
Abstract
Epigenetic variation in peripheral tissues is being widely studied as a molecular biomarker of complex disease and disease-related exposures. To date, few studies have examined differences in DNA methylation associated with attention-deficit hyperactivity disorder (ADHD). In this study, we profiled genetic and methylomic variation across the genome in saliva samples from children (age 7–12 years) with clinically established ADHD (N = 391) and nonpsychiatric controls (N = 213). We tested for differentially methylated positions (DMPs) associated with both ADHD diagnosis and ADHD polygenic risk score, by using linear regression models including smoking, medication effects, and other potential confounders in our statistical models. Our results support previously reported associations between ADHD and DNA methylation levels at sites annotated to VIPR2, and identify several novel disease-associated DMPs (p < 1e–5), although none of them were genome-wide significant. The two top-ranked, ADHD-associated DMPs (cg17478313 annotated to SLC7A8 and cg21609804 annotated to MARK2) are also significantly associated with nearby SNPs (p = 1.2e–46 and p = 2.07e–59), providing evidence that disease-associated DMPs are under genetic control. We also report a genome-wide significant association between ADHD polygenic risk and variable DNA methylation at a site annotated to the promoter of GART and SON (p = 6.71E–8). Finally, we show that ADHD-associated SNPs colocalize with SNPs associated with methylation levels in saliva. This is the first large-scale study of DNA methylation in children with ADHD. Our results represent novel epigenetic biomarkers for ADHD that may be useful for patient stratification, reinforce the importance of genetic effects on DNA methylation, and provide plausible molecular mechanisms for ADHD risk variants.
Similar content being viewed by others

Introduction
Attention-deficit hyperactivity disorder (ADHD) has a substantial heritable component, with genetic factors interacting with early-life environmental exposures to mediate risk1,2. A recent large genome-wide association study (GWAS) meta-analysis of ADHD identified the first genome-wide significant association with common DNA variants3. These data allow for a more precise estimate of the burden of ADHD genetic risk, which is associated with ADHD diagnosis and some of its mechanisms, such as poor executive function4.
The specific mechanisms by which genetic risk factors influence ADHD are not known, although recent evidence supports a role for non-sequence-based (i.e., regulatory) genomic variation in neuropsychiatric phenotypes5,6,7,8. Epigenetic processes, which act to dynamically control gene expression and are known to regulate key neurobiological and cognitive processes in the brain, represent both a potential clue to mechanisms and a possible source of novel biomarker discovery9,10. Epigenetic studies of mental disorders have focused primarily on DNA methylation, the best-characterized and most stable epigenetic modification. It acts to influence gene expression via physical disruption of transcription factor binding and the attraction of methyl-binding proteins that initiate chromatin compaction and gene silencing11. Epigenetic variation can be influenced by environmental exposures. In fact, many of the environmental risk factors associated with ADHD have been associated with changes in DNA methylation in peripheral tissues12,13,14,15,16. Epigenetic changes may also be induced by exposures related to the disease (e.g., medication) or the pathological process itself. Although patterns of DNA methylation are cell-type specific, and generalization from peripheral tissue to brain mechanisms is not appropriate, disease-associated changes in peripheral DNA methylation might serve as useful biomarkers. For instance, DNA methylation changes in peripheral tissues may be indicative of early exposures (caused by, rather than causal of, disease process) or clinical subtypes, or may be predictors of outcome or treatment response17. Importantly, both genetic and environmental etiologies can operate through epigenetic effects, and there is evidence that genetic effects on DNA methylation are relatively conserved across tissues18,19. Thus, an integrated genetic/epigenetic approach is strongly recommended for “second generation” studies of methylation biomarkers18,20. Combining genetic and epigenetic data might also nominate novel mechanistic pathways associated with disease as hypotheses for future study21.
DNA methylation studies relevant to ADHD or ADHD symptoms to date are limited. They include five candidate gene studies on relatively small samples22,23,24,25,26, and four studies targeting specific environmental exposures, such as malnutrition, in relation to ADHD symptoms, by using candidate and epigenome-wide association study (EWAS) approaches27,28,29,30. Three prior EWAS studies are relevant. The first and only study to examine ADHD cases directly, was a small study by our group31. That study of 105 children was able to replicate suggestive findings in MYT1L and VIPR2, also a top hit in an EWAS of environmental exposure28 and a recent twin study32. An EWAS of ADHD symptoms in adult population cohorts reported additional novel candidate sites for exploration, as well as noting that most top-ranked loci were driven by DNA methylation quantitative trait loci (mQTL)33. A third population-based EWAS study7 identified prenatal DNA methylation sites related to later ADHD symptom trajectories, though they did not replicate at the age of 7.
We report here the first large-scale EWAS of children with ADHD and extend our previous study to incorporate analyses of genetic effects in the context of variable DNA methylation, exploring both mQTL and polygenic risk burden derived from GWAS. Given our focus on identifying biomarkers relevant to ADHD, we use DNA derived from saliva in an effort to minimize potential selection bias resulting from clinical differences in rates of child refusal of blood draws.
Methods
Participants and case identification
In a case-finding procedure, families were recruited by soliciting community volunteers with public advertisements and mass mailings. The local Institutional Review Board approved the studies. Parents provided written informed consent; children provided written informed assent. All families completed a multi-informant, multi-method screening process to establish eligibility and diagnostic group assignment for ADHD, non-ADHD, as well as comorbid disorders (Supplementary Materials).
