
Abstract
Alzheimer’s disease (AD) has no cure, but early detection and risk prediction could allow earlier intervention. Genetic risk factors may differ between ethnic populations. To discover novel susceptibility loci of AD in the Japanese population, we conducted a genome-wide association study (GWAS) with 3962 AD cases and 4074 controls. Out of 4,852,957 genetic markers that passed stringent quality control filters, 134 in nine loci, including APOE and SORL1, were convincingly associated with AD. Lead SNPs located in seven novel loci were genotyped in an independent Japanese AD case–control cohort. The novel locus FAM47E reached genome-wide significance in a meta-analysis of association results. This is the first report associating the FAM47E locus with AD in the Japanese population. A trans-ethnic meta-analysis combining the results of the Japanese data sets with summary statistics from stage 1 data of the International Genomics of Alzheimer’s Project identified an additional novel susceptibility locus in OR2B2. Our data highlight the importance of performing GWAS in non-European populations.
Similar content being viewed by others

Introduction
The number of people with dementia is rapidly increasing and is estimated that it will reach 135 million worldwide by 20501. Alzheimer’s disease (AD) is the most common cause of dementia among the elderly and the most frequent multifactorial neurodegenerative disease2,3. To date, there are no curative treatments for patients who already have AD, and available treatments are only able to delay the progression of the disease4. Thus, the disease has become a major global public health issue.
The majority of AD cases are sporadic and diagnosed in people over 65 years of age (late-onset AD: LOAD). LOAD is a heterogeneous disorder with complex interactions between genetic and environmental risk factors, and it is influenced by multiple common variants with low effect sizes5,6. Estimates of genetic heritability range between 60 and 80%7. A large number of genetic factors contribute to the etiopathogenesis and progression of AD. Amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) have been identified as causes of autosomal-dominant AD8. The ε4 polymorphism in the protein encoded by the apolipoprotein E (APOE) gene, located on chromosome 19, is considered to be the strongest genetic risk factor for LOAD9. However, the APOE ε4 effect only accounts for 27.3% of the overall heritability10, and a large proportion of the heritability remains unexplained.
Many attempts have been made to identify additional genetic risk factors. The development of high-throughput genotyping and massively parallel next-generation sequencing has enabled genome-wide association studies (GWAS), which have successfully identified several genetic risk factors that affect the development of AD5,6,11,12. These findings are expected not only to explain some of the heritability of AD, but also to contribute to the understanding of the underlying etiologic, biologic, and pathologic mechanisms of AD.
Trans-ethnic meta-analyses of GWAS data improve the power to detect more common risk variants by increasing the sample size. Many susceptibility loci for AD have been reported, especially for American and European populations6,13. Some ethnicity-specific genetic risk factors have also been reported. Simino et al.14 reported ethnicity-specific genes associated with differences in plasma amyloid-β (Aβ) concentrations between African Americans and European Americans. We also previously reported an ethnicity-specific (Japanese) rare variant in SHRAPIN that is associated with increased risk of AD15. These findings should be addressed in further investigation using ethnicity-specific data sets with larger sample sizes, but there are few reports of large-scale GWAS data for AD in a Japanese population16,17.
Here, we comprehensively examined the genetic architecture of AD based on Japanese GWAS data obtained from a large sample. We discovered FAM47E, a novel ethnicity-specific locus with a significant genome-wide association for AD in the Japanese population. We also found six significant quantitative trait loci (QTLs) correlated with FAM47E SNP genotypes, as well as two AD susceptibility candidates (RECK and TIMP3) through gene-set analysis. Furthermore, we identified an additional novel AD susceptibility locus (OR2B2) through a trans-ethnic meta-analysis. Our data highlight the importance of performing GWAS in ethnicity-specific population data.
