
Abstract
Background
Clozapine is considered to be the most effective antipsychotic medication for schizophrenia. However, it is associated with several adverse effects such as leukopenia, and the underlying mechanism has not yet been fully elucidated. The authors performed a genome-wide association study (GWAS) in a Chinese population to identify genetic markers for clozapine-induced leukopenia (CIL) and clozapine-induced neutropenia (CIN).
Methods
A total of 1879 patients (225 CIL cases, including 43 CIN cases, and 1,654 controls) of Chinese descent were included. Data from common and rare single nucleotide polymorphisms (SNPs) were tested for association. The authors also performed a trans-ancestry meta-analysis with GWAS results of European individuals from the Clozapine-Induced Agranulocytosis Consortium (CIAC).
Results
The authors identified several novel loci reaching the threshold of genome-wide significance level (P < 5 × 10−8). Three novel loci were associated with CIL while six were associated with CIN, and two T cell related genes (TRAC and TRAT1) were implicated. The authors also observed that one locus with evidence close to genome-wide significance (P = 5.08 × 10−8) was near the HLA-B gene in the major histocompatibility complex region in the trans-ancestry meta-analysis.
Conclusions
The associations provide novel and valuable understanding of the genetic and immune causes of CIL and CIN, which is useful for improving clinical management of clozapine related treatment for schizophrenia. Causal variants and related underlying molecular mechanisms need to be understood in future developments.
Similar content being viewed by others

Introduction
Schizophrenia is a severe mental disorder accompanied by considerable morbidity and mortality, which affects ~1% of the population worldwide1. The etiology of schizophrenia is still not well understood. Environmental and genetic factors play important roles in the development of schizophrenia2. The heritability of schizophrenia is estimated to be 60–80%2,3. Antipsychotic medications are commonly used to treat patients with schizophrenia, but responses to these drugs vary widely4. Although antipsychotics can relieve symptoms of psychosis, ~75% of patients discontinue their therapy due to adverse effects in two years5.
Clozapine is considered to be the most effective antipsychotic medication for schizophrenia and has a distinctive pharmacological profile in treatment-resistant schizophrenia6. However, it is also associated with several adverse effects such as weight gain, metabolic dysfunction, cardiovascular disease and leukopenia. Leukopenia is defined as white blood cell (WBC) count less than 4,000 cells per microliter. Neutropenia and agranulocytosis are different types of leukopenia with an absolute neutrophil count (ANC) less than 1,500 and 500 cells per cubic millimeter (mm−3), respectively. Clozapine-induced agranulocytosis (CIA) is a severe leukopenia that may be life-threatening, first reported in Finland in 19747,8. Among patients taking clozapine, the cumulative risk of neutropenia is 3.8% and for agranulocytosis is 0.9%9.
There is evidence of a genetic contribution in the onset of clozapine-induced leukopenia (CIL), neutropenia (CIN) and CIA10; however, the underlying mechanism remains unclear and may involve multiple genes. The HLA (human leukocyte antigen) system, which locates in the major histocompatibility complex (MHC) region, has been reported to be associated with CIL11,12,13. HLA genes such as HLA-B and HLA-DQB1 were reported to be associated with CIL by Clozapine-Induced Agranulocytosis Consortium (CIAC) and many other researchers11,12,13. Previous genome-wide association studies (GWAS) and whole-exome sequencing studies have indicated that other genes such as SLCO1B3, SLCO1B7, UBAP2 and STARD9 also play important roles in the pathophysiology of CIN12. Most studies of CIN have focused on individuals of European ancestry. To generalize the results, some recent studies have begun to investigate patients from different ethnic groups. Ancestry-based differences in alleles may be part of the reason for different prevalence of CIL; for example, Duffy-null genotype has been found to be associated with benign neutropenia in patients with African ancestry14, and some previously reported SNPs have not been replicated in individuals of Japanese ancestry12,15.
Clozapine has been available for Chinese patients since the 1970s. It was once a commonly used antipsychotic drug in China, prescribed for ~25–60% of patients with schizophrenia16,17, and the reported prevalence of CIL in China is 3.92% (0.21% for CIA)16,18. In this study, we report what we believe to be the first GWAS of CIL in a Chinese population, seeking to identify genetic determinants and provide more information on possible underlying etiology.