Medication
Current and lifetime prescription of any psychoactive medication, including any stimulant or non-stimulant preparation, was recorded and statistically controlled. A full frequency list of medications in the sample is given in Table S1.
DNA methylation profiling
An overview of our data quality control (QC) and analysis workflow is provided in Fig. S1. Genomic DNA was isolated from saliva, bisulfite converted, and assessed for DNA methylation on the MethylationEPIC BeadChip (Illumina, Inc.) using a standard protocol. Raw data were imported into Genome Studio v2011.1 (Illumina, Inc.) to investigate sample hybridization quality and to extract signal intensities for each probe (Fig. S2). Data QC measures included manual inspection of beta distributions, curation of control probes using the Illumina BeadArray Controls Reporter, manual inspection of total CpG intensity distributions, sex prediction, outlier sample detection, and comparison of SNP probes on the MethylationEPIC with genotypes, using the lumi34 and minfi35 packages. Data were normalized with lumi34 using smooth quantile normalization. Cell-type profiles and proportions were calculated using reference-free cell-type prediction with the RefFreeEWAS36 package, and beta values were adjusted to account for cell-type proportions in each sample. Methylation values were set as missing if they deviated >4 times the interquartile range from the mean of each probe. Participant samples were further curated to restrict to unrelated children who met full criteria for non-ADHD or ADHD. We excluded a small number of children who only met criteria for the ADHD hyperactive profile, those with subthreshold ADHD (e.g., five symptoms), and those missing information about medication usage, resulting in a final sample of 604 unrelated children. For analyses examining the effects of the ADHD polygenic risk score, only children with European ancestry4 were included (N = 472). We removed probes for the following reasons (Table S2): detection call rate p-value is <0.01 in at least one sample, mapping to multiple genomic locations37, missing from the MethylationEPIC manifest, SNPs underlying the probe37, or non-autosomal probes. Our final dataset included 568,281 probes for analysis.
Genotyping and polygenic score
Salivary DNA samples were genotyped, and the ADHD polygenic risk score (PRS) for each individual calculated as described previously4. Briefly, DNA was hybridized to the PsychCHIP_v1-1 (N = 603,132 SNPs), developed by Illumina, Inc. in collaboration with the Psychiatric Genetics Consortium (PGC). Genotypes were used to determine relatedness38 among samples and population stratification as previously described4. The PRS was constructed using the PGC + iPSYCH meta-analysis3 as the discovery dataset (details in Supplementary Materials).
Analytic models and covariates
Differential global methylation (average methylation across all probes), as well as differentially methylated positions (DMPs) were evaluated using custom R scripts (R 3.5.0, https://github.com/pryabinin/ohsu_adhd_ewas). Briefly, linear models (Eqs. (1)–(5), below) were used to examine associations between DNA methylation and both ADHD status and ADHD PRS. All models used cell-type adjusted beta values36 as the outcome variable, and included covariates for sex, age (in years), the first three genomic principal components (PCs), medication usage (binary variable signifying the history of psychoactive or stimulant medication), and a maternal smoking score12, which are referred to below as the “standard covariates”. The first three genomic PCs were selected based on their association with self-reported ethnicity in our sample. The relationship between ethnicity and genomic PCs in our sample was verified using HapMap samples as a reference. In addition, for the PRS analysis, a covariate was included to account for the number of missing SNPs in the PRS calculation for each patient. Finally, due to sex differences in ADHD prevalence, salient clinical features39, and familial burden not explained by autosomal differences40, a sex interaction term was included for both ADHD status and ADHD PRS (Eqs. (4) and (5) to investigate sex-specific effects:
Differentially methylated region analysis
Testing for differentially methylated regions (DMRs) was performed using the Comb-p software tool41, using the results of each regression model separately. The following Comb-p parameters were used: seed p-value = 0.001, maximum distance between probes = 500 bp, and a minimum of three probes allowed in a DMR. This set of parameters is consistent with previous work42 in the field and recommendations from simulation experiments43.
DNA methylation quantitative trait loci analysis
DNA methylation quantitative trait loci (mQTLs) were calculated using a linear, additive model implemented in the GEM package in R44, where the methylation level at each DMP was regressed on the minor allele count of each SNP. Covariates for sex, age, and the first three genomic PCs were included in the model. For all DMPs with p < 1e–5 (34 total), all 6,374,797 autosomal SNPs were assessed (i.e., both cis- and trans-QTLs were tested; 216,743,098 tests).
Colocalization analysis of mQTLs and ADHD-associated variants
DNA methylation QTLs discovered in our cohort were examined for evidence of colocalization with ADHD-associated variants. All pairs of methylation probes and SNPs within each of 12 ADHD-associated regions (Table S3), determined from the results of the recent PGC + iPSYCH ADHD GWAS meta-analysis3, were evaluated. Statistical analyses were performed using summary data-based Mendelian randomization45 (SMR), as well as the Bayesian colocalization method in the coloc package46 in R.
Functional interpretation and gene set enrichment analyses
Downstream interpretation of the associated DMPs was performed using Genetica (Table S9), a custom R script developed in our lab (https://github.com/bhattp09/genetica) that collates information from publicly available databases (Supplementary Materials) and through the Weizmann Institute of Science (http://www.genecards.org/). Investigations into miRNA profiles were performed using the miRWalk tools47, and seed regions overlapping the hybridized probe sequence was confirmed using the UCSC Genome Browser48.
