
Abstract
Carbonyl stress, a specific form of oxidative stress, is reported to be involved in the pathophysiology of schizophrenia; however, little is known regarding the underlying mechanism. Here, we found that disruption of GLO1, the gene encoding a major catabolic enzyme scavenging the carbonyl group, increases vulnerability to external carbonyl stress, leading to abnormal phenotypes in human induced pluripotent stem cells (hiPSCs). The viability of GLO1 knockout (KO)-hiPSCs decreased and activity of caspase-3 was increased upon addition of methylglyoxal (MGO), a reactive carbonyl compound. In the GLO1 KO-hiPSC-derived neurons, MGO administration impaired neurite extension and cell migration. Further, accumulation of methylglyoxal-derived hydroimidazolone (MG-H1; a derivative of MGO)-modified proteins was detected in isolated mitochondria. Mitochondrial dysfunction, including diminished membrane potential and dampened respiratory function, was observed in the GLO1 KO-hiPSCs and derived neurons after addition of MGO and hence might be the mechanism underlying the effects of carbonyl stress. The susceptibility to MGO was partially rescued by the administration of pyridoxamine, a carbonyl scavenger. Our observations can be used for designing an intervention strategy for diseases, particularly those induced by enhanced carbonyl stress or oxidative stress.
Similar content being viewed by others

Introduction
Schizophrenia is a debilitating psychiatric disorder, which is etiologically heterogeneous1,2,3. Recently, carbonyl stress, a type of oxidative stress, has been reported to be elevated in a subset of cases of schizophrenia4,5. Carbonyl stress is a condition associated with prolific accumulation of advanced glycation end products (AGEs)6. AGEs are damaged proteins, amino acid residues of which are irreversibly modified by reactive carbonyl compounds (RCOs)7,8. This reaction is mediated by the close interaction between free radical oxidation and glycation9. AGE accumulation is known to elicit activation of inflammatory signaling via the receptors for AGEs10,11.
Organisms possess an anti-glycation defense mechanism for eliminating RCOs and suppressing the accumulation of AGEs (Fig. 1A)8,12,13. This defense mechanism, named “glyoxalase cycle”, converts RCOs to harmless aldonates, including d-lactate and glycolate14. The major enzymatic components of this system are glyoxalase I (GLO1) and glyoxalase II (GLO2)13,15. Among them, GLO1 is the rate-limiting enzyme12,15, and inhibition of GLO1 critically increases the accumulation of RCOs16,17,18. Notably, downregulated expression of GLO1 has been reported in a wide range of neuropsychiatric disorders, including autism spectrum disorder, major depressive disorder, depressive state of bipolar disorder, and Alzheimer’s disease19,20,21,22. But the molecular mechanism of GLO1 deficiency in pathogenesis of psychiatric disorders including schizophrenia remains largely unknown.

A Schematic presentation of the biosynthesis of AGEs (left part) and glyoxalase cycle (right part). Reactive carbonyl compounds (RCOs) are produced from glucose and lipids to form AGEs. Accumulated AGEs induce cell toxicity. Glyoxalase I (GLO1) plays a pivotal role in removing RCOs via the glyoxalase cycle. B Western blot of WT- and GLO1 KO-hiPSCs using an antibody against GLO1. C Cell viability analysis in WT- and GLO1 KO-hiPSCs after treatment with 200 µM GO and 200 µM MGO for 24 h. D Analysis of the viability of cells treated with 0, 50, 100, and 200 µM MGO for 24 h. GLO1 KO-hiPSCs showed reduction in cell viability in a concentration-dependent manner. E Analysis of apoptosis in WT- and GLO1 KO-hiPSCs after exposure to 100 µM MGO for 0, 3, and 6 h. Treated GLO1 KO-hiPSCs showed increased caspase activity in a time-dependent manner. F Rescue assay for cell viability after treatment with 100 µM MGO mediated by co-incubation with pyridoxamine (0, 100, 200, and 500 µM) for 24 h. G Rescue assay for apoptosis after exposure to 100 µM MGO mediated by co-incubation with 500 µM pyridoxamine for 6 h. Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; two-way ANOVA, followed by Tukey’s multiple comparison test.
In this study, we aimed to investigate the effects of varying the load of carbonyl stress in neurodevelopment with regard to schizophrenia pathophysiology23,24,25 using GLO1-knockout (KO) human induced-pluripotent stem cells (hiPSCs) and GLO1-KO hiPSC-derived neurons.
Materials and methods
Establishment of hiPSCs and deletion of GLO1
We established hiPSCs from a healthy volunteer (47-year-old male, named THC-10). T cells from peripheral blood were cultured on dishes coated with anti-human CD3 antibody (1:100, BD, Franklin Lakes, NJ, USA) in KBM502 medium (Kohjin Bio, Saitama, Japan). We confirmed that the subject did not harbor any missense variants of the genes relevant for the carbonyl stress pathway, namely, GLO1, GLO2, and AGER (encoding AGE specific receptor), as described previously26. T cells were reprogrammed to hiPSCs using CytoTune®-iPS 2.0 (ID Pharma, Tsukuba, Japan) according to the manufacturer’s instructions. The hiPSCs were maintained under feeder-free conditions using the Cellartis® DEF-CS™ 500 culture system (Takara Bio, Shiga, Japan). GLO1 deletion was generated via genome editing using the CRISPR-Cas9 system. Briefly, cultured hiPSCs were transduced with single guide RNA (5′-CTTGGTACTGGGGTCCGCGTCGG-3′), Guide-it Recombinant Cas9 protein (Takara Bio), and puromycin-resistant vector pDonor-D4 (GeneCopoeia, Rockville, MD, USA) via electroporation (Neon® Transfection system, Thermo Fisher, Waltham, MA, USA). After 24 h, puromycin (Nacalai Tesque, Kyoto, Japan) selection was performed for 24 h. Four wild type (WT) and four GLO1 KO clones were established via single-cell cloning (Fig. S1). The absence of GLO1 at the protein level was confirmed via Western blotting (Fig. 1B) using anti-rabbit GLO1 antibody (Lab-made27). A web-based tool, COSMID (https://crispr.bme.gatech.edu), was used to identify off-target sites, and two possible sites on chromosomes 5 and 7 were identified28. The genomic integrity of these regions was confirmed using the Guide-it mutation detection kit (Takara Bio). All tested clones were negative for mycoplasma. All experimental procedures were approved by the ethics committee of RIKEN (approval no. Wako 3 2019-2). All methods were conducted according to the principles set out in the WMA Declaration of Helsinki and the NIH Belmont Report. The subject provided informed and written consent for participation in the study.