Materials and methods
Subjects
The discovery stage of the study consisted of 3962 AD cases and 4074 controls (2974 AD cases and 3096 controls from the National Center for Geriatrics and Gerontology [NCGG biobank]; 988 AD cases and 978 controls from Niigata University16). The control subjects with normal cognitive function that had subjective cognitive complaints, but normal cognition on the neuropsychological assessment, were categorized as normal controls. The case subjects were diagnosed with a probable or possible AD based on the criteria of the National Institute on Aging Alzheimer’s Association workgroups18,19. The average age was 74.6 years (standard deviation [SD] = 7.5 years) and the female-to-mate ratio was 1.51:1. The independent replication samples were composed of 1216 AD cases and 2446 controls (530 AD cases and 2446 controls from the NCGG biobank, 686 AD cases from the BioBank Japan Project20,21). The average age was 75.5 years (SD = 6.5 years) and the female-to-male ratio was 1.37:1. Genomic DNA was extracted from peripheral blood leukocytes by standard protocols using a Maxwell RSC Instrument (Maxwell RSC Buffy Coat DNA Kit, Promega, USA). All subjects were of Japanese origin and provided informed consent in writing. This study was approved by the ethics committee of each institution.
Genotyping and quality control in the GWAS
Genome-wide genotyping in the discovery stage was performed by using the Affymetrix Japonica Array22 for the NCGG subjects and Affymetix GeneChip 6.0 microarrays for Niigata subjects16. Genotype imputation was conducted by using IMPUTE223 with the 3.5 K Japanese reference panel developed by Tohoku Medical Megabank Organization (https://www.megabank.tohoku.ac.jp/english/) for NCGG subjects and the 1000 Genomes Project reference panel (1000 Genomes Phase 324) for Niigata subjects. We used imputed variants with an INFO score ≥0.4 in the association analysis. Quality control (QC) was performed in each dataset separately after imputation using PLINK software25. We first applied QC filters to the subjects: (1) sex inconsistencies (--check-sex), (2) inbreeding coefficient (--het 0.1), (3) genotype missingness (--missing 0.05), (4) kinship coefficient (--genome 0.2), and (5) exclusion of outliers from the clusters of East Asian populations in a principal component analysis that was conducted together with 1000 Genomes Phase 3 data. We next applied QC filters to the genetic markers (SNPs and Indels): (1) genotyping efficiency or call rate (--geno 0.95), (2) minor allele frequency (--freq 0.001), and (3) Hardy–Weinberg equilibrium (--hwe 0.001). The common autosomal variants with the same effect alleles that passed these QC criteria were assessed with a logistic regression model, adjusting for sex and age with PLINK software (--logistic)25. We also conducted a meta-analysis combining the GWAS summary statistics of the NCGG and Niigata GWAS data for loci detected in the logistic regression model, which was implemented in METASOFT26. The Meta-P values were calculated based on Han and Eskin’s modified random effects model (RE-HE), which is optimized to detect associations under effect heterogeneity26.
Replication study
Of the loci that satisfied P < 5.0 × 10−6 in the GWAS, lead markers were genotyped in the replication study. SNP genotyping was performed with the multiplex PCR-based Invader assay (Third Wave Technologies, Madison, WI). Association analysis in the replication study was performed by using a logistic regression model adjusted for sex and age with PLINK software (--logistic)25. The combined analysis of the GWAS and replication study was verified with the logistic regression method. To examine if the lead SNPs located on the association signals had independent effects, we further performed conditional logistic regression analyses on each lead SNP using association signals of the locus with P < 1.0 × 10−4. Association results were visualized with Q-Q and Manhattan plots created with the R package qqman. Functional motifs around the association signals were also investigated using the HaploReg V4.1 database27 (https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php), including data from the Encyclopedia of DNA Elements (ENCODE)28 and the Roadmap Epigenomics projects29.
Functional annotation
Functional annotation of the GWAS results, including that of genes mapping to the identified risk loci, was conducted using the FUMA web application (https://fuma.ctglab.nl/)30. FUMA requires GWAS summary statistics. Independent significant SNPs in the GWAS summary statistics were identified based on their P values (P < 5.0 × 10−6) and independence from each other (r2 < 0.6 in the 1000 Genomes phase 3 ALL of the reference panel population24) within a 250-kb window. Gene-set and tissue-expression analyses were performed with MAGMA implemented by FUMA30. The MAGMA gene-set analysis assessed over-representation of biological functions based on gene annotations using curated gene sets and gene ontology (GO) terms obtained from the Molecular Signature Database (MsigDB v5.2)31,32. Gene sets with Bonferroni-corrected Pbon < 0.05 were considered to be significantly enriched. The MAGMA tissue-expression analysis was conducted with eQTL data from the Genotype-Tissue Expression project (GTEx v8)33 to identify the tissue specificity of the phenotype.
