
Abstract
Attention-deficit and hyperactivity disorder (ADHD) is a common childhood disorder with a substantial genetic component. However, the extent to which epigenetic mechanisms play a role in the etiology of the disorder is unknown. We performed epigenome-wide association studies (EWAS) within the Pregnancy And Childhood Epigenetics (PACE) Consortium to identify DNA methylation sites associated with ADHD symptoms at two methylation assessment periods: birth and school age. We examined associations of both DNA methylation in cord blood with repeatedly assessed ADHD symptoms (age 4–15 years) in 2477 children from 5 cohorts and of DNA methylation at school age with concurrent ADHD symptoms (age 7–11 years) in 2374 children from 9 cohorts, with 3 cohorts participating at both timepoints. CpGs identified with nominal significance (p < 0.05) in either of the EWAS were correlated between timepoints (ρ = 0.30), suggesting overlap in associations; however, top signals were very different. At birth, we identified nine CpGs that predicted later ADHD symptoms (p < 1 × 10–7), including ERC2 and CREB5. Peripheral blood DNA methylation at one of these CpGs (cg01271805 in the promoter region of ERC2, which regulates neurotransmitter release) was previously associated with brain methylation. Another (cg25520701) lies within the gene body of CREB5, which previously was associated with neurite outgrowth and an ADHD diagnosis. In contrast, at school age, no CpGs were associated with ADHD with p < 1 × 10−7. In conclusion, we found evidence in this study that DNA methylation at birth is associated with ADHD. Future studies are needed to confirm the utility of methylation variation as biomarker and its involvement in causal pathways.
Similar content being viewed by others
Introduction
Attention-deficit and hyperactivity disorder (ADHD) is a common neurodevelopmental disorder characterized by impulsivity, excessive activity, and attention problems. Symptoms often become apparent during school age with a world-wide prevalence of 5–7.5%1. Genetic heritability is estimated between 64 and 88%2,3. Additionally, several environmental factors are suspected to impact ADHD, e.g., prenatal maternal smoking or lead exposure4,5,6,7. However, the genetics and environmental pathways contributing to ADHD risk remain unclear. Possibly, DNA methylation, an epigenetic mechanism regulating gene expression, may mediate genetic or environmental effects.
Several studies have investigated DNA methylation in relation to ADHD diagnoses or symptoms using candidate approaches or epigenome-wide association studies (EWAS) in peripheral blood and saliva tissue8,9. A leading hypothesis concerning the etiology of ADHD suggests that deficiencies in the dopamine system of the brain impact ADHD development4,10. Consequently, candidate studies have focused on genes related to dopamine function. For instance, DNA methylation alterations in DRD411,12,13, DRD512, and DAT112,14 genes have been associated with ADHD, though not consistently15. Beyond the candidate gene approach, three studies tested DNA methylation across the whole genome. One study performed an EWAS with saliva samples in school-aged children using a case–control design16. The study identified differentially methylated probes in VIPR2, a gene expressed in the caudate and previously associated with psychopathology. Another EWAS investigated cord and peripheral blood DNA methylation at birth and at 7 years of age17. At birth, 13 probes located in SKI, ZNF544, ST3GAL3, and PEX2 were associated with ADHD trajectories from age 7 to 15 years, but the methylation status of these probes at age 7 was not associated with ADHD cross-sectionally. An EWAS in adults with ADHD failed to find any differentially methylated sites in peripheral blood18.
Large multi-center epigenome-wide studies, which allow for increased power and generalizability, are lacking for childhood. Here we performed the first epigenome-wide prospective meta-analysis to identify DNA methylation sites associated with childhood ADHD symptoms in cohorts from the Pregnancy And Childhood Epigenetics (PACE) Consortium19. As DNA methylation changes over time20, so could potential associations with ADHD symptoms. On the one hand, one might expect that DNA methylation levels measured around the same time as ADHD symptoms would show the largest associations, as these might represent the immediate effects on symptoms or consequences of ADHD. On the other hand, causes of ADHD may be found early in childhood or even prenatally. Thus methylation levels at birth may be more relevant than later methylation profiles, as suggested by an earlier EWAS21. Since it is unclear when DNA methylation is most relevant for ADHD symptoms, we tested DNA methylation both at birth using cord blood and at school age (age 7–9 years) using DNA derived from peripheral whole blood. In the analyses of cord blood methylation, the aim was to explain ADHD symptoms between ages 4 and 15 years. Many participating cohorts assessed ADHD repeatedly, and we employed a repeated-measures design to increase precision. Furthermore, we utilized data in childhood to examine cross-sectional DNA methylation patterns associated with ADHD symptoms at school age.
Materials and methods
This study comprises a birth methylation EWAS and a school-age methylation EWAS described successively below.
Birth methylation EWAS
Participants
Five cohorts (Avon Longitudinal Study of Parents and Children (ALSPAC)22,23,24, Generation R (GENR)25, INfancia y Medio Ambiente (INMA)26, Newborn Epigenetic Study (NEST)27,28, and Prediction and prevention of preeclampsia and intrauterine growth restriction (PREDO)29) in the PACE consortium had information on DNA methylation in cord blood and ADHD symptoms. These cohorts have a combined sample size of 2477 (Table 1). Participants were mostly of European ancestry, except for NEST, an American cohort that also included participants of African ancestry. In NEST, separate EWAS were conducted for participants identifying as black or white to account for ancestry heterogeneity statistically in a random-effects meta-analysis. We also performed a sensitivity analyses with European ancestry children only. Parents gave informed consent for their children’s participation and local ethics committees approved the study protocols. See Supplementary Information 1 for full cohort descriptions.