Induction of carbonyl stress
Reactive carbonyl compounds were administered extrinsically to the culture medium. Cells were incubated with a major RCO methylglyoxal (MGO; Sigma Aldrich, MO, USA) or a less reactive RCO29 glyoxal (GO; FujiFilm Wako Pure Chemical, Osaka, Japan) for 24 h, unless otherwise noted, before performing the experiments according to previous reports30,31,32.
Cell viability assay
Following incubation of hiPSCs with reactive carbonyl compounds for 24 h, the medium was removed and the cells were stained with Hoechst 33342 (1:2000, Thermo Fisher) in Dulbecco’s phosphate-buffered saline (DPBS). After 5 min of incubation, the staining solution was replaced, and the cells were washed with DPBS. Images were acquired via tile scanning using the Axio Observer (Zeiss, Jena, Germany). Cell number was automatically counted using the Zen software (Zeiss). We performed rescue experiments using pyridoxamine (Sigma Aldrich)33. For hiPSC-derived neurons, cell viability was manually analyzed using the trypan blue exclusion test34. Briefly, the cell suspension was obtained from the cells treated with MGO for 24 h. Suspensions were gently mixed with 0.4% (w/v) trypan blue solution to detect viable cells. The number of live cells was counted using a hemocytometer (Wakenbtech, Kyoto, Japan).
Detection of caspase-3 activity
Apoptosis was evaluated using the APOPCYTO caspase-3 fluorometric assay kit (Medical and Biological Laboratories, Nagoya, Japan) according to the manufacturer’s instructions. Briefly, whole cell lysates were extracted after MGO treatment (0−6 h, 100 µM). These lysates were incubated with DEVD-AMC (7-amino-4-methylcoumarin) in a 96-well microplate for 60 min. DEVD-AMC is a synthetic substrate labeled with four amino acid sequences, which can be cleaved by active caspase-3 to generate free fluorescent AMC. The fluorescence was analyzed using a Varioskan microplate reader (Thermo Fisher). Data were normalized to protein levels determined using the Pierce 660 nm protein assay kit (Thermo Fisher).
Differentiation of hiPSCs to neuronal cell lineage
Neuronal induction was performed as previously described with slight modifications27,35,36. Feeder-free hiPSCs were treated with 2 µM SB431542 (Sigma Aldrich), 3 µM CHIR99021 (FujiFilm Wako Pure Chemical), and 3 µM dorsomorphin (FujiFilm Wako Pure Chemical) in DEF-CS for 5−7 days. Treated hiPSCs were dissociated into single cells using TrypLE Select (Thermo Fisher). Single cells were plated at a density of 1 × 104 cells/mL in uncoated T75 easy flasks (Thermo Fisher) in media hormone mix (MHM) supplemented with 2% B27 supplement (Thermo Fisher), 20 ng/mL bFGF (Peprotech, Cranbury, NJ, USA), 10 ng/mL hLIF (Nacalai Tesque), 10 nM Y-27632 (FujiFilm Wako Pure Chemical), 2 µM SB431542, and 3 µM CHIR99021. The flasks were incubated in a hypoxic and humidified atmosphere (4% O2 and 5% CO2) for 7−10 days. The medium was changed after 5−7 days. Neurospheres, which mainly consist neural stem/progenitor cells35, were formed with passage of time and had been dissociated into single cells after treatment with TrypLE Select27,35. The digestion was quenched with a 0.02% trypsin inhibitor (Sigma Aldrich). The cell suspension was plated onto vessels coated with poly-l-ornithine (15 µg/mL, Sigma Aldrich) and laminin (1:100, Thermo Fisher), and cultured for 3 days in MHM supplemented with 2% B27 supplement, 20 ng/mL BDNF (Peprotech), 10 ng/mL GDNF (Peprotech), 400 µM dibutyryl cAMP (Sigma Aldrich), 200 µM ascorbic acid (Sigma Aldrich), and 1% Cultureone supplement (Thermo Fisher).
Neurite outgrowth assay
Approximately 20−30 neurospheres were allowed to adhere to coated 35 mm glass bottom dishes (Matsunami glass, Osaka, Japan) containing neural induction medium as stated above. For administering carbonyl stress, 50 µM MGO was added to the medium. The cells were fixed for immunocytochemistry after 48 h incubation35. Images were acquired using an Axio Observer (Zeiss) and analyzed using the Zen software (Zeiss). The average neurite length was measured from the edge of the cluster to the most distant end point (10 neurons per neurosphere and 10 neurospheres per given condition). The average migration distance was measured from the edge of the cluster to the nucleus of the most distant neurons (10 neurons per neurosphere and 10 neurospheres per condition).