Methods and materials
Study design and participants
We analyzed genome-wide data of patients with schizophrenia receiving clozapine treatment from a Chinese cohort. Patients were recruited for this study from Wuhu Fourth People’s Hospital and Guangxi Longquan Mountain Hospital from 2007 to 2012. The clinical interviews were conducted by two independent psychiatrists. All patients had diagnoses of schizophrenia according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria. Those patients with treatment-resistant schizophrenia19 were prescribed clozapine and 1,983 patients were included in this study. Those who developed WBC < 4,000 mm−3 during treatment with clozapine were defined as clozapine-induced leukopenia (CIL) cases (n = 242). Of these, 46 cases also developed ANC < 1,500 mm−3, and were thus defined as clozapine-induced neutropenia (CIN) cases. All controls (n = 1,741) received clozapine without developing WBC < 4,000 mm−3 or ANC < 1,500 mm−3.
We also performed a genome-wide meta-analysis of our Chinese sample with individuals of European ancestry from the Clozapine-Induced Agranulocytosis Consortium (CIAC)11,12.
In accordance with the principles in Declaration of Helsinki, the study was approved by the Ethics Committee at the Bio-X Institutes of Shanghai Jiao Tong University and the written informed consent was obtained from each participant. It is confirmed that the research complied with the Guidance of the Ministry of Science and Technology (MOST) for the Review and Approval of Human Genetic Resources.
Procedures and statistical analysis
Details on genotyping and quality control procedures for the genome-wide analysis of Chinese samples are described in Supplementary Material.
We performed primary genome-wide association analysis for CIL To minimize the effect for population stratification, we performed association analysis separately for two sample sets (according to genetic origin criteria), and then combined the summary in the meta-analysis. For the common variants (minor allele frequency [MAF] ≥ 1%), including both genotyped and imputed variants, association analyses were performed by logistic regression using imputed dosage files and the meta-analysis via METAL with a fixed-effects model. Heterogeneity among the sample collections in the meta-analysis was measured with the I2 index and the p value, which was calculated with Cochran’s Q test. For the rare variants, we restricted to the genotyped variants only, association and meta analyses were carried using PLINK with the Cochran–Mantel–Haenszel method.
We conducted secondary analyses on a subset of the more severely affected cases with ANC < 1,500 mm−3 (CIN). Controls were still those who received clozapine without developing WBC < 4,000 mm−3. Those who had a test result with ANC ≥ 1,500 mm−3 and WBC < 4,000 mm−3 were excluded from the secondary analysis. All analyses performed were consistent with methods described above for CIL analysis.
We performed the trans-ancestry meta-analysis of our CIN results with data from the CIAG study11,12 for common variants. We adopted a fixed-effects model implemented in RICOPILI20 for the analysis.
We annotated the genome-wide significant variants and their proxies (r2 ≥ 0.8 in EAS of 1000 Genomes Project Releases Phase 3) to explore putative causal variants and genes underlying these association signals. We also searched for evidence that the common variants were associated with expression of a particular gene in several expression quantitative trait locus (eQTL) datasets: the Genotype-Tissue Expression (GTEx, v7)21, immune cells22 and lymphoblastoid cell lines (LCLs)23. Potential functional effects of the sentinel variants were predicted by the GWAVA (Genome Wide Annotation of VAriants)24, a web-based tool to prioritize the functional variations based on a wide range of annotations of noncoding elements (such as ENCODE/GENCODE, evolutionary conservation and GC-content).
Dual-luciferase reporter assays
An in vitro functional assay was used to investigate allele-dependent effects of the prioritized variants by GWAVA on gene transcription. Experiments were performed with human embryonic kidney (HEK293) cell line. Luciferase reporter constructs were cloned by using the pGL3-promoter vector (Promega, Madison, WI, USA) as a backbone. For each variant, two DNA fragments containing risk or nonrisk allele were produced and all constructs were verified via Sanger sequencing. The pGL3 reporter, risk and nonrisk vectors were transfected into HEK293 cells. Luciferase activities were assessed at 48-h post-transfection according to the manufacturer’s protocols (Promega). The activity of firefly luciferase was normalized to that of Renilla luciferase to control for variations in the transfection efficiency between different wells. All assays were performed in three or more biological replicates in independent experiments, and two-tailed t-tests were performed to analyze statistical differences between experimental groups. Further details are provided in the Supplementary Material.