DNA methylation and quality control (QC)
DNA methylation in cord blood was measured using the Illumina Infinium HumanMethylation450K BeadChip (Table S1). Methylation levels outside of the lower quartile minus 3 × interquartile or upper quartile plus 3 × interquartile range were removed. Each cohort ran the EWAS separately according to a pre-specified harmonized analysis plan. The distribution of the regression estimates and p values were examined for each cohort and pooled results. Deviations from a normal distribution of regression estimates or a higher number of low p values than expected by chance may be signs of residual confounding or the result of a true poly-epigenetic signal. To help in interpretation of the results, we used the BACON method30. BACON analyzes the distribution of regression coefficients and estimates an empirical null distribution. Results can then be compared against the empirical null, which already includes biases, rather than the theoretical null. We excluded CpG probes, which were available in <4 cohorts; <1000 participants; and allosomal probes, due to the complex interpretation of dosage compensation.
ADHD symptoms
ADHD symptoms were measured when children were aged 4–15 years (depending on the cohort) with parent-rated instruments, specifically the Behavior Assessment System for Children31, Child Behavior Checklist (CBCL)32,33, Conners34 and the Development and Well-Being Assessment (DAWBA)35 (Table S2). If a cohort had measured ADHD symptoms repeatedly (three cohorts), we used a mixed model (see “Statistical analysis”). The repeated-measures design increased the precision of the ADHD severity estimate and sample size, since missing data in an assessment can be handled with maximum likelihood. Given the variety of instruments used within and across cohorts, all ADHD scores were z-score standardized to enable meta-analysis.
Statistical analysis
Cohorts with repeated ADHD assessment were analyzed using linear mixed models, with z-scores of ADHD symptoms as the outcome and methylation (in betas, ranging from 0 (unmethylated) to 1 (methylated)) as the main predictor. Each CpG probe was analyzed separately and pooled p values were adjusted for multiple correction using Bonferroni adjustment. We used a random intercept on the participant and batch level, to account for clustering due to repeated measures and batch effects. The following potential confounders were included as fixed effects: maternal age, educational level, smoking status (yes vs no during pregnancy), gestational age, sex, and estimated white blood cell proportions (Bakulski reference estimated with the Houseman method)36. Mixed models were fitted using restricted maximum likelihood. We used R37 with the lme438 package to estimate the models. Cohorts with a single ADHD assessment wave used a model without random effects or batch level only.
Meta-analysis was performed using the Han and Eskin random-effects model39. This model does not assume that true effects are homogeneous between cohorts; however, it does assume that null effects are homogeneous. This modified version of the random effect model has comparable power to a fixed-effects analysis, while better accounting for study heterogeneity, such as ancestry differences, in simulation studies39,40. Genome-wide significance was defined at the Bonferroni-adjustment threshold of p < 1 × 10–7, suggestive significance at p < 1 × 10–5, and nominal significance at p < 0.05.
Follow-up analyses
We performed several lookups of genome-wide significant probes. We used the BECon database41 to check the correlation between peripheral and brain methylation levels in postmortem tissue. To test genetic influence, we interrogated the genome-wide significant probes in MeQTL42 and twin heritability databases43. We also attempted to replicate genome-wide significant probes reported in a previous EWAS from the ALSPAC study17. For replication, we reran the meta-analysis without the ALSPAC cohort. To quantify the variance explained by genome-wide significant probes, we predicted ADHD scores at age 8 years in Generation R by all meta-analytically genome-wide significant probes. We applied 10-fold cross-validation with 100 repetitions to improve generalizability and reduce bias from Generation R, which was part of the discovery.
We examined whether any CpG sites associated with ADHD symptoms are also associated with prenatal maternal stress. As prenatal maternal stress is associated with child psychopathology with mixed evidence of affecting DNA methylation44,45, DNA methylation may be a mediator of adverse prenatal stress effects. We operationalized prenatal maternal stress as in Rijlersdaam et al.45 by using a factor score reflecting life, contextual, personal stress, and interpersonal stress. One modification to the previous definition is that maternal education was not used in the computation of the factor score, as it had been included as covariate in the EWAS model. We first tested whether prenatal stress was associated with ADHD symptoms with an analysis model equivalent to the EWAS model, but instead of DNA methylation, the prenatal risk score was the main predictor. We then tested associations between prenatal stress as predictor and DNA methylation as outcome. p Values were obtained with the lmerTest package46. We estimated the prenatal stress associations only in the Generation R cohort.
Pathway analysis
Pathway enrichment analysis were performed with the missMethylpackage47 on suggestive probes (p < 1 × 10–5). We used as references gene ontology, Kyoto Encyclopedia of Genes and Genomes, and curated gene sets (http://software.broadinstitute.org/gsea/msigdb/collections.jsp#C2) from the Broad Institute Molecular signatures database48. p Values were adjusted using the default procedures by the number of CpGs associated with each gene49 and false discovery rate.
To test enrichment for regulatory features (gene relative position, CpG island relative position, and blood chromatin states), we applied χ2 tests. Enrichment tests were performed for all CpGs, hypomethylated CpGs, and hypermethylated CpGs separately. CpG annotation was performed with the IlluminaHumanMethylation450kanno.ilmn-12.hg19 R package50. Annotation to chromatin states was from the Roadmap Epigenomics Project (https://egg2.wustl.edu/roadmap/web_portal/). See Supplementary Information 2 for full description.