Imaging of mitochondrial membrane potential
Mitochondrial membrane potential was evaluated through JC-1 staining using the JC-1 MitoMP detection kit (Dojindo Laboratories, Kumamoto, Japan). JC-1 changes the fluorescent color from green to red depending on the mitochondrial membrane potential. Reduction in red (healthy mitochondria, 530 nm)/green (unhealthy mitochondria, 590 nm) fluorescence intensity ratio of JC-1 indicates depolarization of mitochondria due to conversion of red fluorescent JC-1 aggregates to green fluorescent JC-1 monomers37.
Cells were incubated with JC-1 solution (final concentration: 2 µmol/L) at 37 °C for 30 min after 24 h incubation with 50 µM MGO in a black/clear bottom microplate (Corning, NY, USA). The wells were washed twice with Hank’s balanced salt solution (HBSS). Then, the medium was replaced with an imaging buffer solution attached to the kit. JC-1 fluorescence was quantified using a Varioskan microplate reader (green: 485 nm (Ex), 535 nm (Em); red: 535 nm (Ex), 595 nm (Em)). Fluorescent images were generated using an Axio Observer microscope.
ATP assay
Intracellular ATP was measured using the Cellno ATP assay reagent (TOYO B-NET, Tokyo, Japan). Briefly, cells were prepared in a white/clear bottom microplate (Thermo Fisher) and treated with 50 µM MGO for 24 h. The next day, 100 µL of assay reagent was added, and the plate was gently shaken for 1 min. After 10 min of incubation, luminescence was measured using a Varioskan microplate reader. Data were normalized to protein levels determined using the micro bicinchoninic acid (BCA) protein assay kit.
Determination of oxygen consumption rate (OCR)
Mitochondrial OCR was measured using a Seahorse XFe96 analyzer (Agilent Technologies, Santa Clara, CA, USA) as previously described with slight modifications38. Briefly, cells were plated in Seahorse XFe96 microplates (Agilent Technologies) and treated with 50 µM MGO for 24 h. On the next day, the cells were pre-incubated in assay medium (Agilent Technologies) supplemented with 25 mM glucose, 4 mM glutamine, and 1 mM sodium pyruvate at 37 °C for 1 h in a CO2-free incubator. Next, the Seahorse XF Mito stress test was performed following sequential injections of oligomycin A (1 μM) from port A, Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (0.5 μM for hiPSCs and 1 μM for neurons39) from port B, and antimycin A (1 μM) plus rotenone (1 μM) from port C. Data were normalized to protein levels determined using the micro BCA protein assay kit.
Immunocytochemistry
For immunocytochemistry, cells were fixed with 4% paraformaldehyde in DPBS. The fixed cells were permeabilized with 0.2% Triton X-100 in DPBS and blocked with 2% skim milk. The cells were incubated with mouse anti methylglyoxal-derived hydroimidazolone (MG-H1; 1:20; Cell Biolabs, San Diego, CA, USA), mouse anti-β-III tubulin (1:500; Billerica, Merck Millipore, MA, USA), and rabbit anti-NeuN antibodies (1:300; Abcam, Cambridge, UK), followed by incubation with secondary antibodies: goat anti-mouse Alexa 488, goat anti-mouse Alexa 594, and goat anti-rabbit Alexa Fluor 488 (1:500; Thermo Fisher, for all). 4′,6-Diamidino-2-phenylindole (1:1000; Roche, Basel, Switzerland) was added as a nuclear marker. Mitotracker Red CMXRos (Thermo Fisher) was used for mitochondrial labeling. After washing with DPBS, the samples were mounted in PermaFluor (Thermo Fisher) or immediately observed using Axio Observer without mounting. Images were analyzed using the Zen software.
Western blotting of isolated mitochondria
Fresh mitochondria were isolated from hiPSCs or neurons using the Qproteome mitochondria isolation kit (Qiagen, Hilden, Germany). Mitochondria were completely dissolved in ice-cold radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail (Sigma Aldrich) and phenylmethanesulfonyl fluoride (Cell Signaling, Beverly, MA, USA) as an inhibitor of serine proteases. Protein samples were loaded onto 10% sodium dodecyl sulfate polyacrylamide gels and separated via electrophoresis. Subsequently, the proteins were transferred onto polyvinylidene difluoride membranes (Merck Millipore). The membranes were blocked with 3% skim milk in Tween Tris Buffered Saline (TTBS) and incubated overnight with either mouse anti-MG-H1 (1:1000; Cell Biolabs), mouse anti-VDAC1 (1:1000; Abcam), rabbit anti-HSP60 (1:20000; Abcam) or goat anti-GAPDH antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C. After primary antibody incubation, the membranes were washed with TTBS and incubated with the following secondary antibodies for 1 h at room temperature: horse radish peroxidase (HRP)-conjugated anti-mouse IgG, HRP-conjugated anti-rabbit IgG, or HRP-conjugated anti-goat IgG (1:3000; GE Healthcare, Little Chalfont, UK, for all). Signals were detected using an Immobilon western chemiluminescent HRP substrate (Merck Millipore) and recorded with FUSION Solo S (Vilber-Lourmat, Collégien, France). Band intensity was analyzed using the ImageJ software (version 1.50i).
Statistical analysis
Statistical comparisons were performed using unpaired t-test (two tails) or two-way analysis of variance (ANOVA), followed by Tukey’s post hoc test using Prism 9 (GraphPad, San Diego, CA, USA). P-values less than 0.05 were considered significant. Data are presented as mean ± standard error of the mean (SEM).