The methylGSA package49 was used to investigate whether DMPs were enriched among genes involved in a particular biological function or process. methylGSA implements an unbiased test of enrichment, accounting for the number of methylation probes within each gene. The default significance threshold suggested by methylGSA (p ≤ 0.001) was used to select DMPs for the enrichment analyses. We first tested for enrichment among brain-expressed genes, using two gene sets defined by the Human Protein Atlas50 (https://www.proteinatlas.org/humanproteome/tissue/brain): genes with brain-enriched expression (408 genes; five-fold higher expression in the brain compared with all other tissues), and genes with brain-elevated expression (1379 genes; five-fold higher expression in the brain compared with the average expression in all other tissues). We next tested for enrichment among all Gene Ontology (GO) categories. GO categories enriched with a false discovery rate-adjusted p ≤ 0.05 were considered significant.
Multiple testing, significance thresholds, and statistical power
Within each EWAS analysis, DNA methylation sites were considered significantly differentially methylated if they met a Bonferroni-corrected p-value threshold of 8.8e–8, which is consistent with a recent recommendation for the genome-wide significance threshold for analyses of EPIC array data51. Following other studies, a p-value threshold of 1e–5 was used to identify suggestive DMPs for preliminary interpretation to guide future hypotheses52. A more inclusive p-value threshold of 0.001 was used to select DMPs for gene set enrichment analyses as suggested by the MethylGSA R package49. In the DMR analysis, a Šidák-corrected p-value < 0.05 was used to determine significance. Finally, a Bonferroni-corrected p-value threshold of 1.8e–10 was used to identify significant mQTLs.
The statistical power to detect an effect size of 1% difference in DNA methylation between cases and controls at a p-value < 8.8e–8 was calculated for all probes included in our analysis, following a previously described method51. Power calculations were done using each probe’s variance across all subjects, and the number for cases and controls in the experiment. The proportion of probes with a particular power threshold (ranging between 0 and 1) is shown in Fig. S3. Our study was well-powered (80%) to detect a 1% difference at ~68% of all sites interrogated.
Results
Sample overview and genome-wide DNA methylation variation
Table 1 provides a demographic and clinical description of the cohort. As intended, the ADHD group had significantly higher clinical symptoms, and as expected, had higher rates of psychiatric comorbidity, and more lifetime exposure to psychiatric medications. In the full sample, the ADHD group had slightly lower family income than controls (p = 0.03), but the difference was not significant in the European-ancestry subsample used for the PRS analyses. As is common in studies of ADHD, the ADHD group had a slightly lower IQ (WISC-IV FSIQ mean difference = 7.1; p = 5.6e–10), but both groups had mean IQ in the average range. The groups were of similar age, although age was included as a covariate as a precaution. Boys were overrepresented in the ADHD group and we adjusted all results for sex.
We calculated a predicted age for each child, based on age-associated DNA methylation sites10. As expected, reported ages and predicted ages were highly correlated (r = 0.58, p = 1.3e–54), providing reassurance as to the validity of the DNA methylation data obtained from saliva. There was no evidence for differential epigenetic age acceleration in the ADHD group (p = 0.98) (Fig. S4). We also tested for a “global” difference in DNA methylation levels between ADHD cases and controls, determined by averaging cell-type-corrected beta values across all 568,281 probes (see the “Methods” section, Eq. (1)), and found no significant difference (p = 0.74). This result indicates that, as expected, ADHD is not associated with any systemic methylomic differences across probes included on the Illumina EPIC array.
DNA methylation associated with ADHD
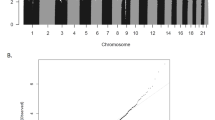
Differential DNA methylation between ADHD cases and controls at individual sites was investigated by regressing methylation levels at each probe on ADHD status (Eq. (2)) with our standard covariates. EWAS results were well-controlled with an inflation factor λ = 1.02 (calculated as the ratio of the median observed log10(p-value) to the median expected log10(p-value))53. A Q–Q plot and Manhattan plot are presented in Fig. S5.
No DMPs passed EWAS significance (p = 8.8e–8), which corrects for all 568,281 probes tested. Table 2 shows the 7 DMPs associated with ADHD at our a priori suggestive significance level of p < 1e–5. Methylation differences between ADHD cases and controls ranged between 0.3% and 1.4%. The top 100 probes associated with ADHD are provided in Table S5 for descriptive purposes. The top-ranked probe, cg17478313 (p = 1.54e–6), shows higher DNA methylation in ADHD cases compared with controls (Δβ = 0.93%) and is located in the promotor region of SLC7A8 (Figs. 1a and 2a). Probe cg21609804 (p = 2.82e–6), also with higher methylation (Δβ = 1.38%) in ADHD cases, is located in the 3′-UTR of MARK2 (Fig. 1b). Additional suggestive DMPs were annotated to PDLIM5, VPS28, ZNF706, and FAM59A.

Genomic diagrams of a SLC7A8 and b MARK2, showing locations of top DM probes and the corresponding mQTL SNP. The MARK2 probe cg21609804 contains a miRNA seed region at the interrogated loci.

