
Abstract
Posttraumatic stress disorder (PTSD) is a chronic and disabling psychiatric disorder prevalent in military veterans. Epigenetic mechanisms have been implicated in the etiology of PTSD, with DNA methylation being the most studied to identify novel molecular biomarkers associated with this disorder. We performed one of the largest single-sample epigenome-wide association studies (EWAS) of PTSD to date. Our sample included 1135 male European–American U.S. veterans who participated in the National Health and Resilience in Veterans Study (NHRVS). DNA was collected from saliva samples and the Illumina HumanMethylation EPIC BeadChip was used for the methylation analysis. PTSD was assessed using the PTSD Checklist. An EWAS was conducted using linear regression adjusted for age, cell-type proportions, first 10 principal components, and smoking status. After Bonferroni correction, we identified six genome-wide significant (GWS) CpG sites associated with past-month PTSD and three CpGs with lifetime PTSD (prange = 10−10–10−8). These CpG sites map to genes involved in immune function, transcription regulation, axonal guidance, cell signaling, and protein binding. Among these, SENP7, which is involved in transcription regulation and has been linked to risk-taking behavior and alcohol consumption in genome-wide association studies, replicated in an independent veteran cohort and was downregulated in medial orbitofrontal cortex of PTSD postmortem brain tissue. These findings suggest potential epigenetic biomarkers of PTSD that may help inform the pathophysiology of this disorder in veterans and other trauma-affected populations.
Similar content being viewed by others


Introduction
Posttraumatic stress disorder (PTSD) is one of the most prevalent psychiatric disorders in veterans and the disorder most closely associated with military experience. PTSD is a chronic and often disabling condition characterized by intrusive symptoms, avoidance, negative cognitions and mood, and alterations in arousal and reactivity. The estimated lifetime prevalence of PTSD in the general population of U.S. veterans is 6.9% [1] and among combat-exposed veterans is as high as 20% [2].
Epigenetic mechanisms may be implicated in the long-term effects of traumatic stress. DNA methylation, the most studied epigenetic modification in humans, plays an important role in the effects of trauma-related events as a risk factor for psychiatric disorders [3,4,5]. There have been several studies examining the epigenetic contribution to PTSD risk at the genome-wide level. A burgeoning body of epigenome-wide association studies (EWAS) examining differentially methylated CpG sites in PTSD have been conducted, with the most promising findings implicating the immune system in peripheral tissue [6,7,8]. However, most of these studies were underpowered and did not adjust for differences in cell-type proportions when studying peripheral tissue, as well as other factors such as technical batch effects or smoking status, which may confound purported associations between these epigenetic markers and PTSD.
An EWAS of PTSD in 96 Australian male Vietnam War veterans found that methylation sites in BRSK1, LCN8, NFG, and DOCK2 genes were significantly associated with the severity of PTSD symptoms [9]. Findings for the DOCK2 gene, which is implicated in Alzheimer’s disease pathology [10], were replicated in an independent sample of 115 African Americans (AAs) from the Grady Trauma Project [9]. Gene enrichment analysis revealed actin cytoskeleton and focal adhesion pathways associated with PTSD. An EWAS study using a larger sample of 473 World Trade Center responders did not identify any genome-wide significant (GWS) methylation sites associated with PTSD. However, enrichment analysis identified several pathways associated with top differentially methylated CpG sites that included synaptic plasticity, oxytocin plasticity, cholinergic synapse, and inflammatory disease pathways [11]. A meta-EWAS study of PTSD in civilian and military cohorts (n = 1147 subjects from seven different cohorts) from the PGC-PTSD consortium Epigenetics workgroup also did not identify GWS associations for PTSD [12], possibly due to the large heterogeneity among study cohorts and type of trauma. A meta-EWAS study of three civilian cohorts (n = 545 subjects) identified two CpG sites significantly associated with current-PTSD mapping to the NRG1 and HGS genes [13]. A recent study in 513 trauma-exposed veterans of the post-9/11 conflicts showed one GWS finding at G0S2 gene, which was replicated in the military cohort from the Consortium PTSD EWAS [14]. Another recent EWAS in 1896 PTSD cases and trauma-exposed controls identified significant differential methylation at 4 CpG sites mapping to AHRR, with supportive evidence from metabolomics data [15]. A summary of these studies is included in Table 1. However, most of these findings from these studies have not been yet replicated in independent datasets and/or have been conducted using a lower resolution array for DNA methylation. Larger samples using arrays with better DNA methylation coverage are needed to identify novel loci associated with PTSD.
In this study, which is one of the largest to date with a comparatively homogenous sample, we examined epigenome-wide DNA methylation signatures of PTSD in a large, nationally representative cohort (n = 1135) of male European–American (EA) U.S. veterans using a high-density array. We conducted an EWAS of both current (past-month) and lifetime PTSD showing replication findings in an independent cohort of veterans and post-EWAS analyses, including symptom-level sensitivity analysis and methylation quantitative trait analysis. We also evaluated the gene expression of our top finding in postmortem brain tissue of an independent sample of individuals with PTSD.
Methods and materials
Study cohort
Our sample included a total of 1,135 EA male U.S. military veterans who participated in the National Health and Resilience in Veterans Study (NHRVS) [16, 17], a nationally representative study of U.S. veterans, conducted in October-December 2011. Participants were recruited from the Knowledge Networks research panel comprising more than 50,000 households that are developed and maintained by Ipsos, a survey research firm. All participants provided informed consent. This study was approved by the Human Subjects Subcommittee of the Veterans Affairs (VA) Connecticut Healthcare System and VA Office of Research & Development. Table 2 illustrates the demographic and clinical characteristics of the sample.