Results
GLO1 KO-hiPSCs showed increased vulnerability to external carbonyl stress with elevation in apoptosis signaling, which was partially rescued by pyridoxamine
To examine the cytotoxicity induced by enhanced carbonyl stress, we evaluated the viabilities of WT and GLO1 KO-hiPSCs. The WT and GLO1 KO-hiPSCs showed almost similar viability under normal culture conditions (Fig. 1C). However, under enhanced carbonyl stress induced by adding 200 µM methylglyoxal (MGO) in the culture medium, the GLO1 KO-hiPSCs showed a drastic decrease in cell number after 24 h, whereas the WT hiPSCs did not show any significant decrease. The addition of 200 µM glyoxal (GO) did not affect the viability of WT and GLO1 KO cells (Fig. 1C). To determine whether lower concentrations of MGO can affect cell viability, we tested a range of MGO concentrations (0, 50, 100, and 200 µM) for 24 h. The toxicity of MGO toward GLO1-hiPSCs was dose-dependent, and the effect of MGO was evident at ≥50 µM (WT vs. GLO1 KO) or at ≥100 μM (GLO1 KO under no MGO vs. GLO1 KO under MGO) (Fig. 1D). These results suggested that GLO1 KO is sensitive to extrinsic carbonyl stress.
To investigate the molecular mechanism underlying the cytotoxic effects of MGO, we evaluated the apoptosis signaling pathways. MGO (100 µM) was administered at different time points (0, 3, and 6 h). Upon MGO treatment, the GLO1 KO-hiPSCs showed elevated apoptotic signaling, as indicated by the increase in caspase-3 activity, in a time-dependent manner (Fig. 1E).
Notably, 500 μM of pyridoxamine treatment significantly, but partially, rescued the cytotoxic damages induced by MGO (100 µM, 24 h) in GLO1 KO-hiPSCs (Fig. 1F). Furthermore, pyridoxamine (500 µM) reversed the elevation of apoptotic signaling elicited by the addition of MGO (100 µM) in the GLO1 KO-hiPSCs (Fig. 1G).
Carbonyl stress reduced neuronal cell viability and impaired neurite extension and migration
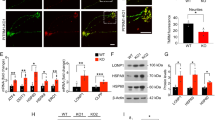
To assess the effects of carbonyl stress on neurodevelopment, we prepared neuronal cells that were differentiated from the neurospheres. Immunocytochemistry analysis showed that more than 95% of the differentiated cells were positive for the neuronal marker βIII-tubulin in both WT and GLO1 KO cells and that their morphologies did not differ (Fig. 2A). The total number of neuronal cells (neurons) differentiated from the GLO1 KO-hiPSCs was similar to that from the WT hiPSCs under normal conditions. However, the number of GLO1 KO-hiPSC-derived neurons was significantly lower than that obtained from the WT after treatment with 200 μM MGO (24 h) (Fig. 2B). These results showed that mild carbonyl stress (GLO1 KO alone) does not affect the viability of derived neurons, although severe stress (GLO1 KO plus 200 μM of MGO) can dampen cell viability in derived neurons, suggesting that GLO1 KO neurons are sensitive to extrinsic carbonyl stress.

A Representative immunofluorescent images of neurons derived from WT- and GLO1 KO-hiPSCs (scale bar: 50 µm). B Cell viability analysis in WT and GLO1 KO neurons after treatment with 0, 25, 50, 100, and 200 µM MGO for 24 h. Cell numbers were similar between WT and GLO1 KO after treatment with 0, 25, 50, and 100 µM MGO. The number of GLO1 KO neurons were significantly lower than that of WT neurons after treatment with 200 µM MGO. C, D Neurite outgrowth analysis in WT and GLO1 KO neurons after treatment with 50 µM MGO for 48 h. Dashed line in C: demarcation of clustered neuronal cell bodies; scale bar: 200 µm. Neurite lengths were significantly lower in GLO1 KO neurons after treatment with 50 µM MGO. E, F Neuronal migration analysis in WT and GLO1 KO neurons after treatment with 50 µM MGO for 48 h. Dashed line in D: demarcation of clustered neuronal cell bodies; scale bar: 100 µm. Neuronal migrations were significantly lower in GLO1 KO neurons treated with 50 µM MGO. Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001***; two-way ANOVA, followed by Tukey’s multiple comparison test.
Next, to analyze neurite extension, we prepared another type of βIII-tubulin-positive neurons, which were differentiated from the neurospheres that had been allowed to adhere to glass bottom dishes. The neurites were visualized using an anti-βIII-tubulin antibody (Fig. 2C). Under normal conditions, the neurite lengths of the WT and GLO1 KO-derived neurons were similar (Fig. 2D). Then, we determined the biological effects of minimal MGO addition (50 μM, 48 h) on the GLO1 KO-hiPSCs (Fig. 1D). After MGO addition, neurite lengths were significantly shorter in the GLO1 KO-derived neurons (432 ± 20 µm) than in the non-treated GLO1 KO-derived (592 ± 32 µm) and MGO-treated WT-derived neurons (545 ± 15 µm) (Fig. 2D). Cell nuclei were visualized using anti-NeuN antibody (Fig. 2E). The neuronal migration distance after MGO addition (50 µM, 24 h) in the GLO1 KO-derived neurons (256 ± 11 µm) was significantly lower than that in the non-treated GLO1 KO-derived (362 ± 11 µm) and MGO-treated WT-derived neurons (354 ± 5.3 µm) (Fig. 2F). These results showed that some extent of carbonyl stress (GLO1 KO plus 50 μM of MGO) can impair neurite extension and neuronal migration.