DNA methylation values (beta values) for the top-ranked ADHD- and ADHD PRS-associated probes.
DNA methylation associated with ADHD polygenic risk
We have previously shown that the ADHD PRS is associated with ADHD status (explaining ~4.5% of the variance) in this sample4. Here we use the ADHD PRS to investigate the relationship between overall ADHD genetic burden and DNA methylation (Eq. (3)). Again, EWAS results for the PRS are well-controlled (λ = 1.08). A Q–Q plot and Manhattan plot are presented in Fig. S6. One probe met our genome-wide significance threshold, cg15472673 (p = 6.71E–8), characterized by reduced DNA methylation with higher PRS (Fig. 2b). The association remains (p = 9.76e–8), when including ADHD status in the regression model, indicating that the effect is not driven by elevated polygenic burden in ADHD cases. This probe is located in a CpG island of a bivariate promoter between the GART and SON genes. None of the SNPs included in the PRS are direct mQTLs (see below) for cg15472673 (all mQTL p-values > 1e–5), indicating that the association with the PRS is not driven by a simple genetic effect on DNA methylation. DNA methylation levels at 12 other probes were associated with the PRS at p < 1.0e–5, 10 of which showed increased methylation with higher PRS (Table 2b). A summary of all findings for ADHD diagnosis and the ADHD PRS is presented in Fig. S7.
Sex-specific variation in DNA methylation
Because we previously reported an association between ADHD and DNA methylation at sites annotated to VIPR2 and MYT1L specifically in boys31, we first examined sex-by-diagnosis interaction effects among all probes annotated to these two genes (239 probes) at an adjusted significance threshold of p < 0.0002 (0.05/239). The two strongest sex-by-diagnosis interactions annotated to VIPR2 were for cg26975193 and cg20998127 (sex-by-diagnosis interaction p = 7.51e–6 and p = 0.000459). Supporting the finding from our previous study, males with ADHD had lower methylation compared with male controls at both sites (Δβ = −0.22%, p = 0.0185; Δβ = −0.51%, p = 3.99e–5). However, among females, ADHD cases had higher methylation levels than controls at cg26975193 (Δβ = 0.35%, p = 0.00597) and were not significantly different from controls at cg20998127 (Δβ = 0.14%, p = 0.384). To ensure replication of the hypomethylation effect in boys with ADHD reported by Wilmot et al.31, we removed the samples included in that previous report (n = 73 that survived QC for the current probe set). Effects among males were consistent at both sites (for cg26975193: Δβ = −0.19%, p = 0.0819; for cg20998127: Δβ = −0.51%, p = 4.14e–4).
For MYT1L, our previously reported finding did not survive multiple-testing correction (minimum sex-by-diagnosis interaction p-value = 0.0039 for cg02870147; males Δβ = −0.49%), although the direction of effect at this site (lower methylation among males with ADHD) was consistent with our previous data, even with prior samples (n = 73) removed (males Δβ = −0.42%). Likewise, no probes annotated to MYT1L showed main effects that passed the Bonferroni threshold (minimum p-value of 0.00395 for cg22140907; Δβ = −0.31%). All results from the analysis of these genes are reported in Table S6.
We next performed an EWAS of sex-specific DMPs associated with both ADHD and the ADHD PRS. No sex interactions were significant at the EWAS-wide threshold, although nine probes show sex-by-diagnosis interactions (Table 2c) and five probes show sex-by-PRS interactions (Table 2d) at our suggestive significance threshold (p < 1e–5).
Differentially methylated regions
The results from all DMP analyses were used to investigate differentially methylated regions (DMRs) using the Comb-p software tool41. A single significant DMR on chromosome 6 was identified (Šidák-corrected p = 3.4e–5), which contained eight probes associated with the ADHD PRS in a sex-specific manner (Fig. S8). Specifically, among females a higher PRS was associated with higher methylation levels, and an opposite (though much weaker) relationship was seen among males. This DMR (chr6: 31148383–31148553) lies within the major histocompatibility complex, ~3 kb upstream of PSORS1C3.
DNA methylation quantitative trait loci
We identified methylation quantitative trait loci (mQTL) associated with all DMPs suggestively associated with (a) ADHD or (b) the ADHD PRS (Table 2). DNA methylation at the two top-ranked ADHD-associated DMPs (cg17478313 annotated to SLC7A8 and cg21609804 annotated to MARK2, Table 2a) was significantly associated with genotypes at nearby SNPs (Fig. 3). The SLC7A8 probe (cg17478313) was associated with SNP rs7141505 (p = 1.2e–46), located in the gene’s promoter region. The MARK2 probe (cg21609804) was associated with an intronic SNP rs928948 (38 kb upstream of the CpG) at p = 2.07e–59 (Fig. 1b). Furthermore, for both mQTLs, the relationship between genotype and DNA methylation level is similar in both ADHD cases and controls (Fig. 3b, d). Also, for both cg17478313 and cg21609804, both ADHD status (p = 2.2e–4 and p = 1.85e–4) and genotype (p = 4.4e–45 and p = 8.9e–58) are associated with methylation levels when included together in the regression models. These results indicate that the mQTL effects are not simply due to allele frequency differences between ADHD cases and controls. An additional 279 SNPs are associated with methylation levels at these two DMPs (31 for cg17478313 and 248 for cg21609804), and pass our experiment-wide mQTL significance threshold (p < 1.8e–10).