Results
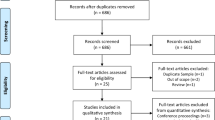
From Jan 1, 2007 to Dec 31, 2012, 1,983 patients with treatment-resistant schizophrenia, who were prescribed clozapine, were enrolled from two Mental Health Centers in China for genome-wide genotyping; 104 (5%) failed quality control (see Supplementary Material), of whom 31 did not meet array QC metrics criteria, 19 did not meet call rate criteria, 23 did not meet identity-by-descent criteria, and 31 were excluded as population outliers after a principal component analysis (see Supplementary Fig. 1). The analyzed sample thus comprised 225 CIL cases (mean [±SD] age, 39.3 ± 12.7 years), 43 CIN cases (37.7 ± 13.3 years) and 1,654 controls (36.2 ± 12.7 years)(see Supplementary Table 1). The sample was drawn from two datasets: Dataset 1: Central Chinese, 155 CIL cases (including 18 CIN cases) and 1,355 controls; Dataset 2: Southern Chinese, 70 CIL cases (including 25 CIN cases and 299 controls). 613,828 genotyped SNPs passed the predefined genotyping quality-control criteria for further analysis (see Fig. 1, Supplementary Material). After imputation and quality control, genotypes for 7,966,570 variants were available for downstream analyses. We performed genome-wide association analysis for 7,936,469 common (MAF ≥ 1%, both genotyped and imputed, see Fig. 2) and 30,101 rare (MAF < 1%, genotyped only, see Supplementary Fig. 2) variants.

QC: quality control; IBD: identity-by-descent; PCA: principal component analysis; HWE: Hardy-Weinberg equilibrium; MAF: minor allele frequency; CIL: clozapine induced leukopenia; CIN: clozapine induced neutropenia; CIAC: Clozapine Induced Agranulocytosis Consortium.

a) the CIL analysis (225 cases and 1,654 controls). b) the CIN analysis (43 cases and 1,654 controls). c) the trans-ancestry GWAS meta-analysis of CIN (43 cases and 1,654 controls) and CIAC (161 cases and 1,196 controls) datasets. -log10P values for analysis are shown.
In the association analysis of 225 CIL cases and 1,654 controls for common variants, one locus near TRAC on chromosome 14 was identified to be associated with CIL genome-wide significantly (five variants, P < 5 × 10−8, see Fig. 2), and the strongest association was at rs377360 (MAF = 18.9 %, OR = 2.19, 95% CI = 1.66–2.89, P = 2.58 × 10−8, see Table 1, Fig. 3 and Supplementary Table 2). As shown in the Q-Q plot, the genomic inflation factor (λ = 0.976) indicated good control of population stratification (see Supplementary Fig. 3). None of the genome-wide significant variants and their proxies (r2 ≥ 0.8 in EAS of 1000 Genomes Project Releases Phase 3) fell within coding regions, except rs3701 which overlaps a 3’ untranslated region (3’-UTR) of TRAC (see Supplementary Table 3). Based on our eQTL analysis, most were associated with gene expression levels of multiple genes (P < 0.05) in whole blood and immune cells (see Supplementary Table 4). The target genes included T cell receptor alpha and delta variable and constant segments coding genes (such as TRAC), and also other genes (such as DAD1), as shown in Supplementary Table 4. For the rare variants, we identified five genome-wide significant variants from two regions (see Supplementary Fig. 2), which were indexed by rs4773794 in DCT on chromosome 13 (MAF = 0.18%, OR = 12.79, P = 3.01 × 10−10), and rs10512698 in EEFSEC on chromosome 3 (MAF = 0.88%, OR = 4.79, P = 1.09 × 10−8). All these lead variants showed significant associations in both datasets (P < 0.05, see Supplementary Table 2) and no evidence for heterogeneity across datasets were observed (Het P > 0.05, see Fig. 4 and Supplementary Fig. 4).

