
Abstract
CACNA1I, a schizophrenia risk gene, encodes a subtype of voltage-gated T-type calcium channel CaV3.3. We previously reported that a patient-derived missense de novo mutation (R1346H) of CACNA1I impaired CaV3.3 channel function. Here, we generated CaV3.3-RH knock-in animals, along with mice lacking CaV3.3, to investigate the biological impact of R1346H (RH) variation. We found that RH mutation altered cellular excitability in the thalamic reticular nucleus (TRN), where CaV3.3 is abundantly expressed. Moreover, RH mutation produced marked deficits in sleep spindle occurrence and morphology throughout non-rapid eye movement (NREM) sleep, while CaV3.3 haploinsufficiency gave rise to largely normal spindles. Therefore, mice harboring the RH mutation provide a patient derived genetic model not only to dissect the spindle biology but also to evaluate the effects of pharmacological reagents in normalizing sleep spindle deficits. Importantly, our analyses highlighted the significance of characterizing individual spindles and strengthen the inferences we can make across species over sleep spindles. In conclusion, this study established a translational link between a genetic allele and spindle deficits during NREM observed in schizophrenia patients, representing a key step toward testing the hypothesis that normalizing spindles may be beneficial for schizophrenia patients.
Similar content being viewed by others


Introduction
Genetic analyses of large patient cohorts have identified multiple genomic loci associated with the risk of schizophrenia. Many of these risk genes encode proteins involved in calcium signaling including CACNA1C, CACNB2, and CACNA1I that may ultimately converge on a common disease mechanism1,2. CACNA1I encodes the pore-forming α1 subunit of voltage-gated calcium CaV3.3 channels that is expressed in a subset of central nervous system neurons including GABAergic neurons of the thalamic reticular nucleus (TRN). In TRN neurons, CaV3.3 channels mediate rebound burst firing in response to transient membrane hyperpolarization that recovers CaV3.3 channels from inactivation3,4. The TRN receives excitatory input from both cortex and thalamocortical cells, while only sending inhibitory input to thalamocortical relay neurons. Rebound burst firing of the TRN mediates oscillatory activities in the thalamocortical circuitry, such as sleep spindles5. Sleep spindles are waxing and waning bursts of 8–15 Hz oscillations (sigma frequency band) detected by electroencephalogram (EEG) and represent a signature of stage 2 non-rapid eye movement (NREM) sleep in humans6. Spindle oscillations have been shown to be important for sleep-dependent memory consolidation in both mice7 and humans8, which underlie normal cognitive functioning.
Marked reductions of spindle activity during NREM have been reported in chronic as well as early course schizophrenia patients with otherwise normal sleep architecture9. While these reports typically involve relatively small sample sizes, spindle deficits throughout the course of schizophrenia, in treatment-naïve patients10, and in first-degree relatives11 implicate abnormal function of thalamocortical circuitry. This abnormality before the onset of the disease is consistent with the finding of reduced thalamic volume and altered thalamocortical connection in ultra-high-risk adolescents for schizophrenia12,13. Further, reduced sleep spindle activity in schizophrenia has been correlated with impaired sleep-dependent cognitive functions14,15,16,17,18. Taken together, these findings suggest that sleep spindle abnormalities in schizophrenic patients are a heritable intermediate phenotype that may underlie cognitive impairment. In this report, we aim to determine whether a patient-derived coding mutation (R1346H) in the schizophrenia-associated gene CACNA1I alters sleep spindle oscillations during NREM in vivo, to investigate potential translational markers across species.
Previous studies show that mice lacking CaV3.3 exhibited a selective reduction in the power density of the sigma frequency band (defined as 10–12 Hz by the authors) only at transitions from NREM to rapid eye movement (REM) sleep in mice19 without any changes in sleep duration20. Sigma frequency analyses indirectly index spindle oscillation and do not detect individual spindles21. More critically, humans, unlike rodents, do not show the same increase in spindle activity just prior to NREM/REM transitions6. In order to address these questions and to investigate the impact of the R1346H mutation throughout NREM in vivo, we generated CaV3.3 knock-out (KO, Cacna1i−/− and Cacna1i+/−) and CaV3.3-RH knock-in mice (RH, Cacna1iRH/RH and Cacna1i+/RH), analyzed TRN neurophysiological function in acutely prepared brain slices, and quantified spindle oscillations using in vivo EEG recordings, explicitly considering spindle occurrence and morphology at 9, 11, 13, and 15 Hz. Here we report altered rebound bursting in TRN neurons and marked abnormalities in sleep spindles in both KO and RH knock-in mice, but not in KO heterozygous. These results highlighted genotype-specific consequences and establish a link between a genetic risk allele and a reduction in sleep spindles reported in schizophrenia patients, representing a key step toward testing the hypothesis that normalizing spindle-dependent impairments in schizophrenia patients are beneficial.
Results
Generation and validation of CaV3.3 knock-in and knock-out mouse models
Previously, we reported that R1346H, a de novo mutation identified in the schizophrenia proband by exome sequencing a cohort of trio samples, reduced the CaV3.3 mediated Ca2+ currents in HEK293 cells22. In this report, we generated both knock-out (KO) and orthologous mutation R1305H knock-in (RH) CaV3.3 mouse models using the CRISPR/Cas9-mediated genome editing approach coupled with pronuclear zygote injection (Fig. 1a, b)23. Loss of functional allele of CaV3.3 was generated by pronuclear injection of Cas9 protein and an sgRNA, resulting in a mouse line with a 10 base-pair genomic deletion that causes a premature stop codon (Fig. 1a). The knock-in mutation was introduced through homologous recombination of a DNA template harboring the R1305H alteration (R1305H in murine CaV3.3 corresponds to R1346H in human channel; Supplementary Fig. 1a), and subsequently sequence-confirmed in the founder animals. We confirmed the lack of CaV3.3 wild-type (Cacna1i+/+) mRNA and channel protein in the homozygous KO animals (Cacna1i−/−; Fig. 1b), and significant reductions of both mRNA and protein levels in the heterozygous KO mice (Cacna1i+/−; Supplementary Fig. 1b–d). In the homozygous knock-in animals (Cacna1iRH/RH), the total amount of CaV3.3 protein is similar to that of the Cacna1i+/+ mice (Fig. 1c, d; whole-cell lysate, Mann Whitney test; p = 0.65). However, the amount of CaV3.3 channel protein was significantly reduced in the crude synaptoneurosomal preparation (Fig. 1c, d; synaptoneurosomal; Mann Whitney test; p = 0.0152) in the Cacna1iRH/RH mice. This synaptoneurosomal preparation is enriched with dendrites and axonal processes including synaptic scaffolding protein PSD95 (Supplementary Fig. 1e). These results suggest that RH mutation does not impact overall channel protein production in neurons but may influence channel trafficking or localization in vivo, similar to previous analyses in human cell lines22.