School-age methylation EWAS
Participants
Nine cohorts (ALSPAC, GENR, HELIX51, and GLAKU52) with a combined sample size of 2374 joined the school-age methylation EWAS (Table 1 and Supplementary Information 1). HELIX consists of six jointly analyzed subcohorts51. All cohorts had participants of European ancestry, except HELIX, which also included participants with a Pakistani background living in the UK and were treated as a separate cohort in the meta-analysis. Again, we accounted for ancestry heterogeneity with a random-effects meta-analysis and also present European ancestry only results as sensitivity analysis. Fifty-three percent of participants in the school-age EWAS were also part of the birth EWAS.
DNA methylation and QC
DNA methylation was measured at ages 7–12 years in peripheral whole blood. The Illumina Infinium HumanMethylation450K BeadChip and Infinium MethylationEPIC Kit (GLAKU) were used to interrogate CpG probes. QC steps were identical to the birth methylation EWAS.
ADHD symptoms
ADHD symptoms were measured at the same age as DNA methylation (age 7–11 years) with the parent-rated measures DAWBA and CBCL (Table S2). Only the assessment closest to the DNA methylation assessment age was analyzed.
Statistical analysis
The statistical model was similar to the model used in the birth methylation EWAS without participant-level random effect. However, cell counts were estimated with the Houseman method using the Reinius reference53. We also added assessment age as covariate. The meta-analysis methods were identical to the birth methylation EWAS.
Follow-up analyses
We did not perform follow-up analyses due to low signal. However, we attempted to replicate six probes identified as suggestive in a previous case–control EWAS in school age16.
Results
Birth cord blood methylation
EWAS quality check
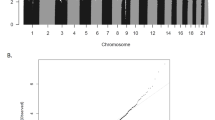
Four out of the six cohorts showed larger number of low p values than expected under the null, as indexed by high λ (Table 1). BACON analysis suggested that the majority of the inflation was due to a true signal, as indicated by inflation values clearly <λ. To test the impact of sample size on λ, we restricted the GENR sample randomly to 900 and 1100 participants, resulting in 812 and 991 participants due to missing covariates. The lambdas were 0.96, 1.21, 1.51 for 812, 991, and 1191 participants. We thus conclude that the overrepresentation of low p values is mostly due to sufficient power to detect associations at higher sample sizes.
The BACON analyses also indicated a trend toward positive/negative regression coefficients in some of the datasets, which might indicate confounding, e.g., by population stratification. To test this, we added principal components of ancestry in GENR and ALSPAC, but these did not meaningfully change results.
We conducted the meta-analysis under the assumption that any such biases will be corrected in the pooled analysis, since they were not homogeneous across cohorts. Indeed, the pooled estimates did not show a trend toward positive or negative regression estimates (Median = +0.02), only an overrepresentation of low p values (λ = 1.86, Fig. 1). The BACON estimates for inflation suggested that these are mostly due to a true signal (Inflation = 1.1).

The diagonal line represents the distribution of the expected p values under the null. Points above the diagonal indicate p values that are lower than expected.
Single probe analysis
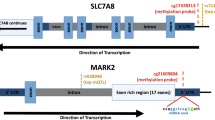
After QC, 472,817 CpG sites remained for the meta-analysis. Results of the cord blood EWAS are shown in Fig. 2. Nine CpG sites showed genome-wide significance (p < 1 × 10–7, Table 2). ADHD symptoms were between 0.16 SD (SE = 0.03) and 0.44 SD (SE = 0.12) higher with 10% lower methylation at these probes. Eight probes out of nine that were available in the BECon database41 are typically methylated in both whole blood and the brain (Figs. 3, S1, and S2). A lookup in the BECon database revealed that the CpG site cg01271805 in the promoter region of gene ERC2 shows variable methylation in three brain regions (BA10, BA20, BA7). Importantly, methylation levels in the brain are moderately correlated with whole-blood methylation (ρ = 0.33–0.46; Fig. 3), suggesting that peripheral cg01271805 methylation levels are a useful marker for brain methylation levels. The other seven genome-wide significant probes showed less consistent correlations between blood and brain tissues and associated genes had less specificity for expression in the brain, based on GTEx54 data. No single-nucleotide polymorphism (SNP) was associated with our nine top CpG probes when accounting for linkage disequilibrium according to the MeQTL database42. Furthermore, all nine probes had a twin heritability <20% in a previous study (Table S3)43. In Generation R, the joint explained variance of ADHD scores at age 8 years by the genome-wide significant probes was 2.0.% (R2 from 10-fold repeated cross-validation). Full EWAS results can be found in Supplementary Data. After adjusting for inflation and bias with BACON, only one CpG remained statistically significant (cg25520701, CREB5, β = −3.54, SE = 0.66, p = 9.59 × 10–8). It should be noted that the BACON adjusted p values rely on statistics from the traditional random effects model. With the traditional model, only cg25520701, cg09762907, and cg22997238 remained genome-wide significant. Thus the difference in p value is not solely the result of adjustment for the inflation but also the use of more conservative tests. When restricting analyses to participants with European ancestry, top hits remained unchanged but one additional CpG site became genome-wide significant: cg10025904 in gene LRRC8B (Table S4).

Red line indicates genome-wide significant (p < 1 × 10–7) and blue line suggestive threshold (p < 1 × 10–5).

Columns 1-4 indicate the locations of the CpG sites. Columns 5-8 show the variability of DNA methylation in three brain regions and blood, with higher values indicating higher variability. Columns 9-11 contain the correlations between brain and blood DNA methylation levels. Columns 12-13 state how much DNA methylation is influenced by cell composition, with higher values indicating higher effect.