Enhanced carbonyl stress elicited mitochondrial dysfunction in GLO1 KO-hiPSCs
As carbonyl stress increased caspase-3 activities (Fig. 1E) and as mitochondria are known to play a pivotal role in apoptotic signaling40 and normal neuronal development41, we investigated the effects of enhanced carbonyl stress on mitochondrial integrity based on the fluorescence intensity ratio of JC-1. We observed that the fluorescence ratios were similar between the WT and GLO1 KO-hiPSCs under normal conditions, although the ratios for the GLO1 KO-hiPSCs showed a decreasing trend after MGO addition (50 µM, 24 h) compared to those under normal condition (P = 0.064) or those for MGO-treated WT hiPSCs (P = 0.068) (Fig. 3A, B). In the ATP assay, MGO treatment (50 µM, 24 h) significantly decreased the intracellular ATP content in the GLO1 KO-hiPSCs when compared to that in the WT-hiPSCs, whereas the treatment did not affect ATP content in the WT-hiPSCs (Fig. 3C).

A, B JC-1 staining of WT- and GLO1 KO-hiPSCs after treatment with 50 µM MGO for 24 h. Scale bar: 50 µm. C Measurement of intracellular ATP contents in WT- and GLO1 KO-hiPSCs treated with 50 µM MGO for 24 h. Data were normalized to protein concentration. D Profile of mitochondrial respiration analysis using Seahorse extracellular flux analyzer. Oligomycin, FCCP, and rotenone + antimycin A were serially administered. OCR oxygen consumption rate. E Plot of OCR in WT- and GLO1 KO-hiPSCs versus measurement time. MGO, treatment with 50 µM MGO for 24 h. Data were normalized to protein concentration. F−I Quantification of basal respiration (F), ATP production (G), maximal respiration (H), and spare capacity (I). GLO1 KO-hiPSCs showed significant reduction in respiratory function after treatment with 50 µM MGO. Data represent mean ± SEM. *P < 0.05, **P < 0.01; two-way ANOVA, followed by Tukey’s multiple comparison test.
Next, we performed a comprehensive mitochondrial respiration analysis by measuring the oxygen consumption rate (OCR) based on four parameters (basal respiration, ATP production, maximal respiration, and spare capacity; Fig. 3D). MGO treatment (50 µM, 24 h) significantly decreased mitochondrial basal respiration, ATP production, maximal respiration, and spare capacity in the GLO1 KO-hiPSCs (Fig. 3E−I), whereas no significant effects were observed in the WT hiPSCs. These results showed that a certain magnitude of carbonyl stress (GLO1 KO plus 50 µM of MGO) can impair overall mitochondrial respiratory functions.
To examine whether mitochondrial dysfunction exists in GLO1-KO hiPSCs under normal conditions, we performed the transcriptome analysis by RNA-seq (Supplemental materials and method), using previously established WT and GLO1-KO hiPSCs26,27. A total of 5098 genes were significantly dysregulated (2579 upregulated and 2519 downregulated, P < 0.01; Fig. S2A). Molecular pathways for oxidative phosphorylation, eukaryotic initiation factor 2 signaling, and mitochondrial dysfunction were significantly enriched for the downregulated genes as evidenced from the Ingenuity Pathway Analysis (IPA) (Fig. S2B). We also found that the broad downregulation of mRNA encoding oxidative phosphorylation complexes in GLO1-KO hiPSCs (Fig. S2C). Given that GLO1 depletion in hiPSCs elicited enhanced carbonyl stress26,27, these results suggest that GLO1-KO hiPSCs have mild mitochondrial dysfunction even under normal conditions, which may stem from enhanced carbonyl stress in GLO1-KO hiPSCs.
Enhanced carbonyl stress elicited mitochondrial dysfunction in GLO1 KO-hiPSC-derived neurons
As MGO treatment (50 µM, 24 h) caused mitochondrial dysfunction in the GLO1 KO-hiPSCs, we performed the same measurements in hiPSC-derived neurons. In JC-1 staining, the mitochondrial membrane potential was similar in WT and GLO1 KO under normal conditions (Fig. 4A, B). However, MGO treatment significantly decreased the mitochondrial membrane potential in the GLO1 KO-hiPSC-derived neurons (Fig. 4B). In the mitochondrial respiration assay based on the OCR, MGO treatment significantly diminished mitochondrial respiration abilities in the GLO1 KO-hiPSC-derived neurons (Fig. 4C). Compared to those in the MGO-treated WT hiPSC-derived neurons and/or GLO1 KO-hiPSC-derived neurons under normal conditions, significant reductions in basal respiration, ATP production, maximal respiration, and spare capacity were observed in the MGO-treated GLO1 KO-hiPSC-derived neurons (Fig. 4D−G).

A, B JC-1 staining of WT and GLO1 KO neurons after treatment with 50 µM MGO for 24 h. Scale bar: 100 µm (A). The ratio of red to green is an indicator of mitochondrial membrane potential (B). C Plot of oxygen consumption rate (OCR) in WT and GLO1 KO neurons using Seahorse extracellular flux analyzer after treatment with 50 µM MGO for 24 h. D−G Quantification of basal respiration (D), ATP production (E), maximal respiration (F), and spare capacity (G). GLO1 KO neurons showed significantly lower respiratory function after treatment with 50 µM MGO. Data represent mean ± SEM. *P < 0.05, **P < 0.01; two-way ANOVA, followed by Tukey’s multiple comparison test.