a The exon/intron structure of mouse Cacna1i gene, and the positions of targeting sgRNAs for generating the CaV3.3 knock-out (KO) and R1305H knock-in animals. The sequences of the targeting sgRNAs and the genomic sequence verification for knock-out and knock-in founder animals are shown underneath. b Western blot from Cacna1i−/− and Cacna1i+/+ whole-brain lysates showing a near complete lack of CaV3.3 protein in the Cacna1i−/− (−/−) samples. Significant reductions of both mRNA and protein levels in the heterozygous KO mice were also found (Supplementary Fig. 1b–d). Bottom panel: quantification of CaV3.3 protein and mRNA levels normalized to Cacna1i+/+ levels (n = 5). c A typical representative western blot showing total CaV3.3 protein levels (top panel) and CaV3.3 protein levels in crude synaptoneurosomal preparations (bottom panel) from brain tissues derived from littermates of Cacna1i+/+ (+/+) and Cacna1iRH/RH (RH/RH). d Quantification of total and synaptoneurosomal CaV3.3 levels in the total brain lysate (n = 8) and crude synaptoneurosomal preparation (n = 6). e A typical western blot showing the developmental progression of CaV3.3 expression in brain tissue across different postnatal days, and at 5 (5m) and 8 months (8m). f Quantification of protein (n = 2 for each time point) and mRNA (n = 2 for each time point except for 5m and 8m that contains n = 3 each) levels during developmental progression. **p < 0.01; error bars represent S.E.M.
Next, we characterized the expression of CaV3.3 over developmental stages in the Cacna1i+/+ brain. As shown in Fig. 1e, f, the expression of CaV3.3 mRNA transcripts and channel protein follow similar patterns where they were barely detectable at birth, started to increase around P10 and peaked around week 5. Such developmental expression profile coincides with the critical period of mouse thalamocortical development24,25,26, suggesting that CaV3.3 may help shape key neuronal connections during development.
Selective reduction of T-type Ca2+ currents in knock-in and knock-out mice
Low-voltage-activated (LVA) and high-voltage-activated (HVA) calcium channels are present in TRN neurons27,28,29. T-type LVA Ca2+ channels are available to open at a holding potential of −100 mV but are inactivated at −60 mV where only HVA Ca2+ channels are available30. As shown in Fig. 2a, b, we isolated T-type Ca2+ currents by subtracting inactivation-resistant HVA Ca2+currents (pre-pulse of −60 mV) from the total amount of Ca2+ currents (pre-pulse of −100 mV). Using this protocol, we report a significant amount of T-type Ca2+ current in Cacna1i+/+ TRN neurons that exhibit maximum currents at −40 mV. This T-type inward current was mostly absent in Cacna1i−/− animals, consistent with previous reports3,19,31. Similarly, the Cacna1iRH/RH mice showed a notable reduction in the peak T-type Ca2+ current density (Fig. 2c, d; Supplementary Table 1), consistent with our previous analyses in cells22 and the reduced level of CaV3.3 protein in the synaptoneurosomal preparation of knock-in animals (Fig. 1d). In a separate cohort, we found similar T-type channel peak current density in the Cacna1i+/+ and heterozygous knock-in (Cacna1iRH/+; Fig. 2e) mice. In contrast, heterozygous knock-out (Cacna1i+/−) mice displayed a significant reduction in the T-type current density (Fig. 2e; for means and p values see Supplementary Table 1), which is consistent with the biochemical analyses (Supplementary Fig. 1b–d). Importantly, there were no differences across all genotypes in the HVA inward current recorded, as is evident from the current–voltage relationship in Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− mice (Fig. 2f) as well as the percent change in peak current densities across the homozygous genotypes (Fig. 2g, Supplementary Table 1) or heterozygous genotypes (Fig. 2h, Supplementary Table 1). Together, these results indicate that there is a selective reduction in T-type Ca2+ conductance in the homozygous Cacna1iRH/RH, together with Cacna1i+/− and Cacna1i−/− mice without any apparent compensatory changes in the overall high-voltage-gated Ca2+ conductance in TRN neurons.