Prenatal stress was associated with ADHD symptoms in childhood. One SD higher prenatal stress was associated with 0.2 SD higher ADHD symptoms (SE = 0.03, p = 2E−13, n = 1121). However, prenatal stress was not associated with any of the genome-wide significant sites (Table S5).
Pathway analysis
Two-hundred forty-nine probes showed suggestive (p < 1 × 10–5) associations and were annotated to 182 unique genes. In gene-based analyses, no pathway survived multiple testing correction.
The 248 suggestive CpGs were enriched in intergenic regions. Of these, hypomethylated CpGs were enriched for 3’-untranslated regions and depleted for TSS200 and first exon regions, open sea, north shelf and south shelf regions, south shore, and islands. Regarding chromatin states, hypomethylated probes showed an enrichment for transcription (Tx and TxWk), quiescent positions, and depletion for transcription start site positions (TSSA, TxFlnk, TxFlnk), bivalent (EnhBiv), and repressor (ReprPC) positions. Hypermethylated probes showed the opposite enrichment/depletion patterns. See Supplementary Information 2 for full results.
Replication of previous EWAS
We attempted to replicate findings for 13 CpGs, at which DNA methylation at birth was associated with ADHD trajectories17. However, no probe survived multiple testing correction (Table S6).
School-age methylation
EWAS quality checks
The regression coefficient distribution showed no signs of errors, but three out of the five cohorts showed a trend toward positive associations in separate analyses (Table 1). The lambda was below 1.11 for all cohorts. BACON suggested no inflation of the test statistics due to confounding or other biases, though the trend toward positive associations remained. The pooled results showed a low lambda (λ = 0.96), no inflation (BACON inflation estimate = 0.92), but a slight overrepresentation of positive associations (BACON bias estimate = 0.14).
Single probe meta-analysis
We associated DNA methylation at school age in whole blood at 466,574 CpG sites with ADHD symptoms at the same age. No CpG reached genome-wide significance (all p > 4.96E−06, Fig. 2). Furthermore, none of the loci at which DNA methylation at birth was significantly associated with ADHD symptoms showed a cross-sectional association at school age (p > 0.33, Table 2). Restricting analyses to children with European ancestry did not change these results.
Replication of previous EWAS
We attempted to replicate the six most suggestive EWAS CpGs of a previous case–control study16. While all but one showed a consistent direction, none of the CpGs were statistically significant (Table S7).
Stability of methylation association across age
The associations between methylation at birth with ADHD symptoms and methylation at school age with ADHD symptoms were largely consistent for nominally significant probes. The regression estimates from CpG sites, with nominally significant associations at birth (p < 0.05, n = 73,057) correlated with the regression estimates of the school-age EWAS (ρ = 0.45). When restricting the school-age methylation EWAS to those cohorts, which were not featured in the birth methylation EWAS (thus excluding overlaps), the correlation remained (ρ = 0.30). Vice versa, when filtering for probes that were nominally significant at school age, 23,770 probes remained of which 4075 overlapped with nominally significant probes at birth. The correlation for this set was very similar, ρ = 0.47 among all cohorts and ρ = 0.35 between independent cohorts.
Discussion
In this population-based study, we performed the first epigenome-wide meta-analysis of ADHD symptoms in childhood, using two DNA methylation assessments (birth and school age), as well as repeated measures of ADHD symptoms. DNA methylation at birth, but not at school age, was associated with later development of ADHD symptoms with genome-wide significance at nine loci. Interestingly, the identified probes showed a pattern of a high average rate of methylation in cord blood, while lower levels of methylation were associated with more ADHD symptoms in childhood. DNA methylation in cord blood reflects the effects of genetics and the intrauterine environment. The results suggest that cord blood DNA methylation is a marker for some of the ADHD risk factors before birth or functions as a potential mediator of these risk factors. While not impossible, reverse causality at this age is unlikely to explain our results, as ADHD only manifests at later stages of development.
We analyzed DNA methylation in cord and peripheral blood, which may not correspond to the methylation status in the brain. While DNA methylation in the periphery may affect behavior via various pathways (e.g., by affecting systemic inflammation), DNA methylation in the brain arguably has the strongest a priori likelihood of representing causal mechanisms. Seven out of eight significant probes did not show consistent correlation between methylation status in whole blood and postmortem brain tissue in a previous study, i.e., DNA methylation levels in blood may not represent brain levels and thus associations with ADHD may be different41. However, methylation levels of cg01271805 in whole blood are associated with methylation levels in various brain regions. Importantly, this probe lies in the promoter region of the gene ERC2, which is highly expressed in brain tissue. ERC2 regulates calcium-dependent neurotransmitter release in the axonal terminal55. Specifically, ERC2 is suspected to increase the sensitivity of voltage-dependent calcium channels to hyperpolarization, resulting in higher neurotransmitter release. SNPs in the ERC2 locus have been suggested to distinguish schizophrenia and bipolar disorder patients56 and to impact cognitive functioning57. ERC2 is especially expressed in Brodmann area 9 of the frontal cortex54. Previous imaging studies have demonstrated differential activation in this area when children with or without ADHD performed various cognitive tasks58,59. The correlation with brain methylation, the location in a promoter, and gene expression in the brain make cg01271805 a plausible candidate locus, where reduced methylation may be mechanistically involved in ADHD development. We hypothesize that lower methylation levels at cg01271805 increases the expression of ERC2, which in turn increases neurotransmitter release, with an adverse impact on the development of ADHD symptoms. Another gene with a genome-wide significant probe and high relevance for neural functioning is CREB5 (cg25520701). CREB5 is expressed in the fetal brain and the prefrontal cortex and has been previously related to neurite outgrowth. Moreover, SNPs in this gene were associated with ADHD in two recent GWAS60,61. Thus it is plausible that differences in DNA methylation at this locus may modify ADHD risk during development. To the best of our knowledge, none of the top CpG sites have been associated with psychopathology before.