The genomic region around SLC7A8 (a), showing locations of cg17478313 and the most significant mQTL rs7141505. Similarly, the genomic region around MARK2 (c) with locations of cg21609804 and the most significant mQTL rs928948. For clarity, only mQTLs with p < 1e–20 in each region are shown. The methylation values (beta values) for both cg17478313 and cg21609804, stratified by mQTL genotype, are also shown (b, d).
For the ADHD PRS, of all the associated DMPs (at p < 1e–5), significant mQTLs were found only for cg04453792 annotated to USP31 (p < 1.8e–10). All mQTL analysis results are provided in Table S7.
To determine whether mQTL variants associated with the top-ranked DMPs (for both ADHD and the ADHD PRS) were also implicated in GWAS for ADHD (i.e., pleiotropic for methylation and ADHD), we examined the results from the PGC ADHD GWAS meta-analysis3 in the regions of SLC7A8, MARK2, GART, SON, USP31, and LOC100130015 (Table 2 and Table S7). We assessed all GWAS SNPs included in our mQTL analysis that were in linkage disequilibrium with these six genes or their putative regulatory regions (20 kb upstream or downstream of each gene, see Supplementary Materials). No genome-wide significant (or suggestive) ADHD-associated SNPs assigned to MARK2 (minimum p = 0.02), SLC7A8 (minimum p = 0.3), GART/SON (minimum p = 0.029), USP31 (minimum p = 0.069), or LOC100130015 (minimum p = 0.011) were seen in the GWAS meta-analysis, indicating there is no evidence of pleiotropy for these mQTLs.
To expand the investigation of pleiotropy, all mQTLs observed in our cohort (not just those associated with DMPs) were tested for colocalization with variants associated with ADHD in the recent ADHD GWAS meta-analysis conducted by the PGC3. Of the 12 ADHD-associated regions identified in the GWAS meta-analysis, 11 contained genome-wide significant cis-mQTLs in our cohort (Table S3). Evidence for colocalization/pleiotropy was found for variants in 5 of the 12 ADHD-associated regions (Table S4). For two of the regions, 12q21.33 and 15q21.1 (Figs. S9 and S10), both the SMR and coloc methods identify the same causal SNPs (rs2279574, SMR p = 2.3e–8, coloc posterior probability = 0.98, and rs1656622, SMR p = 3.6e–5, coloc posterior probability = 0.82). SNP rs2279574 is a missense variant within DUSP6, and rs1656622 lies within an intron of SEMA6D. All significant mQTLs in the 12 ADHD-associated regions are included in Table S10.
Gene set enrichment with DM probes
For exploratory and hypothesis-generating purposes, we report gene set findings. DMPs nominally associated with the ADHD PRS (p < 0.001, the recommended default in the methylGSA package)49 are annotated to 91 genes with elevated expression (five-fold higher-than-average expression in all other tissues, see Methods) in the brain (unadjusted enrichment p-value = 0.0097). Several (Fig. S11) relate to ion channels (e.g., KCNIP1, KCNK10, CACNA1E, and CACNB4) or are involved in cell adhesion (e.g., NCAM2, NRXN1, CNTNAP2, and CDH22), both previously implicated in ADHD or other psychiatric traits54,55,56,57,58. No GO categories were significantly enriched with either ADHD-associated or ADHD PRS-associated DMPs after multiple testing correction. All enrichment analysis results are shown in Table S8.
Discussion
This is the first large EWAS to examine well-characterized ADHD cases and controls, including more ADHD cases than any prior study of DNA methylation in ADHD. It is distinguished by the inclusion of polygenic risk effects—not previously studied in ADHD in an EWAS context—and the examination of sex effects. Furthermore, the inclusion of genetic effects, crucial for interpretation, has rarely been done in ADHD methylation studies. The findings make contributions pertaining to potential peripheral biomarkers for ADHD and ADHD genetic risk, provide additional evidence for the genetic regulation of many disease-associated methylation differences in ADHD, and suggest regulation of DNA methylation as a plausible mechanism for ADHD risk variants identified in GWAS. The study identifies suggestive new candidates to pursue (e.g., VIPR2, MARK2, SLC7A8, SON, and PSORS1C3), and provides new data for considering replications, or lack thereof, with previous epidemiological studies.
The study’s primary limitations are that even with our respectable sample size compared with the literature in this area, the sample is, like prior studies of DNA methylation in ADHD, probably underpowered to identify small effects. Our sample was well powered (80%) to detect methylation differences of 1% between cases and controls at ~68% of sites (Fig. S8); however, many sites show even smaller effects in our dataset and in others33,52. Thus, these findings should be viewed in a discovery context awaiting replication and as a guide for planning future studies. Furthermore, given the cross-sectional nature of this study, it is not possible to examine causality. As mentioned in the introduction, it is possible that the observed methylation differences are not causal of, but are caused by, ADHD disease processes.