Genetic correlations
To estimate the SNP heritability of complex traits and diseases and to estimate the genetic correlation between different phenotypes, we implemented LD Hub34, an online platform for performing LD score regression using GWAS summary statistics data, available at http://ldsc.broadinstitute.org/.
Trans-ethnic meta-analysis
For the trans-ethnic meta-analysis, we used two sets of the ethnic-specific GWAS summary statistics: our Japanese GWAS and the IGAP stage 1 data (21982 AD cases and 41944 controls)6. This trans-ethnic meta-analysis was implemented in METASOFT26. The Meta-P values were calculated based on Han and Eskin’s modified random effects model (RE-HE). Of the loci that satisfied Meta-P (RE-HE) < 5.0 × 10−6 in the trans-ethnic meta-analysis, lead markers were genotyped using the independent replication samples. The combined analysis of our Japanese GWAS and replication study was verified by conducting logistic regression with adjustment for sex and age using PLINK software (--logistic)25.
Quantitative trait locus analysis
We obtained 60 quantitative traits from our NCGG routine blood tests. To examine the relationships between the hematological traits and SNP genotypes, QTL analysis was conducted using linear regression analysis with adjustment for sex and age implemented by PLINK software (--linear)25.
Results
GWAS in the Japanese population
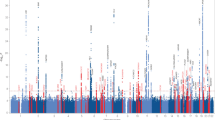
We examined association signals from a Japanese GWAS data set of 3962 AD cases and 4074 controls with genotype imputation (Fig. 1). A total of 4,852,957 genetic markers (single-nucleotide polymorphisms [SNPs] and short insertions and deletions [Indels]) passed stringent QC filters for both genotypes and samples after SNP imputation. The associations were assessed with logistic regression, adjusting for sex and age (see Materials and methods section). A quantile–quantile plot of the genome-wide P values indicated no genomic inflation (λGC = 1.05, Fig. 2a). A total of 134 genetic markers, located within nine genes (APOE, SORL1, FAM47E, PAPOLG, RAB3C, BANK1, LINC01867, LINC00899, and LOC101928561) showed suggestive associations (P < 5.0\(\times 10^{ - 6}\), Fig. 2b). The APOE and SORL1 genes have been reported as AD susceptibility loci in several populations6,11,16 and the remaining seven loci (Supplementary Fig. 1) have not previously been associated with AD in the Japanese population. We also carried out an ethnic meta-analysis combining the GWAS summary statistics of our NCGG and Niigata data on the seven loci using METASOFT. The logistic regression and ethnic meta-analysis showed similar P values for AD associations (Table 1).

Workflow of the study.

a Quantile-quantile plots of the genome-wide P values, b A Manhattan plot of the GWAS, and c Regional association plot of the variants in the FAM47E locus.
Replication study and ethnicity-specific meta-analysis
The lead SNPs located on the seven novel loci (FAM47E, PAPOLG, RAB3C, BANK1, LINC01867, LINC00899, and LOC101928561, Supplementary Fig. 1) were genotyped using an independent Japanese AD case–control cohort of 1216 AD cases and 2446 controls (Table 1). As the lead SNP of LINC00899 (rs8137273; GWAS P = 3.80 × 10−6) showed poor signals in the PCR-based Invader assay, a proxy SNP (rs4073601; GWAS P = 1.90 × 10−5) was used in the subsequent meta-analysis combining results from the GWAS and replication data sets. Of the seven lead SNPs, the FAM47E SNP showed modest evidence of association in the replication study (P < 0.05, Fig. 2c and Table 1), and the subsequent meta-analysis combining results from the GWAS and replication data sets reached genome-wide significance in a logistic regression (n = 11692, P = 5.34 × 10−9, odds ratio = 0.65, 95% CI = 0.57–0.75, Table 1). As the FAM47E SNP genotypes obtained from the GWAS data set were imputed, we genotyped all of samples in the GWAS data set (n = 7562) by PCR-invader assay and evaluated the concordance rate of the imputed SNPs, which provided genotypes of high concordance (0.9963), and obtained similar association for AD with statistical P value of 6.24 × 10−7. The subsequent meta-analysis combining results from the GWAS and replication data sets also reached genome-wide significance in a logistic regression (n = 11229, P = 1.72 × 10−8, odds ratio = 0.65, 95% CI = 0.56–0.76).