While the birth methylation EWAS identified several loci, associating school-age methylation with concurrent ADHD symptoms revealed no genome-wide significant associations. Furthermore, the overall association signal was lower, despite similar sample sizes. None of the probes, which were significantly associated at birth, showed any association when measured at school age. Given that sample sizes were comparable, this difference must come from changes in the epigenome or study heterogeneity, rather than differences in statistical power. In terms of instrument heterogeneity, the school-age EWAS was more homogeneous, almost exclusively using CBCL. Additionally, as both EWAS feature a mix of several cohorts selected based on the same criteria and around half of the participants were represented at both timepoints, study heterogeneity appears to be an unlikely explanation. The stronger signal in the birth EWAS may be considered surprising given that typically two measures are more strongly associated if measured in closer temporal proximity. However, in line with our results Walton et al. also observed in a previous EWAS17 that birth methylation may be a better predictor of later ADHD symptoms than childhood methylation, possibly reflecting sensitive periods. Whether DNA methylation in cord blood has stronger causal effects or is a better marker for early life factors cannot be concluded from the present study. Alternatively, tissue differences between cord blood and whole blood may account for the differences in association pattern. Finally, it is possible that interventions in childhood and other environmental influences reduced the initial epigenetic differences at birth between children with higher and lower ADHD symptoms. Yet, we observed consistency in the associations of methylation at both timepoints with ADHD symptoms. The regression estimates of both EWAS correlated on a genome-wide level.
Strengths of this study include the large sample size, repeated outcome measures, extensive control for potential confounders, and the use of DNA methylation at two different timepoints, enabling us to characterize both prospective and cross-sectional associations with ADHD symptoms. However, several limitations need to be discussed as well. A causal interpretation of our findings is challenged by the possibility of residual confounding and reverse causality. DNA methylation might be a marker for untested adverse environmental factors that could affect ADHD via independent pathways. In addition, children with higher ADHD symptoms may evoke a particular environment, which might shape the epigenome. Furthermore, we had limited ability to infer from our data DNA methylation in the brain during birth, as the BECon database is based on a small sample size, features a limited selection of brain areas, and DNA methylation is measured postmortem. As is typical for (epi-)genetic studies, the effect size of individual top probes was rather small in our study: the joint effect of the genome-wide probes was estimated at 2%. However, the strong genome-wide epigenetic signal suggests a potential for the development of epigenetic scores based on birth methylation, which could lead to early prevention efforts before ADHD symptoms arise. Future studies with larger sample sizes are therefore necessary to detect further methylation sites.
In summary, we identified nine CpG sites for which lower methylation status at birth is associated with later development of ADHD symptoms. The results suggest that DNA methylation in ERC2 and CREB5 may exert an influence on ADHD symptoms, potentially via modification of neurotransmitter functioning or neurite outgrowth.
References
Thomas, R., Sanders, S., Doust, J., Beller, E. & Glasziou, P. Prevalence of attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Pediatrics 135, e994–e1001 (2015).
Bergen, S. E., Gardner, C. O. & Kendler, K. S. Age-related changes in heritability of behavioral phenotypes over adolescence and young adulthood. Twin Res. Hum. Genet. 10, 423–433 (2007).
Larsson, H., Chang, Z., D’Onofrio, B. M. & Lichtenstein, P. The heritability of clinically diagnosed attention deficit hyperactivity disorder across the lifespan. Psychol. Med. 44, 2223–2229 (2014).
Swanson, J. M. et al. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol. Rev. 17, 39–59 (2007).
Marceau, K. et al. Within-family effects of smoking during pregnancy on ADHD: the importance of phenotype. J. Abnorm. Child Psychol. 46, 685–699 (2018).
Daneshparvar, M. et al. The role of lead exposure on attention-deficit/hyperactivity disorder in children: a systematic review. Iran. J. Psychiatry 11, 1–14 (2016).
Lam, J. et al. Developmental pbde exposure and IQ/ADHD in childhood: a systematic review and meta-analysis. Environ. Health Perspect. 125, 086001 (2017).
Barker, E. D., Walton, E. & Cecil, C. A. M. Annual Research Review: DNA methylation as a mediator in the association between risk exposure and child and adolescent psychopathology. J. Child Psychol. Psychiatry 4, 303–322 (2017).
Dall’Aglio, L. et al. The role of epigenetic modifications in neurodevelopmental disorders: a systematic review. Neurosci. Biobehav Rev. 94, 17–30 (2018).
Pappa, I. et al. DRD4 VNTRs, observed stranger fear in preschoolers and later ADHD symptoms. Psychiatry Res. 220, 982–986 (2014).
van Mil, N. H. et al. DNA methylation profiles at birth and child ADHD symptoms. J. Psychiatr. Res. 49, 51–59 (2014).
Xu, Y. et al. Multiple epigenetic factors predict the attention deficit/hyperactivity disorder among the Chinese Han children. J. Psychiatr. Res. 64, 40–50 (2015).
Dadds, M. R., Schollar-Root, O., Lenroot, R., Moul, C. & Hawes, D. J. Epigenetic regulation of the DRD4 gene and dimensions of attention-deficit/hyperactivity disorder in children. Eur. Child Adolesc. Psychiatry 25, 1081–1089 (2016).