a Protocol isolating T-type Ca2+ current in TRN neurons. Low-voltage-activated (LVA) together with high-voltage-activated (HVA) inward currents were elicited in neurons from a holding potential of −100 mV (for 1 s) to depolarizing test potentials ranging from −90 to +20 mV (Δ5 mV), in the presence of K+ and Na+ channel blockers AP-4 and TTX, respectively. HVA only inward currents were measured using the same protocol but from a holding potential of −60 mV. T-type Ca2+ currents from the TRN neurons were then isolated offline, by subtracting the HVA only inward current traces from the ones obtained from the HVA + LVA protocol. b Sample traces from Cacna1i+/+ and Cacna1i−/− animals for voltage steps from −90 to −30 mV are shown for holding potentials at −100 and −60 mV, respectively, as well as the subtracted T-currents. While a significant amount of T-type Ca2+ current can be obtained after the subtraction method in the Cacna1i+/+ neuron, there was a complete lack of subtracted T-type currents in the Cacna1i−/− neuron, validating the assay for T-type Ca2+ current isolation from mouse TRN neurons. c Mean current–voltage relationships (I–V) constructed for the subtracted T-type Ca2+ current density for Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− TRN neurons, showing a dramatic reduction in T-type Ca2+ current in the Cacna1i−/− mice and a moderate reduction in peak current density in the Cacna1iRH/RH mice. d, e Percent change in peak current density for T-type Ca2+ current is plotted from the homozygous (d; p < 0.0001; R2 = 0.563; F (2,32) = 2.181; one-way ANOVA) and heterozygous (e; p = 0.0027; R2 = 0.39; F (2,24) = 1.882; one-way ANOVA) TRN neurons, compared to Cacna1i+/+. f Mean I–V constructed for the HVA only Ca2+ current for Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− TRN neurons. g, h Percent change in HVA only Ca2+ current calculated for the homozygous (g; p = 0.80; R2 = 0.013; F (2,34) = 0.1574; one-way ANOVA) and heterozygous (h; p = 0.58; R2 = 0.0448; F (2,24) = 0.7681; one-way ANOVA) genotypes shows no significant differences in HVA Ca2+ current among the TRN neurons of all genotypes. *p < 0.05; ***p < 0.001; error bars represent S.E.M. All mean values and detailed statistics are listed in Supplementary Table 1.
Reduction of rebound burst firing in TRN neurons of CaV3.3 knock-in and knock-out mice
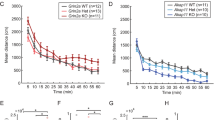
Next, we characterized rebound burst firing and tonic firing in the TRN neurons of knock-in and knock-out mice (Fig. 3, Supplementary Table 1, Supplementary Fig. 2). A hyperpolarizing pulse of 500 ms removes the inactivation of T-type currents and induces oscillatory burst firing upon restoring to the holding potential in TRN neurons (Fig. 3a, Cacna1i+/+, red traces). These rebound bursts had the acceleration–deceleration pattern (Fig. 3a, inset) that was similar across all genotypes. The relationship between the holding potential and the number of bursts (Fig. 3b) or the action potential (AP) frequency of the first burst (Fig. 3c) is displayed across Cacna1i+/+, Cacna1iRH/RH and Cacna1i−/− genotypes. The mean and statistics for comparison are summarized in Supplementary Table 1. While such burst firing was largely absent in the Cacna1i−/− mice (Fig. 3a, b, d), the Cacna1iRH/RH showed an apparent reduction in the average number and frequency of the bursts between −65 and −50 mV (Fig. 3a–c), as well as the maximum number of bursts elicited (Fig. 3d). Notably, Cacna1i−/− neurons showed occasional rebound burst firing at holding potentials more depolarized than −60 mV (Fig. 3b) and that these bursts from Cacna1i−/− animals contained fewer APs, and with reduced frequency (Fig. 3b–e), indicating that such bursts in the Cacna1i−/− neurons had different characteristics.

a Sample traces from Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− mice showing hyperpolarization induced rebound burst firing in a representative TRN neuron from a holding potential of −60 mV. Inset displays a single burst from each genotype at high temporal resolution showing the classic acceleration–deceleration pattern. b Relationship of holding membrane potential and the number of rebound bursts observed in all TRN neurons of Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− animals. In general, Cacna1i+/+ neurons displayed increased number of bursts at more depolarized potentials. Lack of rebound bursting is apparent in Cacna1i−/− TRN neurons, whereas Cacna1iRH/RH TRN neurons show a clear reduction in peak number of rebound burst. c Relationship of holding membrane potential and the first burst frequency observed in TRN neurons of Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− animals. Similar to what is observed in b, in general there was an increase in first burst frequency with more depolarized holding potentials with an apparent reduction in peak burst frequency observed in Cacna1iRH/RH neurons. There was also a dramatic reduction in burst frequency in the Cacna1i−/− neurons that showed some form of rebound bursting. d The maximum number of bursts observed per neuron irrespective of holding potentials are plotted for all genotypes, showing a significant reduction in the Cacna1iRH/RH and Cacna1i−/− TRN neurons (p < 0.0001; R2 = 0.3836; F (4,88) = 13.69; one-way ANOVA). e First burst frequency (p < 0.0001; R2 = 0.4143; F (4,78) = 13.79; one-way ANOVA) and number of action potentials in the first burst (p < 0.0001; R2 = 0.2701; F (4,78) = 7.22; one-way ANOVA), as well as the after hyperpolarization (AHP; p < 0.0001; R2 = 0.4824; F (4,78) = 18.17; one-way ANOVA) observed in the first burst across all genotypes showing significant reductions in Cacna1iRH/RH and Cacna1i−/− animals. f Frequency (p < 0.0001; R2 = 0.361; F (3,48) = 9.04; one-way ANOVA), number of action potentials (p = 0.0001; R2 = 0.3529; F (3,48) = 8.73; one-way ANOVA) in the second burst, and the inter-burst-interval (IBI; p = 0.4027; R2 = 0.0246; F (3,48) = 0.4027; one-way ANOVA) between first and second burst across all genotypes showing significant reductions in Cacna1iRH/RH TRN neurons. Cacna1i−/− TRN neurons never showed repetitive bursting and are therefore excluded. **p < 0.01; ***p < 0.001; in each violin plot for d, e and f black squares represent individual data points and white boxes represent mean value; length of black line represents the interquartile range. All mean values and detailed statistics are listed in Supplementary Table 1.