The most significant SNP of FAM47E was rs920608 (Table 1 and Fig. 2c). Because 11 genetic markers other than rs920608 were included in the locus with P < 1.0 × 10−4, we further examined whether these markers had independent effects using conditional logistic regression analysis on rs920608, but no independent association signals were found (Supplementary Table 1).
The minor allele frequencies (MAFs) of rs920608 in different populations were African (AFR) = 0.11, Mixed American (AMR) = 0.016, European (EUR) = 0.0060, East Asian (EAS) = 0.038, and South Asian (SAS) = 0.021 in the 1000 Genomes Project Phase 3 data24, and AFR = 0.09, AMR = 0.013, EUR = 0.0073, and EAS = 0.043 in the Genome Aggregation Database (gnomAD)35. The MAF of the EAS population (including Japanese) was statistically significantly different from that of each of the other populations (P < 0.05 with Fisher’s exact test, Supplementary Table 2). Furthermore, the HaploReg database showed that this SNP overlaps histone marks associated with enhancers (H3K4me1 and H3K27ac) or promoters (H3K4me3 and H3K9ac) in several brain tissues27 (Fig. 3). These results indicate that the FAM47E locus has the potential to be an East Asian–specific AD susceptibility locus.

Enhancers and promoters in eight brain tissues.
Expression quantitative trait loci
We investigated the effect of novel AD variants on the expression of genes by assessing expression QTLs (eQTLs) using the Genotype-Tissue Expression Portal (GTEx; https://www.gtexportal.org/home/). The A allele for AD risk at rs920608, the top SNP for the FAM47E locus, was associated with increased expression of SCARB2 in the adrenal gland (P = 0.007). The proxies for rs920608 also had eQTLs for FAM47E and SCARB2 in several brain tissues (P < 0.05).
Functional gene annotations and genetic correlations
To gain biological insight from the functional annotation of the GWAS results, we performed gene-set analysis and tissue-expression analysis by using MAGMA36 implemented by FUMA30. The MAGMA gene-set analysis identified six GO terms with Bonferroni-corrected Pbon < 0.05 when applying our ethnicity-specific GWAS summary statistics (Fig. 4a). Of the six, four were associated with amyloid precursor proteins, known pathological hallmarks of AD, and the remaining two were novel proteins associated with negative regulation of metallopeptidase and metalloendopeptidase activities (Fig. 4a). These two GO terms shared four genes (RECK, PICALM, SORL1, and TIMP3), of which two (RECK and TIMP3) have never been reported as AD susceptibility loci. However, both are target genes of microRNA-2137, which has been reported to inhibit cell apoptosis induced by Aβ1–42 and which has a protective role in AD38. Moreover, we examined the tissue specificity of the phenotype with a MAGMA tissue-expression analysis, using RNA sequencing data of 54 tissue types obtained from GTEx v833. Many of top-ranked tissue types observed were regions of the brain, although there were no tissue types significantly associated with AD (Fig. 4b).

a Pbon* represents the Bonferroni-corrected P. b The red lettering indicates brain regions. c Diseases and traits with genetic correlations with AD (P < 0.1) are shown. Those shown with blue symbols represent diseases and traits with P < 0.05.