a) rs377360. b) rs116982346. c) rs73482673. -log10P values are shown for SNPs for the region 500 kb on either side of the marker SNPs. The index SNP is shown in purple, and the r2 values of the other SNPs are indicated by color. The r2 values were established on the basis of 1000 Genomes East Asian (EAS) population data (November 2014). The genes within the relevant regions are annotated and shown as arrows.

a) rs377360. b) rs116982346. c) rs73482673. d) rs11753309. Area of the square represents the weight of each statistical sample; horizontal lines represent OR and 95% CI in two independent datasets. The diamond represents the total 95% CI estimated in meta-analysis. OR, odds ratio; CI, confidence interval.
For the CIN analysis of 43 cases and 1,654 controls, we identified three associated common variants, having MAF between 1% and 5%, that achieved genome-wide significance. These are from two regions and indexed by rs116982346 (MAF = 1.90%, OR = 19.96, P = 2.15 × 10−8) on chromosome 3 and rs73482673 (MAF = 1.10%, OR = 12.05, P = 3.30 × 10−8) on chromosome 9 (see Fig. 3 and Table 1). All these variants showed significant associations in both datasets (P < 0.05, see Supplementary Table 2) and no evidence for heterogeneity across datasets was observed (Het P > 0.05, see Fig. 4). All the genome-wide significant variants and their proxies are in intergenic regions, and the nearest coding genes are TRAT1 (rs116982346) and ELAVL2 (rs73482673)(see Fig. 4). One of them was associated with gene expression level of ELAVL2 in lymphoblastoid cell lines at P < 0.05 (see Supplementary Table 4). We also found several CIN associated rare variants (see Supplementary Fig. 2), which were indexed by rs9808117 in HECW2 on chromosome 2 (MAF = 0.64%, OR = 9.10, P = 9.82 × 10−9), rs373695 in F13A1 on chromosome 6 (MAF = 0.06%, OR = 74.83, P = 1.36 × 10−8), rs7501702 ear MFAP4 on chromosome 17 (MAF = 0.33%, OR = 14.95, P = 1.76 × 10−8) and rs8024434 in NEO1 on chromosome 15 (MAF = 0.36%, OR = 19.39, P = 4.83 × 10−8)(see Supplementary Table 2). Some proxies of the genome-wide significant variants are variants in coding regions, such as 3’-UTR in EPN2 and a missense variant in B9D1 for rs7501702, 3’-UTR in REC114 and a synonymous variant in HCN4 for rs8024434 (see Supplementary Table 3).
To explore the effects of power and heterogeneity, we performed a trans-ancestry GWAS meta-analysis of CIAC (161 cases and 1,196 controls) with CIN analysis (43 cases and 1,654 controls) for common variants. The combined GWAS meta-analysis identified rs11753309 (MAF > 0.05) near HLA-B on chromosome 6 as close to genome-wide significant (OR = 2.95, 95% CI = 2.00–4.36; P = 5.08 × 10−8, Table 1) and no evidence for heterogeneity across ancestries was observed (P > 0.05)(see Fig. 4).
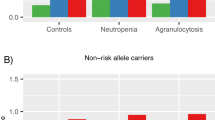
Five prioritized variants (rs377360 and rs4773794 for CIL; rs9808117, rs8024434 and rs7501702 for CIN) by their highest GWAVA scores (see Supplementary Table 5) were tested for transcriptional activity using dual-luciferase reporter assay (see Supplementary Table 6). Luciferase assays showed the DNA segment containing rs377360-T (risk for CIL) increased transcriptional activity in HEK293 cells (49% increase [SD 16.6] comparing to nonrisk allele, P < 0.001, Fig. 5). Consistent with GWAVA prediction, we found rs4773794-G (protective for CIL) was associated with an increase of transcriptional activity in HEK293 cells most significantly (50% increase [SD 27.2] comparing to risk allele, P < 0.001, Fig. 5). For the CIN associated variants, mild or little affections on transcriptional activity were observed (see Fig. 5).