With these strengths and limitations in mind, five main findings are noteworthy. First, our previous report of lower DNA methylation in VIPR2 in never-medicated boys31 was supported in a targeted analysis after removing children used in our prior report, although at a smaller effect size, suggesting that this sex-specific effect is reproducible. Sites annotated to VIPR2 have also been identified as differentially methylated in two other studies of ADHD28,32, but with effects varying in direction. Given that we observe a sex-specific effect in VIPR2 (hypomethylation in male cases, and hypermethylation in female cases) and other studies did not stratify by sex, it is possible that the inconsistencies across studies related to the direction of effect are due to differences in the sex ratio of the samples studied. Other differences across study samples, such as differences in age or specific environmental exposures, may also contribute to the inconsistent direction of effect. The role of VIPR2, a receptor for a small neuropeptide implicated in a wide range of biological functions, needs more exploration. Genetic variation in VIPR2 has been associated with schizophrenia59,60, and in one candidate gene study, with mood disorders61.
Second, we identified suggestive findings for a small number of DNA methylation sites that can provide possible biomarkers for ADHD and associated phenotypes. We note that the top two DMPs are both located in regulatory regions of their respective genes (SLC7A8 and MARK2). SLC7A8 is a sodium-independent solute transporter of neutral amino acids62 and plays a role in metal ion homeostasis and toxicity63 including mercury toxicity64. The DMP annotated to MARK2 is at the seed of two miRNAs, hsa-miR-1199-5p, and hsa-miR6751-3P, suggesting that differential methylation at this locus may indirectly affect gene expression through regulation of miRNAs. The full list of miRNA targets for this sequence using miRWalk47 is provided at https://github.com/pryabinin/ohsu_adhd_ewas. MARK2 is a regulator of cell polarity and microtubule dynamics65,66, is required for neurite outgrowth67, and has recently been implicated in both Alzheimer’s disease and bipolar disorder68.
Both of the top-ranked DMPs are also associated with nearby SNPs, indicating the complexity of the association between DNA methylation in these genes and ADHD. Of note, a variant in MARK2 has previously been associated with blood pressure response to methylphenidate69. This SNP, rs12099085 (27 kb downstream of the top-ranked mQTL), is also an mQTL suggestively associated with methylation at sites within MARK2 (minimum mQTL p = 3.8e–8), indicating that the variant might act through the regulation of DNA methylation.
Third, we identified one DMP significantly associated with polygenic risk for ADHD (ADHD PRS), and another 12 DMPs suggestively associated. The strongest association was at a site in the bivariate promoter for GART and SON, with higher PRS associated with reduced methylation. SON SNPs have been implicated in a GWAS for educational attainment70, which is noteworthy given the known relationship between ADHD and academic underachievement71, as well as the evidence for a shared genetic basis for ADHD and educational attainment72. De novo mutations in SON have been linked to intellectual disability, and gene expression studies suggest that it is a master regulator of genes involved in neurodevelopment73.
Fourth, we found a single DMR, near PSORS1C3, significantly associated with the ADHD PRS in a sex-specific manner. A previous study found that a DMR in this same region was hypomethylated in major depressive disorder (MDD) suicide cases74, although it did not assess sex-specific effects.
Finally, we found significant evidence of colocalization for mQTLs observed in our cohort and ADHD-associated SNPs, suggesting that regulation of DNA methylation may be a mechanism by which many ADHD risk variants operate.
The small number of DMPs and DMRs we observe, and the small effect sizes, are consistent with a recent EWAS meta-analysis of ADHD symptoms in adults33. That study of three population-based cohorts found three suggestively associated DMPs, none of which overlap with the DMPs reported here. The lack of overlap is not surprising, however, given the amount of heterogeneity across cohorts reported in the meta-analysis. That study also failed to see an overlap between ADHD-related DMPs and ADHD GWAS hits from the PGC. However, consistent with our findings, the study found cis-mQTLs for their top-ranked DMP, highlighting the importance of genetic effects.
In conclusion, our findings suggest that further study of DNA methylation in ADHD can be productive to identify biomarkers of illness, and potentially, disease mechanisms, although ultimately multi-position algorithms will likely be necessary to create clinically useful biomarkers10. It also appears likely to be helpful in mapping potential causal routes for genetic influences on ADHD. Future work will examine sex and medication effects, moderators of change and/or stability of DNA methylation over time, map backward to environmental correlates, and evaluate the extent to which identified genes or systems show methylation effects earlier or later in development.
References
Froehlich, T. E. et al. Update on environmental risk factors for attention-deficit/hyperactivity disorder. Curr. Psychiatry Rep. 13, 333–344 (2011).
Palladino, V. S., McNeill, R., Reif, A. & Kittel-Schneider, S. Genetic risk factors and gene-environment interactions in adult and childhood attention-deficit/hyperactivity disorder. Psychiatr. Genet. 29, 63–78 (2019).
Demontis, D. et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 51, 63 (2019).
Nigg, J. T. et al. Working memory and vigilance as multivariate endophenotypes related to common genetic risk for attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 57, 175–182 (2018).
Teroganova, N., Girshkin, L., Suter, C. M. & Green, M. J. DNA methylation in peripheral tissue of schizophrenia and bipolar disorder: a systematic review. BMC Genet. 17, 27 (2016).
Tsankova, N., Renthal, W., Kumar, A. & Nestler, E. J. Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367 (2007).
Walton, E. et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol. Psychiatry 22, 250–256 (2017).
Hamza, M. et al. Epigenetics and ADHD: toward an integrative approach of the disorder pathogenesis. J. Atten. Disord. 23, 655–664 (2019).
Aberg, K. A. et al. A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry 70, 573–581 (2013).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, 3156 (2013).