Adriani, W. et al. Potential for diagnosis versus therapy monitoring of attention deficit hyperactivity disorder: a new epigenetic biomarker interacting with both genotype and auto-immunity. Eur. Child Adolesc. Psychiatry 27, 241–252 (2018).
Ding, K. et al. DAT1 methylation is associated with methylphenidate response on oppositional and hyperactive-impulsive symptoms in children and adolescents with ADHD. World J. Biol. Psychiatry 18, 291–299 (2017).
Wilmot, B., Fry, R., Smeester, L., Musser, E. D. & Mill, J. Methylomic analysis of salivary DNA in childhood ADHD identifies altered DNA methylation in VIPR2. J. Child Psychol. Psychiatry Allied Discip. 57, 152–160 (2016).
Walton, E. et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol. Psychiatry 22, 250–256 (2017).
van Dongen, J. et al. Epigenome-wide association study of attention-deficit/hyperactivity disorder symptoms in adults. Biol. Psychiatry 86, 599–607 (2019).
Felix, J. F. et al. Cohort profile: Pregnancy And Childhood Epigenetics (PACE) consortium. Int. J. Epidemiol. 47, 22u–23u (2018).
Horvath, S. et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 13, R97 (2012).
Walton, E. et al. Epigenetic profiling of ADHD symptoms trajectories: a prospective, methylome-wide study. Mol. Psychiatry 22, 250–256 (2017).
Boyd, A. et al. Cohort profile: The ‘Children of the 90s’-the index offspring of the avon longitudinal study of parents and children. Int. J. Epidemiol. 42, 111–127 (2013).
Fraser, A. et al. Cohort profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int. J. Epidemiol. 42, 97–110 (2012).
Relton, C. L. et al. Data resource profile: Accessible Resource for Integrated Epigenomic Studies (ARIES). Int. J. Epidemiol. 44, 1181–1190 (2015).
Kooijman, M. N. et al. The Generation R Study: design and cohort update 2017. Eur. J. Epidemiol. 31, 1243–1264 (2016).
Guxens, M. et al. Cohort profile: the INMA-INfancia y Medio Ambiente-(environment and childhood) project. Int. J. Epidemiol. 41, 930–940 (2012).
Hoyo, C. et al. Methylation variation at IGF2 differentially methylated regions and maternal folic acid use before and during pregnancy. Epigenetics 6, 928–936 (2011).
Liu, Y. et al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements. Epigenetics 7, 735–746 (2012).
Girchenko, P. et al. Cohort profile: prediction and prevention of preeclampsia and intrauterine growth restriction (PREDO) study. Int. J. Epidemiol. 46, 1380–1381 (2017).
van Iterson, M. et al. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol. 18, 1–13 (2017).
Reynolds, C. R. & Kamphaus, R. W. Behavior assessment system for children: Manual supplement for the clinical report. Second Edition. 7. (Pearson, 2010). Retrieved November 3rd 2020, from http://images.pearsonclinical.com/images/ca/basc2_manual.pdf.
Achenbach, T. M. & Rescorla, L. A. Manual for the ASEBA Preschool Forms and Profiles (University of Vermont, Research Center for Children, Youth, & Families, Burlington, VT, 2000).
Achenbach, T. M. & Rescorla, L. A. Manual for the ASEBA School-Age Forms and Profiles (University of Vermont, Research Center for Children, Youth, & Families, Burlington, VT, 2001).
Conners, C. K., Sitarenios, G., Parker, J. D. A. & Epstein, J. N. The revised Conners’ Parent Rating Scale (CPRS-R): factor structure, reliability, and criterion validity. J. Abnorm. Child Psychol. 26, 257–268 (1998).
Goodman, R., Ford, T., Richards, H., Gatward, R. & Meltzer, H. The development and well-being assessment: description and initial validation of an integrated assessment of child and adolescent psychopathology. J. Child Psychol. Psychiatry 41, 645–655 (2000).
Bakulski, K. M. et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics 11, 354–362 (2016).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, 2016).
Bates, D., Mächler, M., Bolker, B. M. & Walker, S. C. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–51 (2014).
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Hong, J., Lunetta, K. L., Cupples, L. A., Dupuis, J. & Liu, C.-T. Evaluation of a two-stage approach in trans-ethnic meta-analysis in genome-wide association studies. Genet. Epidemiol. 40, 284–292 (2016).
Edgar, R. D., Jones, M. J., Meaney, M. J., Turecki, G. & Kobor, M. S. BECon: a tool for interpreting DNA methylation findings from blood in the context of brain. Transl. Psychiatry 7, e1187 (2017).
Smith, A. K. et al. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. BMC Genomics 15, 145 (2015).
Hannon, E. et al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLoS Genet. 14, 1–27 (2018).
Cecil, C. A. M. et al. Environmental risk, Oxytocin Receptor Gene (OXTR) methylation and youth callous-unemotional traits: a 13-year longitudinal study. Mol. Psychiatry 19, 1071–1077 (2014).
Rijlaarsdam, J. et al. An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: a model approach for replication. Epigenetics 11, 140–149 (2016).
Kuznetsova, A., Brockhoff, P. B. & Christensen, R. H. B. lmerTest Package: tests in linear mixed effects models. J. Stat. Softw. 82, 1–26 (2017).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics 32, 286–288 (2015).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Geeleher, P. et al. Gene-set analysis is severely biased when applied to genome-wide methylation data. Bioinformatics 29, 1851–1857 (2013).
Hansen, K. D. IlluminaHumanMethylation450kanno.ilmn12.hg19: annotation for Illumina’s 450k methylation arrays. R Package Version 02 (Bioconductor, 2014).