To investigate the extent of genetic overlap between our Japanese GWAS data and phenotypes, we also estimated genetic correlations across the different diseases and traits available at LD Hub34 based on LD-score regression. Significant correlations were found for five traits. Two significant positive genetic correlations were observed: with AD in a European population (rG = 0.65, P = 2.3 × 10−3) and with forced vital capacity (rG = 0.32, P = 4.0 × 10−2). Three negative correlations were observed: with age of first birth (a woman’s age at the birth of her first child, rG = −0.34, P = 8.1 × 10−3), squamous cell lung cancer (rG = −0.66, P = 3.7 × 10−2), and total cholesterol (rG = −0.22, P = 4.4 × 10−2; Fig. 4c). Although none were statistically significant after correction for multiple testing, these traits (age of first birth11, forced vital capacity39, squamous cell lung cancer40, and total cholesterol41) were reported in previous studies to correlate with AD. In addition, we estimated overall SNP heritability (\({{h^{\mathrm{2}}}_{{\mathrm{SNP}}}}\)) to be 0.16 (SE = 0.06) in the Japanese GWAS data, although Lee et al.42 estimated the \({{h^{\mathrm{2}}}_{{\mathrm{SNP}}}}\) to be 0.24 (SE = 0.03) in the European GWAS data.
Trans-ethnic meta-analysis
To improve the power to detect additional AD susceptibility loci through a large number of samples, we performed a trans-ethnic meta-analysis combining the results of our Japanese GWAS data with the summary statistics from stage 1 of the International Genomics of Alzheimer’s Project (IGAP; 21982 AD cases and 41944 controls)6. The trans-ethnic meta-analysis was carried out on 2,067,592 genetic markers with Han and Eskin’s modified random effects model (RE-HE) implemented by METASOFT. Of these markers, 26 loci reached a Meta-P (RE-HE) <5.0 × 10−6, of which 18 were located on genes known to contribute to AD risk (APOE, PICALM, BIN1, CLU, CR1, MS4A4A, SORL1, MADD, HLA-DRA, CD2AP, EPHA1, ADAMTS1, SLC24A4, LACTB2, ELL, FERMT2, ZCWPW1, and TSPOAP1, Supplementary Fig. 2). The lead SNPs located on the remaining eight novel loci (MTSS1L, CLEC3B, EFL1, FAM155A, NTM, OR2B2, C1S, and TSPAN14, Supplementary Fig. 2) were genotyped in an independent AD case–control cohort of Japanese samples (Supplementary Table 3). Finally, a subsequent meta-analysis combining results from the GWAS data, replication data, and IGAP data reached genome-wide significance for OR2B2 (rs1497526, Meta-P = 2.14 × 10−8, Table 2). As the lead SNP of OR2B2 (rs1497525) showed poor signals in the PCR-based Invader assay, a proxy SNP (rs1497526) was used in the subsequent meta-analysis combining results from the GWAS and replication data sets, and IGAP data. The proxy SNP was identified by the LDproxy Tool (https://ldlink.nci.nih.gov/?tab=ldproxy, R-squared = 1 in Japanese). As this proxy SNP was imputed in the Japanese GWAS data set, we genotyped all of samples in the GWAS data (n = 7734) by PCR-invader assay and evaluated the concordance rate of the imputed SNPs, which provided genotypes of high concordance (0.9919).
To examine whether OR2B2 is an ethnicity-specific susceptibility locus like FAM47E, we checked the MAF of OR2B2 SNP rs1497526 for several populations in the 1000 Genomes Project Phase 3 data24 (AFR = 0.29, AMR = 0.091, EUR = 0.046, EAS = 0.081, and SAS = 0.076) and gnomAD35 (AFR = 0.24, AMR = 0.076, EUR = 0.032, and EAS = 0.043) and assessed the statistical significance of differences between EAS and each of the other populations. Statistically significant differences were observed between EAS and EUR, between EAS and AFR, and between EAS and AMR (P < 0.05 with Fisher’s exact test, Supplementary Table 2). These results show that OR2B2 could be a common susceptibility locus among some populations. There is no eQTL in the GTEx database at rs1497526.
Quantitative trait locus analysis for FAM47E and OR2B2
We also investigated the relationship between each of the FAM47E and OR2B2 SNP genotypes and the hematological traits. QTL analysis was carried out on 60 blood test results using linear regression analysis with adjustment for sex and age (Supplemental Table 4). Six traits showed suggestive levels of associations with the FAM47E SNP (P < 0.05; I-BIL: indirect bilirubin, D-BIL: direct bilirubin, MPV: mean platelet volume, CRE: creatine, eGFR: estimated glomerular filtration rate, and GLU: glucose) and two traits with the OR2B2 SNP (P < 0.05; TP: total protein, and MPV), although none were statistically significant after correction for multiple testing.