a) rs377360. b) rs4773794. c) rs9808117. d) rs8024434. e) rs7501702. HEK-293 cells were transfected with luciferase reporter constructs for both alleles of each variant. Each construct was transfected five times and assayed in quintuplicate in each experiment. The luciferase activities, normalized with a cotransfected Renilla activities, were expressed by taking the normalized luciferase activity of the vector (pGL3-Promoter) to be 1. Data represent means±SDs. P values were determined by using the t-test. Significance indicators will be written above the bars: *** if p < 0·001, ** if p < 0·01, * if p < 0·05,ns otherwise.
Discussion
To our knowledge, our study is the first GWAS of CIL and CIN in individuals of Chinese ancestry and identified nine novel genome-wide significant loci. Five of the nine index variants identified in our GWAS mapped to the intron of a protein-coding gene, and all except one of the others had a protein-coding gene within 15 kb. The trans-ancestry meta-analysis of the Chinese and European ancestry identified one risk locus which was close to genome-wide significance.
Mechanisms underlying CIL and CIN have not yet been fully elucidated, although an immune reaction has been hypothesized for a long time as implicated10. We identified two T cell related genes in this study, TRAC (T cell receptor alpha constant) and TRAT1 (T Cell Receptor Associated Transmembrane Adaptor 1). The TRAC gene locating on 14q11-12 encodes the constant region of the T cell receptor (TCR) alpha chain. The alpha loci are synthesized by variable, joining, and constant segments. TCR recognizes antigen fragments as peptides bound to MHC molecules, which are important for thymic selection. Deficiency of MHC may cause abnormal immune function and result in a lack of maturation of the corresponding T cells25. Diseases associated with TRAC include TCR alpha/beta deficiency. It has been reported that TRAC mutations cause a human immunodeficiency disorder characterized by a lack of TCR αβ + T cells26. TRAT1 also plays an important role in the immune process. Among the predominantly downregulated cell surface antigens, TRAT1 along with ZAP70, LCK and TCR associated tyrosine kinases are involved in TCR activation, and were all simultaneously downregulated27.
Although we did not identify susceptibility loci reaching the level of genome-wide significance after the trans-ancestry meta-analysis, we found a near-significant risk locus for CIN near HLA-B. HLA-B is a paralog of HLA class I heavy chain, which is anchored in the membrane and plays an important role in the immune system by presenting peptides derived from the lumen of the endoplasmic reticulum. HLA-B was reported as a susceptibility gene of CIA in Caucasian population11, while HLA-B*59:01 was suggested as a risk factor for CIA in the Japanese population28. Most studies of CIL have been conducted in Caucasian populations, and it may not be possible to replicate identified variants across different races and populations for many reasons. However, associations of variants from GWAS have generally added evidence for immune-related mechanisms.
The biological activation or the conversion of stable metabolites to chemically reactive nitrogen ions could represent another causal pathway29,30,31. However, hypotheses of toxicity and immune regulation are not necessarily mutually exclusive. The combination of these reactive metabolites can result in CIL, CIN or CIA through direct toxicity or via initiation of immune mechanisms or both. Clozapine may potentially be oxidized into toxic nitrenium ions, which bind to neutrophils and cause an autoimmune reaction or induce cell death.