PTSD assessment
PTSD was assessed using the PTSD Checklist-Specific (PCL-S), a 17-item self-report measure that assesses DSM-IV symptoms of PTSD related to each respondent’s worst traumatic event assessed using the Trauma History Screen [18]. This assessment allows for individual-level screening for PTSD, aiding in diagnostic assessment of PTSD, and monitoring change in PTSD symptoms. A total symptom severity score was calculated by summing the scores from the 17 items (score range for each item = 1–5); our scores range from 17–85. We also computed symptom cluster scores using a five-factor model of PTSD symptoms that includes re-experiencing, avoidance, emotional numbing, dysphoric arousal (e.g., sleep disturbance), and anxious arousal (e.g., hypervigilance) [19, 20]. Both lifetime and current (i.e., past-month) PTSD symptoms were assessed, with scores ≥ 50 indicative of a positive screen for PTSD [21], which is conservative for general population samples [22]. The median current and lifetime PTSD PCL scores were 20 ± 9.3 (Cronbach’s α = 0.94, and 23 ± 11.2 (Cronbach’s α = 0.94), respectively. A total of 65 (5.7%) veterans met this conservative screening cut-off for lifetime PTSD and 43 (3.0%) for current PTSD. Prevalence for alcohol and drug use disorder, which were assessed using modified self-report version of the MINI Neuropsychiatric Interview [23]; and tobacco use disorder, which was assessed using the Fagerström Test for Nicotine Dependence [24], are also shown in Table 2. Information about the most common index traumas in each group is included in the Supplementary Material.
DNA extraction and preparation for epigenetic analysis
Genomic DNA (500 ng) was extracted from saliva samples using Oragene kits (DNA Genotek, Ottawa, Ontario, Canada) and treated with bisulfite reagents included in the EZ-96 DNA methylation kit (Zymo Research, Orange, CA, USA) following the manufacturer’s standard protocol. Bisulfite-converted DNA samples were then assessed with the Illumina Infinium Human MethylationEPIC BeadChip (Illumina, San Diego, CA, USA), which interrogates DNA methylation of >850,000 loci across the genome at single-nucleotide resolution.
Microarray processing and quality control
Genome-wide DNA methylation assay was conducted at the Yale Center for Genome Analysis (YCGA). Quality control was conducted in R version 3.4.1 and performed based on a recently published pipeline using the ‘minfi’ R package (Bioconductor 1.8.9) [25]. Quality control and normalization are described in the Supplementary Material. After quality control and normalization, a total of 706,573 CpG sites (82% of sites) were left for subsequent analysis.
Since methylation values at CpG sites can be cell-type specific [26], we conducted a cell composition estimation analysis using a modified version of the Housemann method [27, 28]. The relative proportion of each cell type (e.g., CD34, CD14, and buccal cells) was estimated in our heterogeneous peripheral saliva samples, and included in the model. To adjust for possible population stratification (within EAs), a methylation-based principal component (PC) approach [29] was conducted based on sets of CpG sites within 50 kb of SNPs using the 1000 Genomes Project variants with minor allele frequency (MAF) > 0.1 following the Barfield et al. method [29]. The first 10 PCs were included in the model as covariates to adjust for population stratification.
Gene ontology analysis
A gene ontology analysis was conducted using the “gometh” function in the Bioconductor R package missMethyl [30]. This function accounts for the varying number of CpG sites per gene by providing a prior probability for each gene based on gene length, followed by a modified hypergeometric test for overrepresentation of a gene set [31]. We evaluated possible overrepresentation in GO and KEGG pathways among the CpG sites at a false discovery rate (FDR) < 0.05 for current and lifetime PTSD. The FDR was set at 0.05 to adjust for multiple testing.
Methylation quantitative trait loci
Methylation quantitative trait locus (meQTL) analysis was conducted to evaluate the influence of genotype variation on CpG methylation patterns. Single-nucleotide polymorphisms (SNPs) within ±50 kb of the CpG site (hg19 reference genome) were included in the meQTL analysis. Genotyping information, imputation, and quality control methods are included in the Supplementary Material. PLINK 1.9 [32] was used for the linear regression analysis with SNPs located ±50 kb from each GWS CpG site with its corresponding beta value. Covariates included in the linear regression model of the meQTL analysis were age, smoking status, cell type proportion, and PCs. A Bonferroni correction was used to adjust for multiple testing of 12,475 SNPs that were within ±50 kb of the 9 GWS CpG sites (0.05/12,475 = 4.0 × 10−6).
Replication analysis
We conducted a replication analysis in 608 male veterans from the Veterans Aging Cohort Study (VACS), a nationwide multicenter collaborative project designed to understand the role of co-morbid medical and psychiatric diseases on HIV infection (https://medicine.yale.edu/intmed/vacs/). The VACS biobank is comprised of 2,470 participants who were recruited for genetic studies from 2006 to 2007. Participants in the VACS biobank provided informed consent for the genetic study and provided blood samples. Clinical and demographic data were collected within 90-days of the blood sample collection.
Diagnosis of PTSD was derived from the electronic medical record. A total of 112 veterans were diagnosed with PTSD, and 496 veterans without PTSD served as controls. All veterans were men with mean age 49.4 ± 7.6 years old and a majority were African Americans (86%) and HIV-positive. Genomic DNA was extracted from whole blood samples. DNA methylation profiling, quality control, normalization and data analysis are described in Supplementary Material. DNA methylation was assessed using a lower coverage array, the Illumina HumanMethylation 450 K array. Three out of the nine GWS CpG sites were not tested because they are not included in the higher density array used in the discovery dataset.