Discussion
Although trans-ethnic meta-analyses, along with greatly increased sample sizes, have contributed to the identification of many genetic risk factors for AD5,6,11, there are few reports investigating ethnicity-specific associations with AD, especially for the Japanese population16. We succeeded in identifying a novel ethnicity-specific AD susceptibility locus within the FAM47E gene on chromosome 4. The difference in the allele frequency of the genotyped variants among populations could have resulted in the identification of the locus. The MAF of the FAM47E SNP (rs920608) was relatively high in the EAS population compared with the AMR and EUR populations. This SNP has also been shown to overlap histone marks associated with enhancers (H3K4me1 and H3K27ac) or promoters (H3K4me3 and H3K9ac) in several brain tissues from the HaploReg database27. Furthermore, the lead SNP and proxies had eQTLs for FAM47E and SCARB2 in several brain tissues. These results suggest that FAM47E and SCARB2 are likely to be functionally related to AD, although we should verify this using a larger sample size of Japanese AD cases.
The FAM47E locus has been associated with Parkinson’s disease (PD)43,44. Although PD and AD have remarkably different clinical and pathological features, the two diseases are the most common neurodegenerative disorders and show considerable overlap in the development of neurodegeneration. Previous studies have reported some common genetic loci that increase both PD and AD risk. Gregório et al.45 reported that APOE4, a strong risk factor for AD, is also associated with cognitive decline in PD. Allen et al.46 reported that polymorphisms in the glutathione S-transferase omega gene are associated with risk and age at onset of AD and PD. Li et al.47 has reported a common genetic factor in the NEDD9 gene associated with both AD and PD. Thus, the FAM47E locus could be an AD-and-PD–associated locus. However, the associations of the glutathione S-transferase omega and NEDD9 are only shown in relatively small data sets and they should be confirmed in subsequent studies with a large sample size. Furthermore, we found correlations between FAM47E SNP genotypes and six quantitative traits of blood test results, many of which (direct/indirect BIL48, MPV49, CRE50, and GLU51) have been associated with dementias, including AD. These results also support the view that FAM47E is an AD-associated locus.
The functional annotation of the GWAS results can provide biological insight. We detected six statistically significant GO terms using MAGMA36. Four of these GO terms were associated with amyloid precursor proteins, which are known pathological hallmarks of AD. The remaining two were novel and shared four genes including SORL116 and PICALM52,53 which are well established AD genes. These results were not surprising, but they strengthen the credibility of our GWAS findings. Also, while two (RECK and TIMP3) of the four genes have never been reported as AD susceptibility loci, these genes are common targets of microRNA-2137, which has been reported to inhibit cell apoptosis induced by Aβ1–42 and to have protective roles in AD38. These two genes could, therefore, be candidate AD susceptibility genes.
We also identified a novel locus, OR2B2, in a meta-analysis combining the first GWAS data, its replication data, and IGAP data. However, because OR2B2 has never been reported to be associated with AD, and no histone marks associated with enhancers or promoters were enriched in the OR2B2 SNP in several brain tissues, further investigation will be needed to examine whether it has any functional association with AD. We finally assigned all SNPs in the IGAP data to nearest genes, and checked if any FAM47E SNPs and OR2B2 SNPs had AD associations. However, there were no statistically significant associations in the two genes (Supplementary Table 5). There are power limitations and SNP array-based GWAS limitations in the current analyses. Early genome-wide SNP arrays, used for the Niigata subjects in our analysis, were developed based on tag SNPs from reference panels of European populations. Linkage disequilibrium patterns are different among ethnic groups and these arrays provided poor coverage in our Asian population. On the other hand, an ethnic-specific array (the Japonica Array) was used for our NCGG subjects, which should enable a possibly large harvest of ethnic-specific disease signals. When combining the early genome-wide SNP arrays and new ethnic-specific SNP arrays, larger and uniform reference panels will be preferred to improve genotype imputation. In addition, GWAS based on SNP arrays cannot detect rare variants associated with diseases54, and it should be necessary to increase a sample size to increase the power for detecting association signals with small effect sizes. We will perform further investigations with larger sample sizes to further validate the effectiveness of our findings in near future.