We further quantified the properties of rebound burst firing across the homozygous and heterozygous animals and compared to those in the wild-type Cacna1i+/+ mice (Fig. 3d–f). Significant reductions in the maximum AP frequency in the first burst (Fig. 3e, left panel), and the average number of APs in the first burst (Fig. 3e, middle panel) are present in the Cacna1iRH/RH and Cacna1i−/− mice, compared to Cacna1i+/+ mice. In the Cacna1iRH/RH and Cacna1i−/− TRN, we also observed reduced after hyperpolarization potential (AHP) of the first burst, which represents an indirect measure of Ca2+ mediated potassium efflux32 (Fig. 3e, right panel). No significant differences of these properties in heterozygous Cacna1iRH/+ and Cacna1i+/− animals were observed compared to Cacna1i+/+ (Fig. 3e). In terms of repetitive bursting, whenever present, we further analyzed the frequency and the number of APs comprising the second burst across the genotypes (Fig. 3f). Significant reductions in the AP frequency and average number of APs were observed in the second burst of Cacna1iRH/RH mice (Fig. 3f, left and middle panels). No statistical difference was observed in the second burst parameters for heterozygous animals (Cacna1i+/RH and Cacna1i+/−) compared to Cacna1i+/+ animals. Interestingly, inter-burst interval (IBI) was similar across Cacna1i+/+, Cacna1iRH/RH, Cacna1i+/RH, and Cacna1i+/− mice (Fig. 3f, right panel), suggesting that the CaV3.3 channel may not contribute to the molecular mechanisms that regulate the IBI. Finally, there were no notable differences across the genotypes with respect to latency and threshold of APs of the rebound bursts or the passive membrane properties such as the input resistance and resting membrane potentials (Supplementary Table 1) in the TRN neurons.
We used 500 ms depolarization to induce tonic firing of TRN neurons (Supplementary Fig. 2a), and found no apparent difference in the current–frequency relationship (Supplementary Fig. 2b), the maximum firing frequency (Supplementary Fig. 2c), or the Ca2+ independent fast after hyperpolarization of APs (Supplementary Fig. 2d) across Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− TRN neurons. These results showed that tonic firing of TRN neurons remained largely intact under these recording conditions when CaV3.3 function is impaired, consistent with intact HVA Ca2+ current in the mutant animals (Fig. 2f–h). Taken together, our data showed impaired rebound bursting, but not tonic firing, in the TRN neurons of Cacna1i−/− and Cacna1iRH/RH mice while heterozygous Cacna1i+/RH and heterozygous Cacna1i+/− mice are largely comparable to the wild-type Cacna1i+/+ mice in rebound bursting.
Reduction of NREM sleep spindle density in knock-in and knock-out mice
In order to understand the physiological consequences of impaired rebound bursting, we recorded EEG activity in vivo and analyzed power spectral density and spindle activity during the light period (6 h), when nocturnal mice are predominantly asleep, across NREM, REM, and wake periods. In total, out of the 61 mice recorded, 56 mice (17 Cacna1i+/+, 10 Cacna1i+/−, 11 Cacna1i−/−, 8 Cacna1i+/RH, and 10 Cacna1iRH/RH) with at least one EEG channel that passed our quality control criteria (QC+, see Methods) were included in the analysis.
Genotype groups were broadly similar for sleep stage duration (Fig. 4a) and rates of stage transitions (Supplementary Table 2), and the proportion of epochs removed by automated artifact detection, suggesting comparable sleep architecture.

a Sleep architecture showing similar proportion of time spent by Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− mice at the different stages of sleep during the 6 h light cycle analyzed. No changes were observed in sleep transitions as shown in Supplementary Table 2. b Power spectral density in Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− animals during NREM, REM, and wake periods of the 6 h light cycle. Spectral power at each frequency was normalized to total power and is expressed as relative power. Shaded areas show frequency at which significant differences were observed between Cacna1i+/+ and Cacna1i−/− mice. No significant differences were observed between Cacna1i+/+ and Cacna1iRH/RH mice. c Relative sigma power across Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− genotypes normalized to average Cacna1i+/+ values, showing a significant reduction in sigma power of Cacna1i−/− animals. d NREM, REM, and wake average spindle density quantified as spindles per minute for FC = 9, 11, 13, and 15 Hz spindles across Cacna1i+/+, Cacna1iRH/RH, and Cacna1i−/− animals showing a significant reduction in sleep spindles only during NREM in both Cacna1iRH/RH and Cacna1i−/− animals compared to Cacna1i+/+. Inset shows a typical sleep spindle oscillation highlighted. # denotes statistical significance (p < 0.05) between Cacna1i+/+ and Cacna1iRH/RH, and * (p < 0.05) and ** (p < 0.01) denote statistical significance between Cacna1i+/+ and Cacna1i−/− animals. e Spindle density for FC = 11 Hz is normalized to Cacna1i+/+ average. Cacna1iRH/RH and Cacna1i−/− animals show significant reductions in the spindle density during NREM sleep. f, g Characteristics of FC = 11 Hz spindles including duration (f) and integrated spindle activity per spindle (ISAs; g), normalized to the Cacna1i+/+ average values show that in addition to reductions in sleep spindle there is also significant reduction Cacna1iRH/RH and Cacna1i−/− spindle duration and ISAs. **p < 0.01; ***p < 0.001; in each violin plot c, e, f and g black squares represent individual data points and white boxes represent mean value; length of black line represents the interquartile range. h Relative sigma power in NREM (with each point representing the average of two consecutive 10-s epochs) preceding an REM epoch indicates a transient increase during NREM–REM transition (dashed vertical line) in Cacna1i+/+. Significant reduction in sigma power of Cacna1i−/− animals is observed across all NREM epochs. i Average spindle density of FC = 11 Hz spindles for NREM (with each point representing the average of two consecutive 10-s epochs) preceding an REM episode, showing that similar to sigma power there is an increase in mean spindle density during NREM–REM transition (dashed vertical line). Significant reduction of spindle density is observed in both Cacna1iRH/RH and Cacna1i−/− animals during the transition, although no changes in sigma power is observed in the Cacna1iRH/RH mice (d). # (p < 0.05) and ## (p < 0.01) denote statistical significance between Cacna1i+/+ and Cacna1iRH/RH and * (p < 0.05), ** (p < 0.01), and *** (p < 0.001) denote statistical significance between Cacna1i+/+ and Cacna1i−/− animals. Error bars denote S.E.M. All mean values and detailed statistics are listed in Supplementary Table 3.