In this study, we also found several other compelling candidate genes. NEO1 at 15q24.1 encodes a cell surface protein composed of four N-terminal immunoglobulin-like domains, which may be involved in cell growth and differentiation and cell adhesion. Neo1 contributes to the inflammatory response and resolution mechanisms32. Genetic deletion or functional inhibition of Neo1 leads to reduced neutrophil recruitment and shortened the neutrophil lifespan by increasing apoptosis33. DCT at 13q32.1 encodes dopachrome tautomerase and participates in melanocytes pigment biosynthesis with tyrosinase and tyrosinase-related protein 134. Meanwhile it also plays a role in oxidative stress and apoptotic stimuli respondence35,36. An experiment in mice found that the spatiotemporal distribution of DCT expression was related to cortical neurogenesis during embryonic development37. Diseases associated with DCT include microphthalmia and melanoma. Previous studies have proved that a large portion of chromosome, spanning 68 Mb from 13q12 to 13q34, could be linked to susceptibility to schizophrenia38,39. EEFSEC located at 3q21.3, encoding selenocysteine-tRNA specific eukaryotic elongation factor, which is essential for the synthesis of selenoproteins40. Selenoproteins contain several proteins with different functions, some of which play important roles in antioxidant defense preservation and anti-inflammatory regulation41. Polymorphisms or mutations in EEFSEC could account for a variety of disorders, including neuropsychiatric disorders and immune dysfunctions42. We hypothesize that EEFSEC may participate in the mechanism of CIL through selenoproteins. Many selenoproteins participate in redox signaling, redox homeostasis, and antioxidant defense by the action of glutathione peroxidase43. The amount of selenoproteins and glutathione peroxidase decreases under oxidative stress conditions, and the activity of selenium-dependent glutathione peroxidase has been found to be lower in patients treated with clozapine44. Activation of clozapine and/or its metabolites can produce electrophilic nitrenium ions, which might either covalently bind to neutrophils such as glutathione to cause cell death or cause oxidative stress-induced neutrophil apoptosis45,46.
We feel it is reasonable to assume that the risks of CIL and CIN are genetically complex, involving several genes. The application of GWAS and other modern genomic technologies gives us a better understanding of the genetic basis of CIL and CIN. Our study is the first GWAS of CIL and CIN in individuals of Chinese ancestry and identified several novel genome-wide significant loci. However, there are still some limitations in our study. First, many SNPs detected were noncoding variants with unknown effects, which is a common problem with the GWAS approach. There may also be potential Winner’s curse bias in estimating the average genetic effect of a set of rare variants and identifying statistically significant associations. Therefore, further functional studies are needed to verify the related biological mechanisms. Second, since some conditions such as circadian rhythm can affect the amount of peripheral blood neutrophils in patients, other related factors should also be taken into account. Finally, an important challenge is that there is relatively rare incidence of CIN and even rarer incidence of CIA in the Chinese population. In fact, only six CIA cases met the criteria in our study, which may be due to the success of the monitoring system, since once leukopenia is detected, clozapine will be discontinued. It is therefore impossible to know which patients would continue to develop CIA. The rare incidence limits the availability of suitable patients and thus the sample size for research. Therefore, replication of larger sample sizes is critical for clinical applications.
Clozapine is the only effective medication for treatment-resistant schizophrenia, but its use is limited due to the risk of leukopenia, particularly agranulocytosis. Genetic and immune factors are likely to play an important role in determining risk for such adverse reactions. Our results provide novel and valuable understanding of the genetic and immune causes of CIL and CIN. Causal variants and related underlying molecular mechanisms need to be understood in future developments, which will be key to improving diagnostic and therapeutic capabilities in treatment-resistant schizophrenia.
Change history
30 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41398-021-01484-7
References
Kahn, R. S. et al. Schizophrenia. Nat. Rev. Dis. Prim. 2015;1:15067
Burmeister, M, McInnis, M. G. & Zollner, S. Psychiatric genetics: progress amid controversy. Nat. Rev. Genet. 2008;9:527–540.
Nothen, M. M, Nieratschker, V, Cichon, S. & Rietschel, M. New findings in the genetics of major psychoses. Dialogues Clin. Neurosci. 2010;12:85–93.
Huber, C. G, Naber, D. & Lambert, M. Incomplete remission and treatment resistance in first-episode psychosis: definition, prevalence and predictors. Expert Opin. Pharmacother. 2008;9:2027–2038.
Leucht, S. & Heres, S. Epidemiology, clinical consequences, and psychosocial treatment of nonadherence in schizophrenia. J. Clin. Psychiatry. 2006;67:3–8.
Kane, J, Honigfeld, G, Singer, J. & Meltzer, H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch. Gen. Psychiatry. 1988;45:789–796.
Griffith, R. W. & Saameli, K. Letter: Clozapine and agranulocytosis. Lancet. 1975;2:657
Idanpaan-Heikkila, J, Alhava, E, Olkinuora, M. & Palva, I. Letter: Clozapine and agranulocytosis. Lancet. 1975;2:611
Myles, N. et al. Meta-analysis examining the epidemiology of clozapine-associated neutropenia. Acta Psychiatr. Scand. 2018;138:101–109.