Postmortem Brain PTSD Analysis for top signal mapped to SENP7
Given the limited postmortem brain tissue available, we prioritized the replicated SENP7 findings for further examination and validation. Human autopsy brain tissue samples were obtained from the National Center for PTSD Brain Bank and the University of Pittsburgh Medical Center. Fresh frozen samples of prefrontal cortex from medial orbitofrontal cortex (Broadmann area 11, BA11) were collected from each postmortem sample. This brain region was selected as it is an integral hub of the limbic system and plays an important role in PTSD by modulating inhibitory and regulatory functions, among other processes [33]. Quantitative real-time PCR (qRT-PCR) was performed on dissected tissue from 18 subjects with PTSD (mean age, 46.9 ± 12.3 years, 8 females) and 17 matched neurotypical controls (mean age, 48.1 ± 12.7 years, 9 females) of European ancestry. The average postmortem interval (PMI) was 17.1 h in the PTSD cohort and 19.3 h for the controls. There were no significant differences between the PTSD and control samples in age, postmortem interval, pH, or RNA integrity number. Causes of death was similar between PTSD and control samples. In the PTSD sample, the most common cause of death was “natural” (61%), followed by “accidental” (28%). 2 PTSD cases had “suicide” as cause of death. In the control sample, the most common cause of death was also “natural” (76%), followed by “accidental” (24%). None of the control samples have “suicide” as cause of death. qRT-PCR was performed using primers designed to detect the transcript of SENP7 (See Results and Supplementary Material). Fold regulation was calculated by using the –delta delta Ct (2-DDCt) method.
Statistical Analysis
Epigenome-wide analysis study (EWAS) analyses were conducted using the ‘cpg.assoc’ function from the ‘minfi’ R package in which we conducted a series of linear regressions (for current and lifetime PTSD) using age, cell type proportion (CD34, CD14, buccal), first 10 principal components, and smoking status as covariates. Given the heteroscedasticity of beta values, M-values (logit transformation in log2 scale) were calculated and used in all analyses, as recommended by Du et al. [34]. Bonferroni correction was used to adjust for multiple testing (0.05/706,573; p = 7.07 ×10−8). For the sensitivity analysis of PTSD symptoms, a linear regression was conducted using the PTSD symptoms cluster as the outcome, and M value and covariates in the model. For the sensitivity analysis for substance use disorders in our SENP7*cg09657378 finding, we conducted a multi-variable regression analysis, using M-values of SENP7*cg09657378 as the dependent variable and lifetime PTSD, covariates, and either substance use or substance use disorder variables as independent variables (See Supplementary Material).
For the postmortem gene expression analysis, differences in transcriptional changes between PTSD and neurotypical controls were evaluated using Graphpad Prism v7 (Graphpad Software, San Diego, CA) with fold changes calculated using the 2-DDCt method. Mann–Whitney U test followed by Bonferroni correction for multiple comparisons was used to assess statistical differences in fold regulation.
Results
Epigenome-wide association analysis
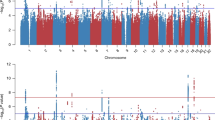
Quantile-quantile plots are shown in Supplementary Fig. 1, with lambda values of 1.006 and 1.020 for current and lifetime PTSD, respectively. After Bonferroni correction, a total of 6 GWS CpG sites were associated with current PTSD (Table 3A). The Manhattan plot is depicted in Fig. 1A. Our top CpG site, cg07672479 (chr14: 102431106, p = 5.49 × 10−10) maps to Dynein Cytoplasmic 1 Heavy Chain 1 (DYNC1H1). Other GWS CpG sites differentially methylated with respect to current PTSD included cg03284870 (chr7:45197346, p = 3.47 × 10−8), cg22500183 (chr17:33914271, p = 4.09 × 10−8), cg06595994 (chr12:51632641, p = 4.68 × 10−8), and cg00770699 (chr11:85370479, p = 7.02 × 10−8) located within the RAMP3 (Receptor Activity Modifying Protein 3), AP2B1 (Adaptor Related Protein Complex 2 Subunit Beta 1), DAZAP2 (DAZ Associated Protein 2), and CREBZF (CREB/ATF BZIP Transcription Factor) genes, respectively. An additional intergenic GWS CpG site associated with current PTSD was cg15559076 (chr11:128109597, p = 6.60 × 10−8). Hypomethylation at five GWS CpG sites (all except cg00770699) was associated with current PTSD, whereas hypermethylation at one site, cg00770699, was associated with current PTSD (Supplementary Fig. 2A).

A Current PTSD, B Lifetime PTSD. The horizontal solid line represents Bonferroni-corrected significance. The dotted line indicates false discovery rate of < 0.05.
Functional annotation of the current-PTSD-associated GWS CpG sites showed that one CpG site (cg07672479; DYNC1H1) is promoter-associated, one is located in the 5′UTR region (cg06595994, DAZAP2), and one in the gene body (cg00770699; CREBZF). Further, cg06595994 is located at an enhancer. A total of five GWS CpG sites (all except cg00770699) have three Dnase hypersensitivity sites, an indicator of open chromatin, whereas cg00770699 has six transcription factor binding sites.
In the lifetime PTSD EWAS analysis, we identified a total of three Bonferroni-corrected GWS CpG sites (Table 3B). The Manhattan plot is depicted in Fig. 1B. These GWS CpG sites included cg09657378 (chr3:101232109, p = 1.71 × 10−8), cg07377876 (chr7:73868114, p = 1.81 × 10−8), and cg19825186 (chr1:207495562, p = 2.08 × 10−8); they map to SENP7 (SUMO Specific Peptidase 7), GTF2IRD1 (GTF2I Repeat Domain Containing 1), and CD55 (CD55 molecule) genes, respectively. Hypomethylation in all three GWS CpG sites was associated with lifetime PTSD (Supplementary Fig. 2B). Functional annotation of these CpG sites showed that two are promoter-associated (cg09657378 and cg19825186), and one is located in the 5′UTR region (cg19825186). Further, cg07377876 is enhancer related and all three CpG sites have three Dnase hypersensitivity sites.