Maitre, L. et al. Human Early Life Exposome (HELIX) study: a European population-based exposome cohort. BMJ Open 8, e021311 (2018).
Strandberg, T. E., Järvenpää, A. L., Vanhanen, H. & McKeigue, P. M. Birth outcome in relation to licorice consumption during pregnancy. Am. J. Epidemiol. 153, 1085–1088 (2001).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012).
GTEx-Portal. GTEx Portal. https://www.gtexportal.org/home/ (2017). Accessed 11 May 2017.
Kiyonaka, S. et al. Physical and functional interaction of the active zone protein CAST/ERC2 and the β-subunit of the voltage-dependent Ca2+ channel. J. Biochem. 152, 149–159 (2012).
Curtis, D. et al. Case-case genome-wide association analysis shows markers differentially associated with schizophrenia and bipolar disorder and implicates calcium channel genes. Psychiatr. Genet. 21, 1–4 (2011).
Hatzimanolis, A. et al. Common genetic variation and schizophrenia polygenic risk influence neurocognitive performance in young adulthood. Am. J. Med Genet. B Neuropsychiatr. Genet. 168, 392–401 (2015).
Epstein, J. N. et al. Differential patterns of brain activation over time in adolescents with and without attention deficit hyperactivity disorder (ADHD) during performance of a sustained attention task. Neuropediatrics 40, 1–5 (2009).
Monden, Y. et al. Right prefrontal activation as a neuro-functional biomarker for monitoring acute effects of methylphenidate in ADHD children: an fNIRS study. Neuroimage Clin. 1, 131–140 (2012).
Franke, B., Neale, B. M. & Faraone, S. V. Genome-wide association studies in ADHD. Hum. Genet. 126, 13–50 (2009).
Klein, M. et al. Genetic markers of ADHD-related variations in intracranial volume. Am. J. Psychiatry 176, 228–238 (2019).
Acknowledgements
We thank all the children and families who took part in this study, as well as the support of hospitals, midwives, and pharmacies. ALSPAC: We are grateful to the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. The UK Medical Research Council (MRC) and Wellcome (Grant ref: 102215/2/13/2) and the University of Bristol provide core support for ALSPAC. This publication is the work of the authors and E.W. will serve as guarantors for the contents of this paper. A comprehensive list of grant funding is available on the ALSPAC website (http://www.bristol.ac.uk/alspac/external/documents/grant-acknowledgements.pdf). Methylation data in the ALSPAC cohort were generated as part of the UK BBSRC funded (BB/I025751/1 and BB/I025263/1) Accessible Resource for Integrated Epigenomic Studies (ARIES, http://www.ariesepigenomics.org.uk). E.B. is supported by an ESRC grant ES/S013229/1. D.C., G.S., and C.L.R. work in a department supported by the UK MRC (MC_UU_00011/5). G.S. is further supported by an MRC New Investigator Research Grant (MR/S009310/1) and the European Joint Programming Initiative “A Healthy Diet for a Healthy Life” (JPI HDHL, NutriPROGRAM project, MRC, MR/S036520/1). GENR: The Generation R Study is conducted by the Erasmus Medical Center in close collaboration with the Erasmus University Rotterdam, Faculty of Social Sciences, the Municipal Health Service Rotterdam area, the Rotterdam Homecare Foundation, Rotterdam, and the Stichting Trombosedienst & Artsenlaboratorium Rijnmond (STAR-MDC), Rotterdam. We gratefully acknowledge the contribution of general practitioners, hospitals, midwives, and pharmacies in Rotterdam. The generation and management of the Illumina 450K methylation array data (EWAS data) for the Generation R Study was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Netherlands. We thank Mr. Michael Verbiest, Ms. Mila Jhamai, Ms. Sarah Higgins, Mr. Marijn Verkerk, and Dr. Lisette Stolk for their help in creating the EWAS database. We thank Dr. A. Teumer for his work on the quality control and normalization scripts. The general design of the Generation R Study is made possible by financial support from Erasmus Medical Center, Rotterdam, Erasmus University Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), and the Ministry of Health, Welfare and Sport. The EWAS data were funded by a grant from the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA; project nr. 050-060-810), by funds from the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, and by a grant from the National Institute of Child and Human Development (R01HD068437). A.N. and H.T. are supported by a grant of the Dutch Ministry of Education, Culture, and Science and the Netherlands Organization for Scientific Research (NWO grant No. 024.001.003, Consortium on Individual Development). A.N. is also supported by a Canadian Institutes of Health Research team grant. The work of H.T. is further supported by a NWO-VICI grant (NWO-ZonMW: 016.VICI.170.200). M.v.I. and M.B.-K. were supported by the Netherlands Organization for Scientific Research (SPINOZA, VICI), and M.B.