr Biol. 2015;25:1389–94.
Evans JA, Leise TL, Castanon-Cervantes O, Davidson AJ. Dynamic interactions mediated by nonredundant signaling mechanisms couple circadian clock neurons. Neuron 2013;80:973–83.
Rohr KE, Pancholi H, Haider S, Karow C, Modert D, Raddatz NJ, et al. Seasonal plasticity in GABAA signaling is necessary for restoring phase synchrony in the master circadian clock network. Elife 2019;8:e49578. https://doi.org/10.7554/eLife.49578.
Mendoza-Viveros L, Chiang CK, Ong JLK, Hegazi S, Cheng AH, Bouchard-Cannon P, et al. miR-132/212 modulates seasonal adaptation and dendritic morphology of the central circadian clock. Cell Rep. 2017;19:505–20.
Dwyer JM, Maldonado-Aviles JG, Lepack AE, DiLeone RJ, Duman RS. Ribosomal protein S6 kinase 1 signaling in prefrontal cortex controls depressive behavior. Proc Natl Acad Sci USA. 2015;112:6188–93.
Shi SQ, White MJ, Borsetti HM, Pendergast JS, Hida A, Ciarleglio CM, et al. Molecular analyses of circadian gene variants reveal sex-dependent links between depression and clocks. Transl Psychiatry. 2016;6:e748.
Soria V, Martinez-Amoros E, Escaramis G, Valero J, Perez-Egea R, Garcia C, et al. Differential association of circadian genes with mood disorders: CRY1 and NPAS2 are associated with unipolar major depression and CLOCK and VIP with bipolar disorder. Neuropsychopharmacology 2010;35:1279–89.
Lee KY, Ahn YM, Kim SH, Kang HG, Joo EJ. Genetic association study of CSNK1E gene in bipolar disorder and circadian characteristics. Nord J Psychiatry. 2018;72:599–604.
Lee KY, Song JY, Kim SH, Kim SC, Joo EJ, Ahn YM, et al. Association between CLOCK 3111T/C and preferred circadian phase in Korean patients with bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1196–201.
Yegin Z, Sarisoy G, Erguner Aral A, Koc H. For whom the circadian clock ticks? Investigation of PERIOD and CLOCK gene variants in bipolar disorder. Chronobiol Int. 2021;38:1109–19.
van Enkhuizen J, Minassian A, Young JW. Further evidence for ClockDelta19 mice as a model for bipolar disorder mania using cross-species tests of exploration and sensorimotor gating. Behav Brain Res. 2013;249:44–54.
Acknowledgements
The work was supported by grants from NIH (NS118026) to R.C.
Author information
Authors and Affiliations
Contributions
RS, AM, and RC drafted the initial version of this article. RS and AM created the initial version of the figures and the table. RC edited the article, the figures and table and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singla, R., Mishra, A. & Cao, R. The trilateral interactions between mammalian target of rapamycin (mTOR) signaling, the circadian clock, and psychiatric disorders: an emerging model. Transl Psychiatry 12, 355 (2022). https://doi.org/10.1038/s41398-022-02120-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02120-8