Sensitivity analysis
To evaluate the relationship between the DNA methylation of GWS CpG sites identified and PTSD symptom clusters (i.e., re-experiencing, avoidance, emotional numbing, dysphoric arousal, anxious arousal) [19, 20], we conducted linear regression analyses using the same covariates as in the EWAS analysis (Fig. 2). For current PTSD, all GWS CpG sites were significantly associated with re-experiencing, with cg07672479 showed the strongest association (p = 3.4 × 10−04). A total of four GWS CpG sites were significantly associated with avoidance (cg07672479, cg22500183, cg1559076, and cg00770699), and five with emotional numbing (all except for cg03284870). Only two GWS CpG sites were significantly associated with dysphoric arousal (cg07672479 and cg15559076), and one with anxious arousal (cg15559076).

A Current PTSD, B Lifetime PTSD. Heatmap colors correspond to the significance (p-value), where darker indicates higher degree of association, lighter indicates a lower degree of association, and white indicates no association.
For lifetime PTSD, all GWS CpG sites were significantly associated with re-experiencing, emotional numbing, and anxious arousal. The strongest associations were cg19825186 for re-experiencing (p = 4.6 × 10−05), cg09657378 for emotional numbing (p = 4.4 × 10−06), and cg19825186 for anxious arousal (p = 2.8 × 10−04). For avoidance, all but cg07377876 were significantly associated, with cg09657378 showing the strongest association (p = 2.9 × 10−04). For dysphoric arousal, all but cg09657378 were significantly associated, with the strongest association for cg19825186 (p = 1.5 × 10−03).
Gene ontology analysis
For lifetime PTSD, the KEGG pathway ‘Viral myocarditis’ was identified (p = 2.45 × 10−06, FDR = 8.15 × 10−04; Supplementary Table 1). For current PTSD, no KEGG pathways were identified. No GO ontologies were enriched at FDR 0.05 for either current or lifetime PTSD.
meQTL analysis for top methylation probes
A total of 30 meQTLs were identified (p-value range: 10−11–10−6) (Supplementary Fig. 3). These mapped to cg15559076, which is nearby the EST1 gene. The most significant meQTL for cg15559076 was at chr11:128025697 (rs10893823; beta = −0.28, p = 7.14 × 10−11).
Replication analysis
For the replication analysis, a total of 608 male veterans (112 PTSD cases, and 496 controls) from the VACS biobank cohort were evaluated. Methylation levels at the SENP7 CpG site (cg09657378, promoter region) were associated (p = 0.02) with PTSD diagnosis. Hypomethylation at cg09657378 was observed in PTSD cases, showing the same directional effect as in the discovery dataset.
Differential expression of SENP7 in Postmortem PTSD Brain Tissue
Postmortem brain tissue from mOFC (Broadmann 11) from subjects with PTSD matched to neurotypical controls of European ancestry (n = 17–18/group) were assayed for gene expression changes by quantitative real-time PCR. Normalized log corrected Ct values were used to calculate fold change. This analysis revealed a significant 1.4 fold decrease in SENP7 expression (P = 0.03; Fig. 3A, B, error bars indicate ± SEM).

Based on the significant observed association and then the replication of the SENP (cg09657378) association in the VACS cohort, we examined gene expression in postmortem brain tissue from medial orbitofrontal cortex (Broadmann 11) in PTSD cases and controls. A Box plots of normalized Ct values for SENP7 in controls (white), and PTSD (blue) individuals. B Log 2-fold change of SENP7 in human mOFC (p < 0.03, n = 35). Error bars indicate ± SEM.
Discussion
To date, EWAS of PTSD have been limited in successfully identifying associated loci, most likely due to lack of power, high heterogeneity of the cohorts studied, and failure to replicate in independent cohorts. In this study, we conducted the largest to date EWAS of PTSD in a single population of U.S. military veterans, and identified novel differentially methylated CpG sites associated with both current and with lifetime PTSD. Specifically, we identified nine GWS CpG associated with PTSD: six were associated with current (past-month) PTSD and three with lifetime PTSD. Of these, a CpG site mapping to SENP7 was replicated in a demographically and clinically distinct cohort of veterans. Follow-up validation in an independent sample of postmortem brain tissue from individuals with PTSD and matched controls showed downregulation of SENP7 in the mOFC.
Our top finding in the EWAS of current PTSD is cg07672479, which is annotated to the DYNC1H1 gene. This gene encodes for cytoplasmic dynein 1, which plays a role in retrograde axonal transport, neuronal migration, and immune function, and is highly expressed in the brain (https://www.proteinatlas.org/ENSG00000197102-DYNC1H1/tissue). Mutations in DYNC1H1 are associated with Charcot-Marie-Tooth disease [35], which is characterized by progressive weakness and atrophy, spinal muscular atrophy [36], and severe intellectual disability [37].
The second GWS CpG site associated with current PTSD included cg03284870, which is located in RAMP3, a gene involved in GPCR signaling; and the third, cg22500183, maps to the AP2B1 gene, which encodes the adaptor protein complex 2, which is implicated in synaptic vesicle pathway, axonal guidance, and immune function. This gene is highly expressed in the brain, endocrine tissues, and male tissues (https://www.protein atlas.org/ENSG00000006125-AP2B1/tissue). Additional GWS CpG sites associated with current PTSD include cg06595994 located in the DAZAP2 gene, which encodes for a protein involved in cell signaling, transcription regulation, and spermatogenesis, cg15559076 located in an intergenic region, and cg00770699 which maps to CREBZF involved in MAPK-Erk pathway, which is implicated in the long-term formation of fear-related memories [38].