EEG power spectral density
During wakefulness, we did not observe differences in relative EEG power spectral density among genotypes, for any frequency tested (0.5–20 Hz in 0.5 Hz bins; Fig. 4b). In contrast, during NREM we observed significantly (p < 0.01) reduced power in the sigma range, with a maximal difference at 11 Hz, in Cacna1i−/− mice compared to Cacna1i+/+ (Fig. 4b, c), whereas the homozygous knock-in mice (Cacna1iRH/RH) were similar to Cacna1i+/+. Sigma (11–15 Hz) band relative power was also reduced in Cacna1i−/− mice compared to Cacna1i+/+ (p = 0.005; Fig. 4c), whereas Cacna1i+/− (p = 0.13), Cacna1i+/RH (p = 0.39), and Cacna1iRH/RH (p = 0.86) groups were similar to Cacna1i+/+ (heterozygous mice data displayed in Supplementary Fig. 3a). In addition to reduced sigma power, Cacna1i−/− mice had increased power at lower frequencies, in particular delta range (3–5 Hz, Fig. 4b, p = 0.001); heterozygous Cacna1i+/− mice also showed a small increase in this frequency range (p = 0.044) relative to Cacna1i+/+ (Supplementary Fig. 4). Neither Cacna1i+/RH (p = 0.92) nor Cacna1iRH/RH (p = 0.83) mice exhibited increased delta frequency oscillation power. Finally, the increase in NREM delta frequency observed in Cacna1i−/− was also present during REM, as was the decrease in sigma power, albeit to a lesser extent (Fig. 4b).
Spindle detection
We next applied a wavelet-based algorithm to detect spindles directly, centering on four target frequencies (FC = 9, 11, 13, and 15 Hz). Spindle density (counts per minute) was only modestly correlated with relative sigma power (e.g. during NREM, r = 0.25, p = 0.06 for FC = 13 Hz spindles). As expected, when detecting spindles across all stages with similar detection parameters, we observed markedly lower spindle densities during REM sleep and wake compared to NREM sleep, across all groups and target frequencies (Fig. 4d). For example, FC = 11 Hz spindle density in Cacna1i+/+ mice was 3.02 spindles per minute during NREM compared to 0.55 during REM and 0.12 during wakefulness (Supplesmentary Table 3).
Compared to wild type, both Cacna1i−/− and Cacna1iRH/RH groups showed large (−0.9 and −0.66 standard deviation units, respectively, for FC = 11 Hz spindles) statistically significant reductions in spindle density during NREM (Fig. 4d, e, the mean and statistical comparison are summarized in Supplementary Table 3). These genotype-associated differences were strongest for FC = 11 Hz spindles (p = 0.002 and p = 0.027; Fig. 4e) although broadly similar effects were observed for FC = 13 Hz spindles (p = 0.002 and p = 0.025) and 15 Hz spindles (p = 0.011 and p = 0.028) (Fig. 4d, Supplementary Fig. 5), but not slower FC = 9 Hz spindles (p = 0.17 and p = 0.14; Supplementary Fig. 5). Analyses restricted to stable epochs (those not contiguous with a stage transition, i.e. NREM epochs flanked by other NREM epochs) yielded similar results. In contrast, no significant differences in NREM spindle density were observed for the Cacna1i+/− and Cacna1i+/RH mice compared to Cacna1i+/+ (Supplementary Figs. 5 and 6), which is consistent with the wild-type-like rebound bursting in TRN neurons of the heterozygous animals (Fig. 3d–f).
In contrast to NREM, there were no significant differences in spindle densities among genotypes during REM and wake periods when focusing on stable REM and wake epochs not flanking stage transitions (Supplementary Table 3). Thus, the spindle deficits observed in both Cacna1i−/− and Cacna1iRH/RH mice appear to be specific to NREM and consistent with the spectral analyses in Fig. 4b.
Spindle density at NREM–REM transitions
Relative to stable NREM sleep, rodents show a characteristic peak19,33 of sigma and spindle activity approximately 30 s prior to NREM/REM transitions, followed by an immediate reduction in sigma power and spindling during subsequent REM (Fig. 4h, i). As expected in Cacna1i+/+ mice, we observed these peak activities of both sigma and spindle density during NREM/REM transtion, for all values of FC (Fig. 4h, i, Supplementary Fig. 7). The reductions in spindle density described above, for Cacna1i−/− and Cacna1iRH/RH mice during all NREM, were pronounced prior to NREM/REM transitions (Fig. 4i). Analyzed in 20-s intervals (i.e. pairs of adjacent epochs), Cacna1i−/− and Cacna1iRH/RH mice showed reductions (p = 0.007 and p = 0.001, respectively, for the second interval (30–50 s) prior to the transition; p = 0.0002 and p = 0.006 for the first interval (10–30 s) prior) in FC = 11 Hz spindle density compared to Cacna1i+/+. Standardizing densities to have 0 mean and unit variance within the Cacna1i+/+ group, the means for Cacna1i−/− and Cacna1iRH/RH mice were −2.8 and −1.8 SD units, respectively, indicative of very large effect sizes. As before, heterozygous mice failed to show any differences in spindle density prior to NREM–REM transitions (Supplementary Fig. 7).
Genotype differences in NREM spindle morphology
In addition to changes in spindle density, homozygous knock-in and knock-out mice also showed notable differences in spindle morphology. For FC = 11 Hz NREM spindles, Cacna1i−/− and Cacna1iRH/RH groups exhibited significantly reduced spindle durations (Fig. 4f; p = 1×10−5 and p = 0.001, respectively; Supplementary Table 4) and spindle-wise measures of integrated spindle activity (ISAs, reflecting both spindle duration and amplitude; Fig. 4g; p = 0.001 and p = 0.004; Supplementary Table 4). That is, beyond differences in the number of spindles detected, those that were detected were significantly smaller in Cacna1i−/− and Cacna1iRH/RH mice compared to Cacna1i+/+. No changes in the spindle (FC = 11 Hz) duration or ISAs were observed in heterozygous Cacna1i+/− and Cacna1i+/RH mice (Supplementary Fig. 8).