Conclusions
We conducted a Japanese genome-wide association study of AD with a large sample size and a trans-ethnic meta-analysis with Caucasian GWAS data, and identified novel susceptibility loci for AD. Further investigation using well-powered GWAS in specific ethnic groups would likely identify additional genetic risk factors associated with AD, which causes a great deal of suffering for patients and their families and is also a leading cause of death in many countries. We anticipate that the identification of the genetic architecture of AD susceptibility and related pathways will help us to understand the pathogenesis of LOAD. In turn, this will contribute to innovative medical and pharmaceutical approaches that advance the development of precision medicine for this common but serious disorder.
Code availability
We used the open source program languages R (version 3.4.1) and Ruby (version 2.4.0) to analyse the data and create the plots. Code is available upon request from the corresponding authors.
References
Robinson, L., Tang, E. & Taylor, J. P. Dementia: timely diagnosis and early intervention. BMJ 350, h3029 (2015).
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002).
Prince, M. et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63–75 (2013).
Lee, G. et al. Predicting Alzheimer’s disease progression using multi-modal deep learning approach. Sci. Rep. 9, 1952 (2019).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430 (2016).
Liu, C. C. et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 (2013).
Ridge, P. G. et al. Assessment of the genetic variance of late-onset Alzheimer’sdisease. Neurobiol. Aging 41, e213–200 (2016).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Bis, J. C. et al. Correction: whole exome sequencing study identifies novel rare and common Alzheimer’s-Associated variants involved in immune response and transcriptional regulation. Mol. Psychiatry 25, 1901–1903 (2020).
Jun, G. R. et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 13, 727–738 (2017).
Simino, J. et al. Whole exome sequence-based association analyses of plasma amyloid-beta in African and European Americans; the Atherosclerosis Risk in Communities-Neurocognitive Study. PLoS ONE 12, e0180046 (2017).
Asanomi, Y. et al. A rare functional variant of SHARPIN attenuates the inflammatory response and associates with increased risk of late-onset Alzheimer’s disease. Mol. Med. 25, 20 (2019).
Miyashita, A. et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS ONE 8, e58618 (2013).
Hirano, A. et al. A genome-wide association study of late-onset Alzheimer’s disease in a Japanese population. Psychiatr. Genet. 25, 139–146 (2015).
McKhann, G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011).
Albert, M. S. et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 270–279 (2011).
Hirata, M. et al. Cross-sectional analysis of BioBank Japan clinical data: A large cohort of 200,000 patients with 47 common diseases. J. Epidemiol. 27, S9–S21 (2017).
Nagai, A. et al. Overview of the BioBank Japan Project: study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Kawai, Y. et al. Japonica array: improved genotype imputation by designing a population-specific SNP array with 1070 Japanese individuals. J. Hum. Genet. 60, 581–587 (2015).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Ward, L. D. & Kellis, M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881 (2016).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Watanabe, K., Umicevic Mirkov, M., de Leeuw, C. A., van den Heuvel, M. P. & Posthuma, D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 10, 3222 (2019).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Zheng, J. et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Sandhir, R., Gregory, E. & Berman, N. E. Differential response of miRNA-21 and its targets after traumatic brain injury in aging mice. Neurochem. Int. 78, 117–121 (2014).
Feng, M. G. et al. MiR-21 attenuates apoptosis-triggered by amyloid-beta via modulating PDCD4/ PI3K/AKT/GSK-3beta pathway in SH-SY5Y cells. Biomed. Pharmacother. 101, 1003–1007 (2018).
Kim, Y. et al. Reduced forced vital capacity is associated with cerebral small vessel disease burden in cognitively normal individuals. Neuroimage Clin. 25, 102140 (2020).
Greco, A. et al. Molecular inverse comorbidity between Alzheimer’s Disease and lung cancer: new insights from matrix factorization. Int. J. Mol. Sci. 20, 3114 (2019).
Shepardson, N. E., Shankar, G. M. & Selkoe, D. J. Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch. Neurol. 68, 1239–1244 (2011).
Lee, S. H. et al. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum. Mol. Genet. 22, 832–841 (2013).