For lifetime PTSD, three GWS CpG sites were identified: cg09657378, annotated to the SENP7 gene; cg073777876, annotated to the GTF2IRD1; and cg19825186 annotated to the CD55 gene. SENP7 encodes for an enzyme that deconjugates SUMO2 and SUMO3, which are involved in transcription regulation. Of note, this gene has been previously identified in genome-wide association studies (GWAS) of risk-taking behavior (n = 315,894) and alcohol consumption (n = 414,343) [39]. The second gene, GTF2IRD1, is also implicated in transcription regulation and has been identified in GWAS of neuroticism (n = 329,821) [40]. Lastly, CD55 is a gene involved in the innate immune response, specifically in the complement cascade. Gene enrichment analysis of GWS CpG sites of lifetime PTSD at FDR < 0.05 identified the “viral myocarditis” KEGG pathway, implicating immune function and cardiovascular disorders.
Half of the GWS CpG sites for current PTSD are only present in the high-density DNA methylation array (cg06595994, cg15559076, and cg00770699), showing that use of this array increased ability to identify significant CpG sites associated with current PTSD. To determine whether changes in methylation at these CpG sites are influenced by genotype, we integrated the genotype data and identified 30 meQTLs (p-value range: 10−11–10−6) mapping to cg15559076. The closest-mapped gene is ETS1, implicated in immune regulation. SNPs at this gene have been also identified in GWAS of celiac disease [41].
When comparing EWAS findings of current and lifetime PTSD, we found some overlap (Supplementary Table 2). All GWS CpG sites from the lifetime PTSD EWAS showed suggestive association in current-PTSD EWAS (p-value range: 10−5–10−4). In the current-PTSD EWAS, 4 out of 6 GWAS CpG sites showed significance in the lifetime PTSD EWAS (p-value range: 10−6–10−3). The other two CpG sites (cg22500183, AP2B1; cg06595994, DAZAP2) did not show any nominal p-value in the lifetime PTSD EWAS. This suggests that differential methylation at these sites may not persist over time.
For replication, we examined our GWS hits an independent veteran cohort of European- and African-American ancestry. We observed a significant replication of the association of SENP7 gene methylation (cg09657378, p = 0.02) with PTSD diagnosis, with the same direction effect of hypomethylation of SENP7 associated with PTSD. Of note, this association was independent of substance use or substance use disorder comorbidity in the discovery cohort (See Supplementary Table 3). Evaluating postmortem brain from the mOFC of an independent cohort of individuals with PTSD and matched controls, we found evidence of downregulation of SENP7 in PTSD. cg09657378 is located in the promoter region. While it is generally expected that hypomethylation in the promoter region is associated with gene activation, recent studies have found that its effects may be gene-specific, leading to either increased or decreased gene expression [42]. Future studies should evaluate whether differential methylation directly influences SENP7 gene expression and examine its effects on the behavioral level.
Overall, results of the current study are consistent with previous EWAS studies of PTSD implicating differential methylation of genes implicated in the immune system. Our results also revealed novel genes involved in axon guidance, transcription regulation, and cell signaling. They further suggest that associations between DNA methylation of these CpG sites and PTSD may be driven by specific PTSD symptomatology. Specifically, for current PTSD, all GWS CpG sites were associated with re-experiencing (i.e., intrusive thoughts, nightmares, and flashbacks of the traumatic event), and are considered the most characteristic set of PTSD symptoms. However, for dysphoric and anxious arousal symptoms, the association is present in cg15559076, an intergenic region but with enhancer activity, and cg07672479, which is annotated to DYNC1H1, which is implicated in retrograde axonal transport, neuronal migration, and immune function, and is highly expressed in the brain. These PTSD symptom clusters, which are characterized by sleep disturbance, concentration difficulties, and hypervigilance, are common to many anxiety-related disorders and are not specific to PTSD. In lifetime PTSD, all GWS CpG sites are associated with re-experiencing, but also numbing and dysphoric arousal symptoms. Differential methylation at cg09657379 and cg19825186 were observed in relation to avoidance symptoms, whereas cg07377876 and cg19825186 in dysphoric arousal. Notably, one might speculate of an apparent distinction between current and lifetime PTSD: while current PTSD is associated with genes implicated in neuroplasticity (i.e., response to stress), lifetime PTSD is associated with traits related to poor impulse control (i.e., the likelihood of exposing oneself to stressful situations). However, further studies are needed to evaluate the specificity of these associations and the extent to which epigenetic markers reflect the causes or consequences of PTSD symptoms.
To the best of our knowledge, this is the largest EWAS of PTSD to date that evaluated epigenetic markers associated with both current and lifetime PTSD in a veteran cohort. Study strengths are the investigation of a relatively large and homogeneous cohort, the use of a high-density DNA methylation array, evaluation of both current and lifetime PTSD as well as PTSD symptom clusters; integration of genotype data; replication in an independent veteran cohort; and postmortem brain validation of the top replicated EWAS hit.