-K. was supported by the European Research Council (AdG 669249). J.F.F. has received funding from the European Joint Programming Initiative “A Healthy Diet for a Healthy Life” (JPI HDHL, NutriPROGRAM project, ZonMw the Netherlands no.529051022). This project received funding from the European Union’s Horizon 2020 research and innovation program (733206, LifeCycle; 633595, DynaHEALTH). GLAKU: We thank all the research nurses, research assistants, and laboratory personnel involved in the GLAKU study. The study has been supported by Academy of Finland, University of Helsinki, Hope and Optimism Initiative, Finnish Foundation for Pediatric Research, Sigrid Juselius Foundation, Jalmari and Rauha Ahokas Foundation, Signe and Ane Gyllenberg Foundation, Yrjo Jahnsson Foundation, Juho Vainio Foundation, Emil Aaltonen Foundation, and Ministry of Education and Culture, Finland. INMA: A full roster of the INMA Project Investigators can be found at http://www.proyectoinma.org/presentacion-inma/listado-investigadores/en_listado-investigadores.html. S.A. is funded by a Juan de la Cierva-Incorporación fellowship (IJCI-2017-34068) awarded by the Spanish Ministerio de Economía, Industria y Competitividad. M.G. is funded by a Miguel Servet fellowship (MS13/00054, CP18/00018) awarded by the Spanish Institute of Health Carlos III. ISGlobal is a member of the CERCA Programme, Generalitat de Catalunya. J.J. is funded by a Miguel Servet fellowship (MS14/00108, CP14/00108) awarded by the Spanish Institute of Health Carlos III. HELIX: The authors would like to thank all practitioners and researchers in the six countries who took part in this study. The authors would like to thank Sonia Brishoual, Angelique Serre, and Michele; Grosdenier (Poitiers Biobank, CRB BB-0033-00068, Poitiers, France) for biological sample management; and Professor Frederic Millot (Principal Investigator), Elodie Migault, Manuela Boue, and Sandy Bertin (Clinical Investigation Center, Inserm. CIC1402, CHU de Poitiers, Poitiers, France) for planning and investigational actions. The authors would like to thank Veronique Ferrand-Rigalleau, Céline Leger, and Noella Gorry (CHU de Poitiers, Poitiers, France) for administrative assistance (EDEN). The authors would like to thank Silvia Fochs, Nuria Pey, Cecilia Persavente, and Susana Gross for field work, sample management, and overall management in INMA. The authors would like to thank Georgia Chalkiadaki and Danai Feida for biological sample management, to Eirini Michalaki, Mariza Kampouri, Anny Kyriklaki, and Minas Iakovidis for field study performance and to Maria Fasoulaki for administrative assistance (RHEA). The authors would also like to thank Ingvild Essén for thorough field work, Heidi Marie Nordheim for biological sample management, and the Norwegian Mother, Father and Child cohort study (MoBa) administrative unit. The MoBa cohort study is supported by the Norwegian Ministry of Health and Care Services and the Ministry of Education and Research. The contribution of the Spanish National Genotyping Center (CEGEN-PRB2) is also acknowledged. NEST: The NEST cohort has been supported by the National Institute of Environmental Health Sciences (R01ES016772 [to C.H.], R21ES014947 [to C.H.], P30ES025128 [to C.H., D.D.J., R.M.], R01MD011746 [to C.H., S.K.M.], P30ES011961 pilot project [to S.K.M.], P01ES022831 [to S.K.M., B.F.F.]), the US Environmental Protection Agency (RD-83543701 [to S.K.M., B.F.F.]), the National Institute of Diabetes and Digestive and Kidney Diseases (R01DK085173 [to C.H., S.K.M.]), the National Institute of Aging (R21AG041048 [to B.F.F.]), and the Duke Cancer Institute [to C.H., S.K.M.]. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or the United States Environmental Protection Agency (US EPA). Further, USEPA does not endorse the purchase of any commercial products or services mentioned in the publication. PREDO: The PREDO Study has been funded by the Academy of Finland, EraNet Neuron, EVO (a special state subsidy for health science research), University of Helsinki Research Funds, the Signe and Ane Gyllenberg Foundation, the Emil Aaltonen Foundation, the Finnish Medical Foundation, the Jane and Aatos Erkko Foundation, the Novo Nordisk Foundation, the Päivikki and Sakari Sohlberg Foundation, and the Sigrid Juselius Foundation granted to members of the Predo study board. Methylation assays were funded by the Academy of Finland. The PREDO study would not have been possible without the dedicated contribution of the PREDO study group members: A-K Pesonen (Department of Psychology and Logopedics, University of Helsinki; Finland), A Aitokallio-Tallberg, A-M Henry, VK Hiilesmaa, T Karipohja, R Meri, S Sainio, T Saisto, S Suomalainen-Konig, V-M Ulander, T Vaitilo (Department of Obstetrics and Gynaecology, University of Helsinki and Helsinki University Central Hospital, Helsinki, Finland), L Keski-Nisula, Maija-Riitta Orden (Kuopio University Hospital, Kuopio Finland), E Koistinen, T Walle, R Solja (Northern Karelia Central Hospital, Joensuu, Finland), M Kurkinen (Päijät-Häme Central Hospital, Lahti, Finland), P.Taipale. P Staven (Iisalmi Hospital, Iisalmi, Finland), and J Uotila (Tampere University Hospital, Tampere, Finland). We also thank all the research nurses, research assistants, and laboratory personnel involved in the PREDO study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neumann, A., Walton, E., Alemany, S. et al. Association between DNA methylation and ADHD symptoms from birth to school age: a prospective meta-analysis. Transl Psychiatry 10, 398 (2020). https://doi.org/10.1038/s41398-020-01058-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-01058-z
This article is cited by
-
Gestational epigenetic age and ADHD symptoms in childhood: a prospective, multi-cohort study
Molecular Psychiatry (2024)
-
Cortico-striatal differences in the epigenome in attention-deficit/ hyperactivity disorder
Translational Psychiatry (2024)
-
Epigenome-wide association study identifies neonatal DNA methylation associated with two-year attention problems in children born very preterm
Translational Psychiatry (2024)
-
Genetic Variations in Elements of the Oxytocinergic Pathway are Associated with Attention/Hyperactivity Problems and Anxiety Problems in Childhood
Child Psychiatry & Human Development (2024)
-
DNA methylation as a potential mediator of the association between indoor air pollution and neurodevelopmental delay in a South African birth cohort
Clinical Epigenetics (2023)