MG-H1, an AGE derived from MGO, accumulated in the mitochondria of GLO1 KO-hiPSCs and derived neurons under external carbonyl stress
To investigate the mechanism of mitochondrial dysfunction, we assessed the accumulation of AGEs elicited by MGO treatment. In the WT hiPSCs, MG-H1 immuno-positive staining co-localized partially with the mitochondrial marker MitoTracker® (Fig. 5A). We further isolated mitochondria from hiPSCs and hiPSC-derived neurons. The purification of isolated mitochondria was confirmed using antibodies against voltage-dependent anion channel 1 (VDAC1) and heat shock protein family D (Hsp60) member 1 (HSP60), both of which are well-established mitochondrial markers (Fig. 5B). Compared to that in non-treated KO and MGO-treated WT-hiPSCs, MG-H1 protein modification increased markedly in the GLO1 KO-hiPSCs after MGO treatment (50 µM, 24 h), (Fig. 5C, D). Compared to that in the non-treated GLO1 KO-derived neurons and the WT-derived neurons, increase in protein modification with MG-H1 was also observed in the GLO1 KO-hiPSC-derived neurons after MGO treatment (Fig. 5E). Note here that the bands for VDAC1 can be single or double depending on experimental conditions42,43,44, which may be due to protein modification(s), alternative splicing, proteolytic breakdown, or other reasons. These data suggested that mitochondrial dysfunction in hiPSCs and derived neurons was partially induced under external carbonyl stress via protein modification with MG-H1.

A Immunofluorescence images of MG-H1 (green) in mitochondria labeled with Mitotracker® (red) in WT-hiPSCs (scale bar: 20 µm). MG-H1 immuno-positive staining was partially localized in mitochondria. B Representative Western blot for VDAC1, HSP60, and GAPDH in lysates from whole cell and isolated mitochondria. C Western blotting for MG-H1 and VDAC1 in mitochondria isolated from WT- and GLO1 KO-hiPSCs after treatment with 50 µM MGO for 72 h. D Densitometric analysis of the Western blot shown in C. Significant increase in MG-H1 modification was observed in mitochondria from GLO1 KO-hiPSCs after treatment with 50 µM MGO. E Western blotting for MG-H1 and VDAC1 in mitochondria isolated from WT- and GLO1 KO-derived neurons after treatment with 50 µM MGO for 24 h. Mitochondria from GLO1 KO-derived neurons showed increase in MG-H1 modification upon MGO exposure. Data represent mean ± SEM. **P < 0.01; two-way ANOVA, followed by Tukey’s multiple comparison test.
Discussion
Accumulating evidence shows the involvement of enhanced carbonyl stress in schizophrenia pathophysiology5,45,46. This study showed that hiPSCs and derived neurons harboring deletion in GLO1, an important gene responsible for the clearance of carbonyl compounds, elicited biological dysfunction when extrinsic carbonyl stress was induced with MGO treatment. Our results suggested that a certain threshold of carbonyl stress, induced by a combination of genetic defects that increase susceptibility to oxidative stress and external factors, may play a substantial role in schizophrenia pathophysiology.
Reports have shown that MG-H1, an AGE derivative from MGO, accumulates in the mitochondria of Caenorhabditis elegans upon disruption of glyoxylase I activity47. Therefore, we immunologically evaluated hiPSCs using an antibody against MG-H1. As revealed by perturbed OCRs and accumulation of MG-H1-modified proteins, mitochondrial dysfunction due to carbonyl stress was thought to lead to decrease in cell viability, increase of caspase-3 activity, and dampening of neurite extension and cell migration. Mounting evidence points to the relationship between mitochondrial dysfunction and the pathophysiology of mental disorders48,49.
Although not investigated in this study, another possible mechanism of carbonyl stress-mediated cell damage may include the elevation of inflammatory signaling via the receptors for AGEs (RAGE)11. MG-H1 has high affinity for RAGE, resulting in the activation of nuclear factor (NF)κB signaling50. NFκB increases the release of proinflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)−1β, and IL-6, and production of reactive oxygen species51,52. These inflammatory signaling pathways lead to mitochondrial impairments, which in turn induce inflammatory responses, yielding a vicious cycle of mitochondrial failure53.
hiPSCs and differentiated neurons derived from patients with schizophrenia demonstrated several abnormal phenotypes in vitro, indicating that they can be used for addressing neurodevelopmental abnormalities associated with schizophrenia35,54. We observed that MG-H1 immuno-positive staining was co-localized with mitochondria in hiPSCs. This finding prompted us to focus on mitochondrial protein, although other cellular components were also immuno-positive. MGO is a freely membrane permeable compound and reacts with multiple proteins, lipids, and nucleic acids55. The multiple mitochondrial proteins from GLO1 KO-hiPSCs and derived neurons showed increased accumulation of MG-H1 upon MGO stress. MGO is known to modify arginine, lysine, and cysteine residues of various proteins56. Rosca et al.57 found several protein dots positive for anti-MGO-derived imidazole AGE antibody in two-dimensional electrophoresis using renal mitochondria from diabetic rats. They identified seven mitochondrial electron transport chain proteins from these immunoreactive spots. Future studies are warranted to identify MG-H1-modified proteins in hiPSCs and derived neurons.
Information regarding whether amelioration of carbonyl stress improves schizophrenia symptoms is critical. Pyridoxamine, one of the three forms of vitamin B6 (pyridoxine, pyridoxal, and pyridoxamine), is a carbonyl scavenger that inhibits protein glycation under carbonyl stress via metal ion chelation and decomposition of carbonyl intermediates58. One open-label trial in which patients with schizophrenia suffered from enhanced carbonyl stress, reported that a pyridoxamine add-on treatment was partially effective45. Our findings also showed that pyridoxamine administration could rescue dampened cell viability and apoptosis in GLO1 KO-hiPSCs under external carbonyl stress. Hence, our data suggested that therapeutic intervention with carbonyl scavengers during neural development provides a beneficial prevention method for those who have multiple inherent risk factors relevant to oxidative stress. In addition, our data indicated that mitochondria may be a therapeutic target for those who have deleterious carbonyl stress burden.