To confirm our findings, replication in independent well-powered samples and in more heterogeneous samples in terms of sex and race, are warranted. This study is further limited by the lack of gene expression data in the discovery cohort, and the use of saliva, a peripheral tissue, for DNA methylation profiling since DNA methylation patterns are tissue-specific [20]. Nevertheless, several studies comparing the DNA methylation patterns across different peripheral tissues and the brain consistently show the highest correlation between saliva and brain [28, 43, 44]. However, the changes observed in peripheral tissue may not necessarily reflect those in the brain. Further, PTSD not only impacts the brain, but also the numerous other bodily systems. Examination of epigenetic changes in peripheral tissue may thus be informative in identifying biological pathways implicated in PTSD other than the brain, for example, the immune system. Accumulating evidence in peripheral tissue from previous EWAS of PTSD and other studies show alterations in the immune function [6,7,8]. Our findings suggest that these changes could occur system-wide, but may also be gene-level specific as has been shown previously [45]. Further, if personalized epigenetics results are ever to have treatment implications, they need to derive from accessible tissues such as blood or saliva, despite the limitations of this approach.
Conclusions
We identified nine GWS DNA methylation sites associated with current or lifetime PTSD in U.S. military veterans—six for current PTSD and three for lifetime PTSD. These mapped to genes involved in immune function, axon guidance, transcription regulation, and cell signaling pathways, and showed differential associations with PTSD symptom clusters. Further, alterations of SENP7 in PTSD were replicated in an independent cohort of veterans and validated in postmortem brain analysis. Future research should focus on integrating brain and peripheral methylomics and transcriptomics datasets, together with GWAS data, to identify molecular biomarkers of PTSD. Our findings provide new insights into the epigenetic architecture underlying the complex etiology of PTSD.
Code availability
Code will be available upon request.
References
Smith SM, Goldstein RB, Grant BF. The association between post-traumatic stress disorder and lifetime DSM-5 psychiatric disorders among veterans: data from the National Epidemiologic Survey on Alcohol and Related Conditions-III (NESARC-III). J Psychiatr Res. 2016;82:16–22. https://doi.org/10.1016/j.jpsychires.2016.06.022
Seal KH, Metzler TJ, Gima KS, Bertenthal D, Maguen S, Marmar CR. Trends and risk factors for mental health diagnoses among Iraq and Afghanistan veterans using Department of Veterans Affairs health care, 2002-2008. Am J public health. 2009;99:1651–8. https://doi.org/10.2105/AJPH.2008.150284
Montalvo-Ortiz JL, Gelernter J, Hudziak J, Kaufman J. RDoC and translational perspectives on the genetics of trauma-related psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2016;171B:81–91. https://doi.org/10.1002/ajmg.b.32395
Montalvo-Ortiz JL, Bordner KA, Carlyle BC, Gelernter J, Simen AA, Kaufman J. The role of genes involved in stress, neural plasticity, and brain circuitry in depressive phenotypes: convergent findings in a mouse model of neglect. Behav Brain Res. 2016;315:71–74. https://doi.org/10.1016/j.bbr.2016.08.010
Klengel T, Pape J, Binder EB, Mehta D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology. 2014;80:115–32. https://doi.org/10.1016/j.neuropharm.2014.01.013
Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, et al. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci USA. 2010;107:9470–5. https://doi.org/10.1073/pnas.0910794107
Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, et al. Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am J Med Genet B: Neuropsychiatr Genet: Off Publ Int Soc Psychiatr Genet. 2011;156B:700–8. https://doi.org/10.1002/ajmg.b.31212
Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW, et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci USA. 2013;110:8302–7. https://doi.org/10.1073/pnas.1217750110
Mehta D, Bruenig D, Carrillo-Roa T, Lawford B, Harvey W, Morris CP, et al. Genomewide DNA methylation analysis in combat veterans reveals a novel locus for PTSD. Acta Psychiatr Scandinavica. 2017;136:493–505. https://doi.org/10.1111/acps.12778
Cimino PJ, Sokal I, Leverenz J, Fukui Y, Montine TJ. DOCK2 is a microglial specific regulator of central nervous system innate immunity found in normal and Alzheimer’s disease brain. Am J Pathol. 2009;175:1622–30. https://doi.org/10.2353/ajpath.2009.090443
Kuan PF, Waszczuk MA, Kotov R, Marsit CJ, Guffanti G, Gonzalez A, et al. An epigenome-wide DNA methylation study of PTSD and depression in World Trade Center responders. Transl psychiatry. 2017;7:e1158 https://doi.org/10.1038/tp.2017.130
Ratanatharathorn A, Boks MP, Maihofer AX, Aiello AE, Amstadter AB, Ashley-Koch AE, et al. Epigenome-wide association of PTSD from heterogeneous cohorts with a common multi-site analysis pipeline. Am J Med Genet Part B, Neuropsychiatr Genet. 2017;174:619–30. https://doi.org/10.1002/ajmg.b.32568
Uddin M, Ratanatharathorn A, Armstrong D, Kuan PF, Aiello AE, Bromet EJ, et al. Epigenetic meta-analysis across three civilian cohorts identifies NRG1 and HGS as blood-based biomarkers for post-traumatic stress disorder. Epigenomics. 2018;10:1585–601. https://doi.org/10.2217/epi-2018-0049
Logue MW, Miller MW, Wolf EJ, Huber BR, Morrison FG, Zhou Z, et al. An epigenome-wide association study of posttraumatic stress disorder in US veterans implicates several new DNA methylation loci. Clin Epigenetics. 2020;12:46 https://doi.org/10.1186/s13148-020-0820-0
Smith AK, Ratanatharathorn A, Maihofer AX, Naviaux RK, Aiello AE, Amstadter AB, et al. Epigenome-wide meta-analysis of PTSD across 10 military and civilian cohorts identifies methylation changes in AHRR. Nat Commun. 2020;11:5965 https://doi.org/10.1038/s41467-020-19615-x
Stefanovics EA, Potenza MN, Pietrzak RH. The physical and mental health burden of obesity in U.S. veterans: results from the National Health and Resilience in Veterans Study. J Psychiatr Res. 2018;103:112–9. https://doi.org/10.1016/j.jpsychires.2018.05.016
Fuehrlein, BS, Kachadourian, LK, DeVylder, EK, Trevisan, LA, Potenza, MN, Krystal, JH, et al. Trajectories of alcohol consumption in U.S. military veterans: Results from the National Health and Resilience in Veterans Study. The Am. J. Addictions. 2018. https://doi.org/10.1111/ajad.12731.