Legge, S. E. & Walters, J. T. Genetics of clozapine-associated neutropenia: recent advances, challenges and future perspective. Pharmacogenomics. 2019;20:279–290.
Goldstein, J. I. et al. Clozapine-induced agranulocytosis is associated with rare HLA-DQB1 and HLA-B alleles. Nat. Commun. 2014;5:4757
Legge, S. E. et al. Genome-wide common and rare variant analysis provides novel insights into clozapine-associated neutropenia. Mol. Psychiatry. 2017;22:1509
Valevski, A. et al. HLA-B38 and clozapine-induced agranulocytosis in Israeli Jewish schizophrenic patients. Eur. J. Immunogenet. 1998;25:11–13.
Legge, S. E. et al. A genome-wide association study in individuals of African ancestry reveals the importance of the Duffy-null genotype in the assessment of clozapine-related neutropenia. Mol. Psychiatry. 2019;24:328–337.
Saito, T. et al. Transethnic replication study to assess the association between clozapine-induced agranulocytosis/granulocytopenia and genes at 12p12.2 in a Japanese Population. Biol. Psychiatry. 2017;82:e9–e10.
Si, T. M. et al. Use of clozapine for the treatment of schizophrenia: findings of the 2006 research on the china psychotropic prescription studies. Clin. Psychopharmacol. Neurosci. 2012;10:99–104.
Tang, Y. L. et al. Clozapine in China. Pharmacopsychiatry. 2008;41:1–9.
de With, S. A. J, Pulit, S. L, Staal, W. G, Kahn, R. S. & Ophoff, R. A. More than 25 years of genetic studies of clozapine-induced agranulocytosis. Pharmacogenomics J. 2017;17:304–311.
Kane, J. M. et al. Clinical guidance on the identification and management of treatment-resistant schizophrenia. J Clin Psychiatry 80, 18com12123 (2019).
Lam, M. et al. RICOPILI: rapid imputation for COnsortias PIpeLIne. Bioinformatics. 2020;36:930–933.
Carithers, L. J. & Moore, H. M. The Genotype-Tissue Expression (GTEx) Project. Biopreservation Biobanking. 2015;13:307–308.
Ishigaki, K. et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat. Genet.2017;49:1120-+
Grundberg, E. et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 2012;44:1084
Ritchie, G. R. S, Dunham, I, Zeggini, E. & Flicek, P. Functional annotation of noncoding sequence variants. Nat. Methods. 2014;11:294–U351.
La Gruta, N. L, Gras, S, Daley, S. R, Thomas, P. G. & Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 2018;18:467–478.
Morgan, N. V. et al. Mutation in the TCRalpha subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRalphabeta+ T cells. J. Clin. Invest. 2011;121:695–702.
Wang, M, Windgassen, D. & Papoutsakis, E. T. Comparative analysis of transcriptional profiling of CD3+, CD4+ and CD8+ T cells identifies novel immune response players in T-cell activation. BMC Genomics. 2008;9:225
Saito, T. et al. Pharmacogenomic study of clozapine-induced agranulocytosis/granulocytopenia in a Japanese population. Biol. Psychiatry. 2016;80:636–642.
Flanagan, R. J. & Dunk, L. Haematological toxicity of drugs used in psychiatry. Hum. Psychopharmacol. 2008;23:27–41.
Maggs, J. L, Williams, D, Pirmohamed, M. & Park, B. K. The metabolic formation of reactive intermediates from clozapine, a drug associated with agranulocytosis in man. J. Pharm. Exp. Ther. 1995;275:1463–1475.
Pirmohamed, M. & Park, K. Mechanism of clozapine-induced agranulocytosis: current status of research and implications for drug development. CNS Drugs. 1997;7:139–158.
Ravichandran, K. S. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–455.
Schlegel, M. et al. Inhibition of neogenin fosters resolution of inflammation and tissue regeneration. J. Clin. Invest. 2018;128:4711–4726.
Abdel-Malek, Z. et al. Contribution of melanogenic proteins to the heterogeneous pigmentation of human melanocytes. J. Cell Sci. 1993;106:1323–1331.