Finally, whether enhanced carbonyl stress is a cause contributing to the development of schizophrenia or is a secondary state-dependent measure following deterioration and/or relapse of the schizophrenic process accompanied by neuroinflammation remains unclear46,59,60. We showed here that at least genetic defects in GLO1 that are reported in schizophrenia5,61,62 may result in vulnerability to extrinsic oxidative stress and abnormal neurodevelopment. Further careful analyses are necessary to clarify the causal and mechanistic relationship between carbonyl stress and schizophrenia pathology.
References
Balan, S., Toyoshima, M. & Yoshikawa, T. Contribution of induced pluripotent stem cell technologies to the understanding of cellular phenotypes in schizophrenia. Neurobiol. Dis. 131, 104162 (2019).
van Os, J., Kenis, G. & Rutten, B. P. The environment and schizophrenia. Nature 468, 203–212 (2010).
Austin, J. C. & Honer, W. G. Psychiatric genetic counselling for parents of individuals affected with psychotic disorders: a pilot study. Early Inter. Psychiatry 2, 80–89 (2008).
Miyashita, M. et al. Clinical features of schizophrenia with enhanced carbonyl stress. Schizophr. Bull. 40, 1040–1046 (2014).
Arai, M. et al. Enhanced carbonyl stress in a subpopulation of schizophrenia. Arch. Gen. Psychiatry 67, 589–597 (2010).
Lyons, T. J. & Jenkins, A. J. Glycation, oxidation, and lipoxidation in the development of the complications of diabetes: a carbonyl stress hypothesis. Diabetes Rev. 5, 365–391 (1997).
Miyata, T., Saito, A., Kurokawa, K. & de Strihou, Cv. Y. Advanced glycation and lipoxidation end products: reactive carbonyl compounds‐related uraemic toxicity. Nephrol. Dialysis Transplant. 16, 8–11 (2001).
Mostafa, A. A. et al. Plasma protein advanced glycation end products, carboxymethyl cysteine, and carboxyethyl cysteine, are elevated and related to nephropathy in patients with diabetes. Mol. Cell. Biochem. 302, 35–42 (2007).
Traverso, N. et al. Mutual interaction between glycation and oxidation during non-enzymatic protein modification. Biochim. et. Biophys. Acta 1336, 409–418 (1997).
Sharma, C., Kaur, A., Thind, S. S., Singh, B. & Raina, S. Advanced glycation End-products (AGEs): an emerging concern for processed food industries. J. Food Sci. Technol. 52, 7561–7576 (2015).
Roorda, M. M. Therapeutic interventions against accumulation of advanced glycation end products (AGEs). Glycative Stress Res. 4, 132–143 (2017).
Gaffney, D. O. et al. Non-enzymatic lysine lactoylation of glycolytic enzymes. Cell Chem. Biol. 27, 206–213.e206 (2020).
Szwergold, B. A hypothesis: moderate consumption of alcohol contributes to lower prevalence of type 2 diabetes due to the scavenging of alpha-dicarbonyls by dietary polyphenols. Rejuvenation Res. 21, 389–404 (2018).
Xue, M., Rabbani, N. & Thornalley, P. J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 22, 293–301 (2011).
Sousa Silva, M. et al. The glyoxalase pathway: the first hundred years… and beyond. Biochem. J. 453, 1–15 (2013).
Shinohara, M. et al. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 101, 1142–1147 (1998).
Thornalley, P. J. Glyoxalase I−structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 31, 1343–1348 (2003).
Kawatani, M. et al. The identification of an osteoclastogenesis inhibitor through the inhibition of glyoxalase I. Proc. Natl Acad. Sci. USA 105, 11691 (2008).
Fujimoto, M. et al. Reduced expression of glyoxalase-1 mRNA in mood disorder patients. Neurosci. Lett. 438, 196–199 (2008).
Lv, H. et al. 20S proteasome and glyoxalase 1 activities decrease in erythrocytes derived from Alzheimer’s disease patients. Neural Regen. Res. 15, 178–183 (2020).
Kovač, J., Podkrajšek, K. T., Lukšič, M. M. & Battelino, T. Weak association of glyoxalase 1 (GLO1) variants with autism spectrum disorder. Eur. Child Adolesc. Psychiatry 24, 75–82 (2015).
Gabriele, S. et al. The GLO1 C332 (Ala111) allele confers autism vulnerability: family-based genetic association and functional correlates. J. Psychiatr. Res. 59, 108–116 (2014).
Owen, M. J., O’Donovan, M. C., Thapar, A. & Craddock, N. Neurodevelopmental hypothesis of schizophrenia. Br. J. Psychiatry 198, 173–175 (2011).
Weinberger, D. R. Future of days past: neurodevelopment and schizophrenia. Schizophrenia Bull. 43, 1164–1168 (2017).
Birnbaum, R. & Weinberger, D. R. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 18, 727–740 (2017).
Ohnishi, T. et al. Investigation of betaine as a novel psychotherapeutic for schizophrenia. EBioMedicine 45, 432–446 (2019).
Toyoshima, M. et al. Enhanced carbonyl stress induces irreversible multimerization of CRMP2 in schizophrenia pathogenesis. Life Sci. Alliance 2, e201900478 (2019).
Cradick, T. J., Qiu, P., Lee, C. M., Fine, E. J. & Bao, G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids 3, e214 (2014).
Pampati, P. K., Suravajjala, S. & Dain, J. A. Monitoring nonenzymatic glycation of human immunoglobulin G by methylglyoxal and glyoxal: a spectroscopic study. Anal. Biochem. 408, 59–63 (2011).
Chen, Y.-J. et al. Tenuigenin protects cultured hippocampal neurons against methylglyoxal-induced neurotoxicity. Eur. J. Pharmacol. 645, 1–8 (2010).