Carlson EB, Smith SR, Palmieri PA, Dalenberg C, Ruzek JI, Kimerling R, et al. Development and validation of a brief self-report measure of trauma exposure: the Trauma History Screen. Psychol Assess. 2011;23:463–77. https://doi.org/10.1037/a0022294
Pietrzak RH, Tsai J, Harpaz-Rotem I, Whealin JM, Southwick SM. Support for a novel five-factor model of posttraumatic stress symptoms in three independent samples of Iraq/Afghanistan veterans: a confirmatory factor analytic study. J Psychiatr Res. 2012;46:317–22. https://doi.org/10.1016/j.jpsychires.2011.11.013
Harpaz-Rotem I, Tsai J, Pietrzak RH, Hoff R. The dimensional structure of posttraumatic stress symptomatology in 323,903 U.S. veterans. J Psychiatr Res. 2014;49:31–36. https://doi.org/10.1016/j.jpsychires.2013.10.020
Weathers, F, Litz BT, Herman DS, Huska JA, Keane TM. In: Paper presented at the Annual Meeting of the International Society for Traumatic Stress Studies (San Antonio, TX., 1993).
Disorder, NCFPS Using the PTSD Checklist for DSM-IV (PCL) 2021. http://www.ptsd.va.gov/professional/assessment/adult-sr/ptsd-checklist.asp.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59:22–33. quiz 34–57
Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom KO. The Fagerstrom Test for Nicotine Dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br J Addict. 1991;86:1119–27. https://doi.org/10.1111/j.1360-0443.1991.tb01879.x
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9. https://doi.org/10.1093/bioinformatics/btu049
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31 https://doi.org/10.1186/gb-2014-15-2-r31
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13:86 https://doi.org/10.1186/1471-2105-13-86
Smith AK, Kilaru V, Klengel T, Mercer KB, Bradley B, Conneely KN, et al. DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am J Med Genet Part B, Neuropsychiatr Genet. 2015;168B:36–44. https://doi.org/10.1002/ajmg.b.32278
Barfield RT, Almli LM, Kilaru V, Smith AK, Mercer KB, Duncan R, et al. Accounting for population stratification in DNA methylation studies. Genet Epidemiol. 2014;38:231–41. https://doi.org/10.1002/gepi.21789
Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 2016;32:286–8. https://doi.org/10.1093/bioinformatics/btv560
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14 https://doi.org/10.1186/gb-2010-11-2-r14
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7 https://doi.org/10.1186/s13742-015-0047-8
Weston CS. Posttraumatic stress disorder: a theoretical model of the hyperarousal subtype. Front Psychiatry. 2014;5:37 https://doi.org/10.3389/fpsyt.2014.00037
Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, et al. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010;11:587 https://doi.org/10.1186/1471-2105-11-587
Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89:308–12. https://doi.org/10.1016/j.ajhg.2011.07.002
Fiorillo C, Moro F, Yi J, Weil S, Brisca G, Astrea G, et al. Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum Mutat. 2014;35:298–302. https://doi.org/10.1002/humu.22491
Willemsen MH, Vissers LE, Willemsen MA, van Bon BW, Kroes T, de Ligt J, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet. 2012;49:179–83. https://doi.org/10.1136/jmedgenet-2011-100542
Cestari V, Rossi-Arnaud C, Saraulli D, Costanzi M. The MAP(K) of fear: from memory consolidation to memory extinction. Brain Res Bull. 2014;105:8–16. https://doi.org/10.1016/j.brainresbull.2013.09.007
Karlsson Linner R, Biroli P, Kong E, Meddens SFW, Wedow R, Fontana MA, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51:245–57. https://doi.org/10.1038/s41588-018-0309-3
Luciano M, Hagenaars SP, Davies G, Hill WD, Clarke TK, Shirali M, et al. Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat Genet. 2018;50:6–11. https://doi.org/10.1038/s41588-017-0013-8
Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. https://doi.org/10.1038/ng.543
Wang F, Xu H, Zhao H, Gelernter J, Zhang H. DNA co-methylation modules in postmortem prefrontal cortex tissues of European Australians with alcohol use disorders. Sci Rep. 2016;6:19430 https://doi.org/10.1038/srep19430
Murata, Y, Fujii, A, Kanata, S, Fujikawa, S, Ikegame, T, Nakachi, Y, et al. Evaluation of the usefulness of saliva for DNA methylation analysis in cohort studies. Neuropsychopharmacol Rep. 2019. https://doi.org/10.1002/npr2.12075.
Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl psychiatry. 2019;9:47 https://doi.org/10.1038/s41398-019-0376-y
Hess JL, Tylee DS, Barve R, de Jong S, Ophoff RA, Kumarasinghe N, et al. Transcriptome-wide mega-analyses reveal joint dysregulation of immunologic genes and transcription regulators in brain and blood in schizophrenia. Schizophrenia Res. 2016;176:114–24. https://doi.org/10.1016/j.schres.2016.07.006
Hammamieh, R., Chakraborty, N., Gautam, A. et al. Whole-genome DNA methylation status associated with clinical PTSD measures of OIF/OEF veterans. Transl Psychiatry. 2017;7:e1169. https://doi.org/10.1038/tp.2017.129.