In summary, both Cacna1i−/− and Cacna1iRH/RH mice showed marked deficits in spindle density and morphology compared to wild-type mice, while no significant changes were observed in the heterozygous knock-out and knock-in mice (Supplementary Fig. 3). Major genotype-dependent differences in spindle activity were specific to NREM sleep, with no clear pattern of significant effects during REM or wake. While most pronounced just prior to NREM/REM transition, these spindle deficits were significant when considering all NREM in the sleep period. Importantly, the deficits in Cacna1IRH/RH mice were only revealed by analyses that directly detected and characterized individual spindles, rather than traditional spectral analyses based on average sigma power.
Discussion
CACNA1I has been implicated by both genome-wide common variant association2 and de novo exonic variant analyses34 as a genetic risk factor for schizophrenia. However, its connection to the disease is elusive. This study generated mice harboring the de novo mutation identified in a schizophrenic patient (Cacna1iRH/RH) and investigated its biological impact. We found T-type Ca2+ current density was significantly reduced in the TRN neurons of the Cacna1iRH/RH mice, without compensatory changes in high voltage activated Ca2+ currents (Fig. 2), lending confidence in addressing CaV3.3-specific function in these mice. The reduction in CaV3.3 currents led to robust functional deficits in TRN excitability (Fig. 3), and impaired spindle oscillations in vivo (Fig. 4).
While the previous study on CaV3.3 KO mice reported sigma power deficits during NREM/REM transition19, this report demonstrated sleep spindle occurrences and morphology deficits throughout NREM as well as during NREM/REM transition in mice harboring the RH mutation and lacking CaV3.3 (Fig. 4), thus directly connecting CACNA1I to sleep spindle deficits reported in schizophrenia patients9,10,11,16,17,18 during NREM. Given that humans, unlike rodents, do not show the same increase in spindle activity just prior to NREM/REM transitions6, our new findings strengthened the inferences we can make on spindle oscillation between species.
Whereas no significant deficits in sigma power were observed in Cacna1iRH/RH mice, our automated spindle detection algorithm revealed a robust deficit in sleep spindle density through NREM sleep and immediately prior to NREM/REM transitions. Unlike sigma power analyses, which assumes statistical stationarity of time series and captures tonic/baseline sigma activity, the spindle detection explicitly recapitulates the definition of a spindle as a temporally limited, non-stationary burst of increased, phasic activity relative to the baseline. Thus, it can be expected that spindle density may better capture transient changes in sigma activity (i.e. discrete and relatively obvious spindle events) compared to spectral power statistics computed across a longer time period, as seen in human EEG analyses6. On the other hand, sigma power may better capture activity related to very low amplitude or short spindles that are missed by our current thresholding factor needed to detect spindles as discrete events. There was no alteration in the overall sleep architecture or sleep transitions between sleep stages, during the light cycle of the mutant mice, indicating loss or reduction of CaV3.3 function may not directly shape the onset of sleep stages. Indeed, the larger human studies looking at sleep spindles in schizophrenia16 did not observe any significant differences in macro-architecture between cases and controls, but did observe differences in spindle activity. This shows that the CaV3.3 model can be utilized to understand consequences of specific deficits in spindle oscillations during sleep.
Our results across homozygous and heterozygous mice consistently show that when there is a significant deficit in rebound bursting, there is a deficit in sleep spindle density (Figs. 3 and 4; Supplementary Figs. 5 and 6), suggesting that rebound bursting correlates and predicts sleep spindle deficits during NREM sleep in vivo in these mice. Comparing the heterozygous Cacna1i+/− and the homozygous Cacna1iRH/RH animals, it is unclear why both models produced reduced T-type Ca2+ current density (Fig. 2), yet only Cacna1iRH/RH model had TRN rebound bursting deficits (Fig. 3). One explanation for such discrepancy may lie in the subcellular location of the CaV3.3 channels, as higher CaV3.3 density in distal dendrites3 is critical for rebound bursting. It may be possible that the CaV3.3 channel density in the distal dendrites of the Cacna1i+/− mice is comparable to wild-type mice despite reduced mRNA levels, whereas that in the Cacna1iRH/RH mice is much reduced and unable to support normal rebound bursting. This effect induced by the RH mutation may be caused by certain deficits such as trafficking impairment as suggested previously by our work in human cell lines22. Further analyses in CaV3.3 trafficking in vivo and dendritic excitability of TRN neurons could test this hypothesis. Regardless of mechanism, differential rebound bursting patterns in Cacna1iRH/RH and Cacna1i+/− animals indicating that simple CaV3.3 haploinsufficiency alone is not enough to induce the rebound bursting, or spindle deficits in mice. Furthermore, sigma frequency power analyses can detect deficits in homozygous KO animals but not in Cacna1iRH/RH mice (Fig. 4), suggesting differences in spindling between the two genotypes.
Taken together, these results indicate that spindle activity may serve as an in vivo indicator for TRN function in mice, and that Cacna1iRH/RH animals would allow studies that could not be done in Cacna1i+/− or Cacna1−/−. For example, the ability of pharmacological or genetic perturbations on or upstream of CaV3.3 to normalize the spindle deficits can only be tested in Cacna1iRH/RH but not in Cacna1i+/− (that do not show spindle deficits) or Cacna1−/− (that shows spindle deficits but do not have the CaV3.3 channel), as the CaV3.3 channel is required for rebound burst firing in TRN neurons19,31 underlying sleep spindle oscillations. As such, Cacna1iRH/RH provide a valuable model not only to dissect potential disease mechanism and spindle biology but also to evaluate pharmacological and genetic modulations.