Di Loreto, S. et al. Methylglyoxal induces oxidative stress-dependent cell injury and up-regulation of interleukin-1β and nerve growth factor in cultured hippocampal neuronal cells. Brain Res. 1006, 157–167 (2004).
Kikuchi, S. et al. Neurotoxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. J. Neurosci. Res. 57, 280–289 (1999).
Nagai, R., Murray, D. B., Metz, T. O. & Baynes, J. W. Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 61, 549–559 (2012).
Giordano, G., Hong, S., Faustman, E. M. & Costa, L. G. in In Vitro Neurotoxicology: Methods and Protocols (eds Costa, L. G., Giordano, G. & Guizzetti, M.) (Humana Press, 2011).
Toyoshima, M. et al. Analysis of induced pluripotent stem cells carrying 22q11.2 deletion. Transl. Psychiatry 6, e934 (2016).
Fujimori, K. et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589 (2018).
Sivandzade, F., Bhalerao, A. & Cucullo, L. Analysis of the mitochondrial membrane potential using the cationic JC-1 dye as a sensitive fluorescent probe. Bio-Protocol 9, e3128 (2019).
Futamura, Y. et al. Bioenergetic and proteomic profiling to screen small molecule inhibitors that target cancer metabolisms. Biochim. Biophys. Acta Proteins Proteom. 1867, 28–37 (2019).
Lorenz, C. et al. Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell 20, 659–674 (2017). e659.
Wang, C. & Youle, R. J. The role of mitochondria in apoptosis*. Annu Rev. Genet. 43, 95–118 (2009).
Rangaraju, V. et al. Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J. Neurosci. 39, 8200–8208 (2019).
Doki, T. et al. Mitochondrial localization of PABPN1 in oculopharyngeal muscular dystrophy. Lab. Investig. 99, 1728–1740 (2019).
Habarou, F. et al. Biallelic mutations in LIPT2 cause a mitochondrial lipoylation defect associated with severe neonatal encephalopathy. Am. J. Hum. Genet. 101, 283–290 (2017).
Rodríguez, L. R. et al. Oxidative stress modulates rearrangement of endoplasmic reticulum-mitochondria contacts and calcium dysregulation in a Friedreich’s ataxia model. Redox Biol. 37, 101762 (2020).
Itokawa, M. et al. Pyridoxamine: a novel treatment for schizophrenia with enhanced carbonyl stress. Psychiatry Clin. Neurosci. 72, 35–44 (2018).
Son, S. et al. Enhanced carbonyl stress and disrupted white matter integrity in schizophrenia. Schizophr. Res. https://doi.org/10.1016/j.schres.2020.08.007. (2020).
Morcos, M. et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 7, 260–269 (2008).
Ide, M. et al. Excess hydrogen sulfide and polysulfides production underlies a schizophrenia pathophysiology. EMBO Mol. Med. 11, e10695 (2019).
Rajasekaran, A., Venkatasubramanian, G., Berk, M. & Debnath, M. Mitochondrial dysfunction in schizophrenia: pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 48, 10–21 (2015).
Xue, J. et al. The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 53, 3327–3335 (2014).
Cai, W. et al. Uric acid induces endothelial dysfunction by activating the HMGB1/RAGE signaling pathway. BioMed. Res. Int. 2017, 1–11 (2017).
Murphy, MichaelP. Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 18, 145–146 (2013).
Malhotra, J. D. & Kaufman, R. J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal 9, 2277–2293 (2007).
Brennand, K. J. et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225 (2011).
Allaman, I., Bélanger, M. & Magistretti, P. J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 9, 23–23 (2015).
Polykretis, P., Luchinat, E., Boscaro, F. & Banci, L. Methylglyoxal interaction with superoxide dismutase 1. Redox Biol. 30, 101421 (2020).
Rosca, M. G. et al. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am. J. Physiol. Ren. Physiol. 289, F420–F430 (2005).
Voziyan, P. A., Metz, T. O., Baynes, J. W. & Hudson, B. G. A post-Amadori inhibitor pyridoxamine also inhibits chemical modification of proteins by scavenging carbonyl intermediates of carbohydrate and lipid degradation. J. Biol. Chem. 277, 3397–3403 (2002).
Ohnuma, T. et al. Carbonyl stress and microinflammation-related molecules as potential biomarkers in schizophrenia. Front. Psychiatry 9, 82 (2018).
Byun, K. et al. Advanced glycation end-products produced systemically and by macrophages: a common contributor to inflammation and degenerative diseases. Pharm. Ther. 177, 44–55 (2017).
Toyosima, M. et al. Schizophrenia with the 22q11.2 deletion and additional genetic defects: case history. Br. J. Psychiatry 199, 245–246 (2011).
Balan, S. et al. 22q11.2 deletion carriers and schizophrenia-associated novel variants. Br. J. Psychiatry 204, 398–399 (2014).
Acknowledgements
This research was supported by the Japan Agency of Medical Research and Development (AMED) under grant numbers JP20dm0107083 (to T.Y.), and by the Japan Society for the Promotion of Science (JSPS) KAKENHI grant number 18H04042 (to T.Y), 20K21743 (to Y.O), 19H04026 (to Y.O) and 18K07616 (to M.T.). This study was also supported by the RIKEN Junior Research Associate Program (to T.H.). We thank the Support Unit for Bio-Material Analysis and Animal Resources Development, Research Resources Division, RIKEN Center for Brain Science, for animal care and sequencing services.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hara, T., Toyoshima, M., Hisano, Y. et al. Glyoxalase I disruption and external carbonyl stress impair mitochondrial function in human induced pluripotent stem cells and derived neurons. Transl Psychiatry 11, 275 (2021). https://doi.org/10.1038/s41398-021-01392-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01392-w