Acknowledgements
The National Health and Resilience in Veterans Study is supported by the U.S. Department of Veterans Affairs National Center for Posttraumatic Stress Disorder. Preparation of this report is supported by the Department of Veterans Affairs’ grant 1IK2CX002095, the NARSAD Young Investigator Grant, and the National Institute on Drug Abuse grant R21DA050160 to JLMO, and the National Institute on Drug Abuse grant R01 DA038632 to KX. The members of the Traumatic Stress Brain Research Group include Matthew J. Friedman MD, PhD (Director), Victor E. Alvarez MD, David Benedek MD, Christopher Brady PhD, Dianne Cruz MS, David A Davis PhD, Ronald S. Duman PhD, Matthew J. Girgenti PhD, Paul E. Holtzheimer MD, Bertrand R. D. Huber MD, Terence M. Keane PhD, Neil Kowell MD, John H. Krystal MD, Mark W. Logue PhD, Ann McKee MD, Brian Marx PhD, Deborah Mash PhD, Mark W. Miller PhD, William K. Scott PhD, Thor Stein MD, PhD, Robert Ursano MD, Douglas E. Williamson PhD, Erika J. Wolf PhD, and Keith A. Young PhD.
Author information
Authors and Affiliations
Consortia
Contributions
R.H.P., J.G., and J.L.M.O. conceived and designed the study. J.L.M.O. obtained the data, performed the bioinformatic analyses, and wrote the manuscript. M.G., K.X., X.Z., S.G., and H.Z., conducted additional bioinformatic analyses. J.L.M.O., Z.C., and M.G. prepared figures and tables. R.H.P., J.G., and J.L.M.O. coordinated the study and supervised the manuscript preparation. All authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The following competing interests for JHK: (1) Consultant: Note: The Individual Consultant Agreements listed below are less than $10,000 per year: AstraZeneca Pharmaceuticals; Biogen, Idec, MA; Biomedisyn Corporation; Bionomics, Limited (Australia); Concert Pharmaceuticals, Inc.; Heptares Therapeutics, Limited (UK); Janssen Research & Development; L.E.K. Consulting; Otsuka America Pharmaceutical, Inc.; Spring Care, Inc.; Sunovion Pharmaceuticals, Inc.; Takeda Industries; Taisho Pharmaceutical Co., Ltd; Scientific Advisory Board; Bioasis Technologies, Inc.; Biohaven Pharmaceuticals; Blackthorn Therapeutics, Inc.; Broad Institute of MIT and Harvard; Cadent Therapeutics; Lohocla Research Corporation; Pfizer Pharmaceuticals; Stanley Center for Psychiatric Research at the Broad Institute; (2) Stock: ArRETT Neuroscience, Inc.; Blackthorn Therapeutics, Inc.; Biohaven Pharmaceuticals Medical Sciences; Spring Care, Inc. Stock Options: Biohaven PharmaceuticalsMedical Sciences; (3) Income Greater than $10,000: Editorial BoardEditor—Biological Psychiatry; Patents and Inventions: Seibyl JP, Krystal JH, Charney DS. Dopamine and noradrenergic reuptake inhibitors in the treatment of schizophrenia. US Patent #:5,447,948. September 5, 1995; Vladimir, Coric, Krystal, John H, Sanacora, Gerard—Glutamate Modulating Agents in the Treatment of Mental Disorders US Patent No. 8,778,979 B2 Patent Issue Date: July 15, 2014. US Patent Application No. 15/695,164: Filing Date: 09/05/2017; Charney D, Krystal JH, Manji H, Matthew S, Zarate C., Intranasal Administration of Ketamine to Treat Depression United States Application No. 14/197,767 filed on March 5, 2014; United States application or Patent Cooperation Treaty (PCT) International application No. 14/306,382 filed on June 17, 2014; Zarate, C, Charney, DS, Manji, HK, Mathew, Sanjay J, Krystal, JH, Department of Veterans Affairs “Methods for Treating Suicidal Ideation”, Patent Application No. 14/197.767 filed on March 5, 2014 by Yale University Office of Cooperative Research; Arias A, Petrakis I, Krystal JH. Composition and methods to treat addiction; Provisional Use Patent Application no.61/973/961. April 2, 2014. Filed by Yale University Office of Cooperative Research; Chekroud, A., Gueorguieva, R., & Krystal, JH. “Treatment Selection for Major Depressive Disorder” [filing date 3rd June 2016, USPTO docket number Y0087.70116US00]. Provisional patent submission by Yale University; Yoon G, Petrakis I, Krystal JH. Compounds, Compositions, and Methods for Treating or Preventing Depression and Other Diseases. U. S. Provisional Patent Application No. 62/444,552, filed on January10, 2017 by Yale University Office of Cooperative Research OCR 7088 US01; Abdallah, C, Krystal, JH, Duman, R, Sanacora, G. Combination Therapy for Treating or Preventing Depression or Other Mood Diseases. U.S. Provisional Patent Application No. 047162-7177P1 (00754) filed on August 20, 2018 by Yale University Office of Cooperative Research OCR 7451 US01. JG is named as an inventor on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. Other co-authors declare no completing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Montalvo-Ortiz, J.L., Gelernter, J., Cheng, Z. et al. Epigenome-wide association study of posttraumatic stress disorder identifies novel loci in U.S. military veterans. Transl Psychiatry 12, 65 (2022). https://doi.org/10.1038/s41398-022-01822-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-01822-3
This article is cited by
-
Differential methylation of linoleic acid pathway genes is associated with PTSD symptoms – a longitudinal study with Burundian soldiers returning from a war zone
Translational Psychiatry (2024)
-
Tet Enzyme-Mediated Response in Environmental Stress and Stress-Related Psychiatric Diseases
Molecular Neurobiology (2023)