Ainger, S. A. et al. DCT protects human melanocytic cells from UVR and ROS damage and increases cell viability. Exp. Dermatol. 2014;23:916–921.
Sendoel, A, Kohler, I, Fellmann, C, Lowe, S. W. & Hengartner, M. O. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature. 2010;465:577–583.
Jiao, Z. et al. Dopachrome tautomerase (Dct) regulates neural progenitor cell proliferation. Dev. Biol. 2006;296:396–408.
Brzustowicz, L. M. et al. Linkage of familial schizophrenia to chromosome 13q32. Am. J. Hum. Genet. 1999;65:1096–1103.
Detera-Wadleigh, S. D. & McMahon, F. J. G72/G30 in schizophrenia and bipolar disorder: review and meta-analysis. Biol. Psychiatry. 2006;60:106–114.
Dobosz-Bartoszek, M. et al. Crystal structures of the human elongation factor eEFSec suggest a non-canonical mechanism for selenocysteine incorporation. Nat. Commun. 2016;7:12941
Labunskyy, V. M, Hatfield, D. L. & Gladyshev, V. N. Selenoproteins: molecular pathways and physiological roles. Physiol. Rev. 2014;94:739–777.
Hornig, M. The role of microbes and autoimmunity in the pathogenesis of neuropsychiatric illness. Curr. Opin. Rheumatol. 2013;25:488–795.
Touat-Hamici, Z, Legrain, Y, Bulteau, A. L. & Chavatte, L. Selective up-regulation of human selenoproteins in response to oxidative stress. J. Biol. Chem. 2014;289:14750–14761.
Miljevic, C. et al. Lipid status, anti-oxidant enzyme defence and haemoglobin content in the blood of long-term clozapine-treated schizophrenic patients. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2010;34:303–307.
Husain, Z. et al. Increased FasL expression correlates with apoptotic changes in granulocytes cultured with oxidized clozapine. Toxicol. Appl Pharm. 2006;214:326–334.
Ip, J, Wilson, J. X. & Uetrecht, J. P. Testing the hypothesis that vitamin C deficiency is a risk factor for clozapine-induced agranulocytosis using guinea pigs and ODS rats. Chem. Res Toxicol. 2008;21:869–873.
Acknowledgements
This work was supported by the National Key R&D Program of China [2016YFC1306900, 2017YFC0908105, 2019YFA0905400], the Natural Science Foundation of China [U1804284, 32070679, 82071500, 81960258, 81871055, 81871051, 81701321], Shanghai Key Laboratory of Psychotic Disorders [13dz2260500], Shanghai Municipal Science and Technology Major Project [2017SHZDZX01, 2018SHZDZX05], Shanghai Municipal Commission of Science and Technology [18DZ2260200], the National Program for Support of Top-Notch Young Professionals to Y.S., Taishan Scholar Program of Shandong Province [tsqn201812153], Natural Science Foundation of Shandong Province [ZR2019YQ14], Shanghai Hospital Development Center [SHDC12017X12, SHDC12016115], Shanghai Mental Health Centre Clinical Research Center Special Project for Big Data Analysis [CRC2018DSJ01-4], Shanghai Youth Top-notch Talent Support Program to J.C., Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support, Innovative research team of high-level local universities in Shanghai and Shanghai Clinical Research Center for Mental Health [19MC1911100], R.S. is part-funded by: i) the National Institute for Health Research (NIHR) Biomedical Research Centre at the South London and Maudsley NHS Foundation Trust and King’s College London; ii) a Medical Research Council (MRC) Mental Health Data Pathfinder Award to King’s College London; iii) an NIHR Senior Investigator Award; iv) the National Institute for Health Research (NIHR) Applied Research Collaboration South London (NIHR ARC South London) at King’s College Hospital NHS Foundation Trust.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, J., Yang, P., Zhang, Q. et al. Genetic risk of clozapine-induced leukopenia and neutropenia: a genome-wide association study. Transl Psychiatry 11, 343 (2021). https://doi.org/10.1038/s41398-021-01470-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01470-z
This article is cited by
-
Sex-based influential factors for dental caries in patients with schizophrenia
BMC Psychiatry (2023)