Schubeler, D. Function and information content of DNA methylation. Nature 517, 321–326 (2015).
Elliott, H. R. et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin. Epigenetics. 6, 1 (2014).
Liu, C. et al. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 23, 422–433 (2018).
Papale, L. A., Seltzer, L. J., Madrid, A., Pollak, S. D. & Alisch, R. S. Differentially methylated genes in saliva are linked to childhood stress. Sci. Rep. 8, 10785 (2018).
Houtepen, L. C. et al. Childhood adversity and DNA methylation in two population-based cohorts. Transl. Psychiatry 8, 266 (2018).
Li, Y. et al. Lead exposure during early human development and dna methylation of imprinted gene regulatory elements in adulthood. Environ. Health Perspect. 124, 666–673 (2016).
Ladd-Acosta, C. & Fallin, M. D. The role of epigenetics in genetic and environmental epidemiology. Epigenomics 8, 271–283 (2016).
Islam, S. A. et al. Integration of DNA methylation patterns and genetic variation in human pediatric tissues help inform EWAS design and interpretation. Epigenetics Chromatin. 12, 1 (2019).
Gutierrez-Arcelus, M. et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 11, e1004958 (2015).
Lappalainen, T. & Greally, J. M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 18, 441–451 (2017).
Rakyan, V. K., Down, T. A., Balding, D. J. & Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 12, 529–541 (2011).
van Mil, N. H. et al. DNA methylation profiles at birth and child ADHD symptoms. J. Psychiatr. Res. 49, 51–59 (2014).
Park, S. et al. Associations between serotonin transporter gene (SLC6A4) methylation and clinical characteristics and cortical thickness in children with ADHD. Psychol. Med. 45, 3009–3017 (2015).
Xu, Y. et al. Multiple epigenetic factors predict the attention deficit/hyperactivity disorder among the Chinese Han children. J. Psychiatr. Res. 64, 40–50 (2015).
Dadds, M. R., Schollar-Root, O., Lenroot, R., Moul, C. & Hawes, D. J. Epigenetic regulation of the DRD4 gene and dimensions of attention-deficit/hyperactivity disorder in children. Eur. Child Adolesc. Psychiatry 25, 1081–1089 (2016).
Heinrich, H. et al. Attention, cognitive control and motivation in ADHD: linking event-related brain potentials and DNA methylation patterns in boys at early school age. Sci. Rep. 7, 3823 (2017).
Rijlaarsdam, J. et al. Prenatal unhealthy diet, insulin-like growth factor 2 gene (IGF2) methylation, and attention deficit hyperactivity disorder symptoms in youth with early-onset conduct problems. J. Child Psychol. Psychiatry 58, 19–27 (2017).
Peter, C. J. et al. DNA Methylation signatures of early childhood malnutrition associated with impairments in attention and cognition. Biol. Psychiatry 80, 765–774 (2016).
Sengupta, S. M., Smith, A. K., Grizenko, N. & Joober, R. Locus-specific DNA methylation changes and phenotypic variability in children with attention-deficit hyperactivity disorder. Psychiatry Res. 256, 298–304 (2017).
Gervin, K., Nordeng, H., Ystrom, E., Reichborn-Kjennerud, T. & Lyle, R. Long-term prenatal exposure to paracetamol is associated with DNA methylation differences in children diagnosed with ADHD. Clin. Epigenetics. 9, 77 (2017).
Wilmot, B. et al. Methylomic analysis of salivary DNA in childhood ADHD identifies altered DNA methylation in VIPR2. J. Child Psychol. Psychiatry 57, 152–160 (2016).
Chen, Y. C. et al. Neuroanatomic, epigenetic and genetic differences in monozygotic twins discordant for attention deficit hyperactivity disorder. Mol. Psychiatry 23, 683–690 (2018).
van Dongen J., et al. Epigenome-wide association study of attention-deficit/hyperactivity disorder symptoms in adults. Biol. Psychiatry. 86, 599–607 (2019).
Du, P., Kibbe, W. A. & Lin, S. M. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008).
Aryee, M. J. et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Houseman, E. A. et al. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinform. 17, 259 (2016)
McCartney, D. L. et al. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genomics Data. 9, 22–24 (2016).
Conomos, M. P., Reiner, A. P., Weir, B. S. & Thornton, T. A. Model-free estimation of recent genetic relatedness. Am. J. Hum. Genet. 98, 127–148 (2016).
Mowlem, F., Agnew-Blais, J., Taylor, E. & Asherson, P. Do different factors influence whether girls versus boys meet ADHD diagnostic criteria? Sex differences among children with high ADHD symptoms. Psychiatry Res. 272, 765–773 (2019).
Martin, J. et al. A genetic investigation of sex bias in the prevalence of attention-deficit/hyperactivity disorder. Biol. Psychiatry 83, 1044–1053 (2018).
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 28, 2986–2988 (2012).
Roberts, S. et al. Longitudinal investigation of DNA methylation changes preceding adolescent psychotic experiences. Transl. Psychiatry 9, 69 (2019).