Spectral analyses of sigma power have often been used in studies of sleep spindle neurocircuitry, as a surrogate index of spindle activity. For example, in three mouse studies, overall NREM sigma power was unchanged across EEG recording sessions when comparing experimental groups of mice, whereas it differed prior to NREM/REM transitions19,35,36. In contrast, our primary analyses of sleep employed a spindle detection pipeline based on a wavelet-based algorithm6, enabling us to quantify the density, morphology, and oscillation frequencies of individual sleep spindles at single Hertz resolution. As described in human EEG studies6, faster or slower spindles can exhibit different properties and functional associations. Several observations support the validity of our spindle detection procedure. First, we observed a markedly higher incidence of spindles during NREM sleep, whereas spindle-like oscillations were rarely detectable during REM periods and wakefulness during the light cycle (Fig. 4). Second, in accordance with previous reports in rodents33,37,38, we observed peak spindle density around 11 Hz and a steep increase in NREM spindle density immediately preceding a transition to REM (Fig. 4). Third, as would be predicted, based on the previous observation of reduced sigma power in Cacna1i−/− mice just before NREM/REM transitions19, we observed a sharp reduction in spindle density in Cacna1i−/− mice prior to NREM–REM transitions in addition to a reduction in sigma power.
Quantifying discrete spindle events may index TRN dysfunction more sensitively compared to average sigma band power. In our study, we observed spindle deficits not just at NREM/REM transitions in Cacna1i−/− mice, but across all NREM sleep in both Cacna1i−/− mice and Cacna1iRH/RH mice, while analyses of sigma power alone did not differentiate Cacna1iRH/RH animals. Whereas drastic manipulations of the circuit—like the total loss of rebound bursting in Cacna1i−/− mice—can lead to noticeable differences in the overall sigma power (Figs. 3 and 4), more subtle changes—such as reduced rebound bursting in Cacna1iRH/RH mice —may impact TRN function that is not reflected in overall sigma power. These results highlight the importance of individual sleep spindle detection.
We show here that the CACNA1I risk variant induces spindle deficits, which corroborated with two recent studies of PTCHD1 and GRIA1, two genes implicated by neuropsychiatric genetics, that abnormal sleep spindle oscillation may underlie neurodevelopmental disorders such as autism and schizophrenia39,40,41. Moreover, the immunoreactivity of parvalbumin (PV) was found to be drastically reduced in the postmortem TRN neurons of schizophrenia and bipolar patients40, lending support for altered TRN function in schizophrenia pathophysiology. Several clinical studies reported reduced sleep spindle density in schizophrenia patients, drug-naive patients, and first-degree relatives of patients9, suggesting that sleep spindle impairments be an endophenotype in schizophrenia and are correlated with sleep-dependent cognitive impairement14. Clearly, behavioral tests of these Cacna1i genetic models should be next investigated to address the question of behavioral correlates of spindle deficits. In particular, sleep-dependent learning or phenotypes would be a focus of next-stage investigations. Nonetheless, our results from the Cacna1iRH/RH mice linked a risk variant in schizophrenia with a quantifiable EEG endophenotype marker in NREM reported in schizophrenia patients, providing reverse and forward translational opportunities to understand deficits of sleep spindle and TRN function in schizophrenia6,9 and serving as a key step toward testing the therapeutic hypothesis that normalizing sleep spindles by targeting CaV3.3 and/or TRN function may lead to novel strategies for treating schizophrenia patients.
Methods
All methods are listed in the Supplementary Methods section. The spindle analysis pipeline and a schematic of wavelet-based spindle detection are illustrated in Supplementary Fig. 9 and Supplementary Fig. 10 respectively.
References
Cross-Disorder Group of the Psychiatric Genomics C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013).
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Destexhe, A., Contreras, D., Steriade, M., Sejnowski, T. J. & Huguenard, J. R. In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J. Neurosci. 16, 169–185 (1996).
Huguenard, J. R. & Prince, D. A. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J. Neurosci. 14, 5485–5502 (1994).
Halassa, M. M. et al. State-dependent architecture of thalamic reticular subnetworks. Cell 158, 808–821 (2014).
Purcell, S. M. et al. Characterizing sleep spindles in 11,630 individuals from the National Sleep Research Resource. Nat. Commun. 8, 15930 (2017).
Latchoumane, C. V., Ngo, H. V., Born, J. & Shin, H. S. Thalamic spindles promote memory formation during sleep through triple phase-locking of cortical, thalamic, and hippocampal rhythms. Neuron 95, 424–435 e426 (2017).
Cairney, S. A., Guttesen, A. A. V., El Marj, N. & Staresina, B. P. Memory consolidation is linked to spindle-mediated information processing during sleep. Curr. Biol. 28, 948–954 e944 (2018).
Manoach, D. S., Pan, J. Q., Purcell, S. M. & Stickgold, R. Reduced sleep spindles in schizophrenia: a treatable endophenotype that links risk genes to impaired cognition? Biol. Psychiatry 80, 599–608 (2016).
Poulin, J., Daoust, A. M., Forest, G., Stip, E. & Godbout, R. Sleep architecture and its clinical correlates in first episode and neuroleptic-naive patients with schizophrenia. Schizophr. Res. 62, 147–153 (2003).
Forest, G. et al. Attention and non-REM sleep in neuroleptic-naive persons with schizophrenia and control participants. Psychiatry Res. 149, 33–40 (2007).
Lunsford-Avery, J. R. et al. Sleep dysfunction and thalamic abnormalities in adolescents at ultra high-risk for psychosis. Schizophr. Res. 151, 148–153 (2013).
Cao, H. et al. Cerebello-thalamo-cortical hyperconnectivity as a state-independent functional neural signature for psychosis prediction and characterization. Nat. Commun. 9, 3836 (2018).
Baran, B. et al. Spared and impaired sleep-dependent memory consolidation in schizophrenia. Schizophr. Res. 199, 83–89 (2018).
Goder, R. et al. Impairment of sleep-related memory consolidation in schizophrenia: relevance of sleep spindles? Sleep. Med. 16, 564–569 (2015).