Nalls, M. A. et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993 (2014).
Redensek, S., Trost, M. & Dolzan, V. Genetic determinants of Parkinson’s disease: can they help to stratify the patients based on the underlying molecular defect? Front. Aging Neurosci. 9, 20 (2017).
Gregorio, M. L. et al. Impact of genetic variants of apolipoprotein E on lipid profile in patients with Parkinson’s disease. Biomed. Res. Int. 2013, 641515 (2013).
Allen, M. et al. Glutathione S-transferase omega genes in Alzheimer and Parkinson disease risk, age-at-diagnosis and brain gene expression: an association study with mechanistic implications. Mol. Neurodegener. 7, 13 (2012).
Li, Y. et al. Evidence that common variation in NEDD9 is associated with susceptibility to late-onset Alzheimer’s and Parkinson’s disease. Hum. Mol. Genet. 17, 759–767 (2008).
Vasantharekha, R., Priyanka, H. P., Swarnalingam, T., Srinivasan, A. V. & ThyagaRajan, S. Interrelationship between Mini-Mental State Examination scores and biochemical parameters in patients with mild cognitive impairment and Alzheimer’s disease. Geriatr. Gerontol. Int. 17, 1737–1745 (2017).
Koc, E. R., Uzar, E., Cirak, Y., Parlak Demir, Y. & Ilhan, A. The increase of mean platelet volume in patients with Alzheimer disease. Turk. J. Med. Sci. 44, 1060–1066 (2014).
Smith, R. N., Agharkar, A. S. & Gonzales, E. B. A review of creatine supplementation in age-related diseases: more than a supplement for athletes. F1000Res 3, 222 (2014).
Crane, P. K. et al. Glucose levels and risk of dementia. N. Engl. J. Med. 369, 540–548 (2013).
Harold, D. et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093 (2009).
Lee, J. H. et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch. Neurol. 68, 320–328 (2011).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Acknowledgements
We thank the NCGG Biobank and Department of Molecular Genetics at Niigata University for providing the study materials, clinical information, and technical support. Some of the samples and data used for this research were provided from the BioBank Japan Project, which is supported by the Japan Agency for Medical Research and Development (AMED). We also thank ELSS editors and Keith A. Boroevich for English corrections. This study was supported by the grants from AMED (grant number JP18kk0205009 to S.N., grant number JP18kk0205012 to S.N., and grant number JP19dk0207045 to T.I., S.N., and K.O., and grant number JP20km040550 to K.O.); The Japan Foundation for Aging and Health and Takeda Science Foundation (to D.S.); The Research Funding for Longevity Sciences from the National Center for Geriatrics and Gerontology (29-45 to K.O., and 30-29 to D.S.); and a grant for Research on Dementia from the Japanese Ministry of Health, Labour, and Welfare (to K.O.).
Author information
Authors and Affiliations
Contributions
D.S. developed the statistical method, performed the analyses, and wrote the manuscript; R.M. and Y.A. performed experimental analyses and contributed to data acquisition; S.A., T.M., and S.H. provided technical assistance; A.M., N.H., and T.I. managed the DNA samples for the cohort at Niigata University and performed genotyping; G.T. and K.K. performed the imputation analysis on the Japonica data; S.N. managed the samples from the NCGG biobank and contributed to data acquisition; D.S. and K.O. designed the study. K.O. supervised the project. All authors contributed to and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shigemizu, D., Mitsumori, R., Akiyama, S. et al. Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Transl Psychiatry 11, 151 (2021). https://doi.org/10.1038/s41398-021-01272-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01272-3
This article is cited by
-
SR-TWAS: leveraging multiple reference panels to improve transcriptome-wide association study power by ensemble machine learning
Nature Communications (2024)
-
Genetic architecture of brain age and its causal relations with brain and mental disorders
Molecular Psychiatry (2023)
-
Identification of candidate DNA methylation biomarkers related to Alzheimer’s disease risk by integrating genome and blood methylome data
Translational Psychiatry (2023)
-
Genetics of Alzheimer’s disease: an East Asian perspective
Journal of Human Genetics (2023)
-
A global view of the genetic basis of Alzheimer disease
Nature Reviews Neurology (2023)