Mallik, S. et al. An evaluation of supervised methods for identifying differentially methylated regions in Illumina methylation arrays. Brief Bioinform. 20, 2224–2235 (2018)
Pan, H., Holbrook, J. D., Karnani, N. & Kwoh, C. K. Gene, Environment and Methylation (GEM): a tool suite to efficiently navigate large scale epigenome wide association studies and integrate genotype and interaction between genotype and environment. BMC Bioinforma. 17, 299 (2016).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Dweep, H. & Gretz, N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat. Methods 12, 697 (2015).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Ren, X. & Kuan, P. F. methylGSA: a bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics 35, 1958–1959 (2019).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Mansell, G. et al. Guidance for DNA methylation studies: statistical insights from the Illumina EPIC array. BMC Genomics. 20, 366 (2019).
Hannon, E. et al. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 10, 19 (2018).
Perrier, F. et al. Identifying and correcting epigenetics measurements for systematic sources of variation. Clin. Epigenetics. 10, 38 (2018).
Rivero, O. et al. Impact of the ADHD-susceptibility gene CDH13 on development and function of brain networks. Eur. Neuropsychopharmacol. 23, 492–507 (2013).
Lesch, K. P. et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J. Neural Transm. (Vienna). 115, 1573–1585 (2008).
Liu, Q. R. et al. Addiction molecular genetics: 639,401 SNP whole genome association identifies many “cell adhesion” genes. Am. J. Med Genet. B Neuropsychiatr. Genet. 141b, 918–925 (2006).
Mooney, M. A. et al. Pathway analysis in attention deficit hyperactivity disorder: An ensemble approach. Am. J. Med Genet. B Neuropsychiatr. Genet. 171, 815–826 (2016).
Askland, K., Read, C., O’Connell, C. & Moore, J. H. Ion channels and schizophrenia: a gene set-based analytic approach to GWAS data for biological hypothesis testing. Hum. Genet. 131, 373–391 (2012).
Vacic, V. et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature 471, 499–503 (2011).
Levinson, D. F. et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry 168, 302–316 (2011).
Soria, V. et al. Differential association of circadian genes with mood disorders: CRY1 and NPAS2 are associated with unipolar major depression and CLOCK and VIP with bipolar disorder. Neuropsychopharmacology 35, 1279–1289 (2010).
Pochini, L., Scalise, M., Galluccio, M. & Indiveri, C. Membrane transporters for the special amino acid glutamine: structure/function relationships and relevance to human health. Front. Chem. 2, 61 (2014).
Simmons-Willis, T. A., Koh, A. S., Clarkson, T. W. & Ballatori, N. Transport of a neurotoxicant by molecular mimicry: the methylmercury–l-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2. Biochem J. 367, 239–246 (2002).
Stamova, B. et al. Correlations between gene expression and mercury levels in blood of boys with and without autism. Neurotox. Res. 19, 31–48 (2011).
Drewes, G., Ebneth, A., Preuss, U., Mandelkow, E.-M. & Mandelkow, E. MARK, a Novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 89, 297–308 (1997).
Ducharme, N. A. et al. MARK2/EMK1/Par-1Bα phosphorylation of Rab11-Family interacting protein 2 is necessary for the timely establishment of polarity in Madin-Darby canine kidney cells. Mol. Biol. Cell. 17, 3625–3637 (2006).
Biernat, J. et al. Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol. Biol. Cell. 13, 4013–4028 (2002).
Drange, O. K. et al. Genetic overlap between alzheimer’s disease and bipolar disorder implicates the MARK2 and VAC14 Genes. Front Neurosci. 13, 220 (2019).
Mick, E., McGough, J. J., Middleton, F. A., Neale, B. & Faraone, S. V. Genome-wide association study of blood pressure response to methylphenidate treatment of attention-deficit/hyperactivity disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 466–472 (2011).
Lee, J. J. et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 50, 1112–1121 (2018).
Fleming, M. et al. Educational and health outcomes of children treated for attention-deficit/hyperactivity disorder. JAMA Pediatr. 171, e170691 (2017).
Shadrin, A. A. et al. Novel loci associated with attention-deficit/hyperactivity disorder are revealed by leveraging polygenic overlap with educational attainment. J. Am. Acad. Child Adolesc. Psychiatry 57, 86–95 (2018).
Kim, J. H. et al. De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am. J. Hum. Genet. 99, 711–719 (2016).
Murphy, T. M. et al. Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl. Psychiatry 7, e989 (2017).
Acknowledgements
This study was supported by the Abracadabra Foundation of Portland Oregon and the National Institute of Mental Health (R01MH099064).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mooney, M.A., Ryabinin, P., Wilmot, B. et al. Large epigenome-wide association study of childhood ADHD identifies peripheral DNA methylation associated with disease and polygenic risk burden. Transl Psychiatry 10, 8 (2020). https://doi.org/10.1038/s41398-020-0710-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-0710-4
This article is cited by
-
Gestational epigenetic age and ADHD symptoms in childhood: a prospective, multi-cohort study
Molecular Psychiatry (2024)
-
Cortico-striatal differences in the epigenome in attention-deficit/ hyperactivity disorder
Translational Psychiatry (2024)
-
Epigenome-wide association study identifies neonatal DNA methylation associated with two-year attention problems in children born very preterm
Translational Psychiatry (2024)
-
ADHD genetic burden associates with older epigenetic age: mediating roles of education, behavioral and sociodemographic factors among older adults
Clinical Epigenetics (2023)
-
DNA methylation is associated with prenatal exposure to sulfur dioxide and childhood attention-deficit hyperactivity disorder symptoms
Scientific Reports (2023)