Manoach, D. S. et al. Sleep spindle deficits in antipsychotic-naive early course schizophrenia and in non-psychotic first-degree relatives. Front. Hum. Neurosci. 8, 762 (2014).
Schilling, C. et al. Fast sleep spindle reduction in schizophrenia and healthy first-degree relatives: association with impaired cognitive function and potential intermediate phenotype. Eur. Arch. Psychiatry Clin. Neurosci. 267, 213–224 (2017).
Wamsley, E. J. et al. Reduced sleep spindles and spindle coherence in schizophrenia: mechanisms of impaired memory consolidation? Biol. Psychiatry 71, 154–161 (2012).
Astori, S. et al. The Ca(V)3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc. Natl Acad. Sci. USA 108, 13823–13828 (2011).
Tatsuki, F. et al. Involvement of Ca(2+)-dependent hyperpolarization in sleep duration in mammals. Neuron 90, 70–85 (2016).
Pellegrini, C., Lecci, S., Luthi, A. & Astori, S. Suppression of sleep spindle rhythmogenesis in mice with deletion of CaV3.2 and CaV3.3 T-type Ca(2+) channels. Sleep 39, 875–885 (2016).
Andrade, A. et al. A rare schizophrenia risk variant of CACNA1I disrupts CaV3.3 channel activity. Sci. Rep. 6, 34233 (2016).
Lander, E. S. The Heroes of CRISPR. Cell 164, 18–28 (2016).
Barkat, T. R., Polley, D. B. & Hensch, T. K. A critical period for auditory thalamocortical connectivity. Nat. Neurosci. 14, 1189–1194 (2011).
Fox, K. A critical period for experience-dependent synaptic plasticity in rat barrel cortex. J. Neurosci. 12, 1826–1838 (1992).
Hensch, T. K. Critical period mechanisms in developing visual cortex. Curr. Top. Dev. Biol. 69, 215–237 (2005).
Liu, X. B., Murray, K. D. & Jones, E. G. Low-threshold calcium channel subunit Ca(v) 3.3 is specifically localized in GABAergic neurons of rodent thalamus and cerebral cortex. J. Comp. Neurol. 519, 1181–1195 (2011).
Sun, Q. Q., Huguenard, J. R. & Prince, D. A. Neuropeptide Y receptors differentially modulate G-protein-activated inwardly rectifying K+ channels and high-voltage-activated Ca2+ channels in rat thalamic neurons. J. Physiol. 531, 67–79 (2001).
Talley, E. M. et al. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J. Neurosci. 19, 1895–1911 (1999).
Huguenard, J. R. & Prince, D. A. A novel T-type current underlies prolonged Ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J. Neurosci. 12, 3804–3817 (1992).
Lee, S. E. et al. Rebound burst firing in the reticular thalamus is not essential for pharmacological absence seizures in mice. Proc. Natl Acad. Sci. USA 111, 11828–11833 (2014).
Cueni, L. et al. T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat. Neurosci. 11, 683–692 (2008).
Vyazovskiy, V. V., Achermann, P., Borbely, A. A. & Tobler, I. The dynamics of spindles and EEG slow-wave activity in NREM sleep in mice. Arch. Ital. Biol. 142, 511–523 (2004).
Gulsuner, S. et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013).
Wimmer, R. D. et al. Sustaining sleep spindles through enhanced SK2-channel activity consolidates sleep and elevates arousal threshold. J. Neurosci. 32, 13917–13928 (2012).
Winsky-Sommerer, R. et al. Normal sleep homeostasis and lack of epilepsy phenotype in GABA A receptor alpha3 subunit-knockout mice. Neuroscience 154, 595–605 (2008).
Cirelli, C. & Tononi, G. Cortical development, electroencephalogram rhythms, and the sleep/wake cycle. Biol. Psychiatry 77, 1071–1078 (2015).
Kim, D., Hwang, E., Lee, M., Sung, H. & Choi, J. H. Characterization of topographically specific sleep spindles in mice. Sleep 38, 85–96 (2015).
Ang, G. et al. Absent sleep EEG spindle activity in GluA1 (Gria1) knockout mice: relevance to neuropsychiatric disorders. Transl. Psychiatry 8, 154 (2018).
Steullet, P. et al. The thalamic reticular nucleus in schizophrenia and bipolar disorder: role of parvalbumin-expressing neuron networks and oxidative stress. Mol Psychiatry 23, 2057–2065 (2017).
Wells, M. F., Wimmer, R. D., Schmitt, L. I., Feng, G. & Halassa, M. M. Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/-) mice. Nature 532, 58–63 (2016).
Acknowledgements
The current work is supported by the following grants: R01 MH115045 (to J.Q.P.), R21 NS110355 (to J.Q.P.), U54 NS108874 (to J.Q.P), R01 HG005827 (to S.M.P.), R01 NS098505 (to G.F.), R01 MH097104 (to G.F.), IK2 BX002130 and I01 BX004500 (to J.M.M.), I01 BX001356 (to R.E.B.), I01 BX002774 (to R.E.S.), R01 MH039683 (to R.E.B.), T32 HL007901 and IK2 BX004905 (to D.S.U.), P01 HL095491 (to R.E.S.) and Stanley Center for Psychiatric Research. D.S.U., J.M.M., R.E.S. and R.E.B. are Research Health Scientists at VA Boston Healthcare System, West Roxbury, MA. The contents of this work do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. We thank Carlton Goold, Daniel Curtis, Robert W. McCarley, Radhika Basheer, and Fumi Katsuki for their contributions to this project.
Author information
Authors and Affiliations
Contributions
Q.Z., J.M.H., X.M. and T.B.N. generated the mutant mouse lines. L.Y., D.B.-N. and A.A. designed and performed cellular and molecular characterizations. L.Y. and M.F. performed and analyzed biochemical studies. A.G., V.G.L.-H., M.A.A.-G. and M.F. performed slice electrophysiology studies, and A.G. and M.F. analyzed the data. D.S.U., J.M.M., S.C., K.I., R.E.S., R.E.B. R.E.S. and S.M.P. contributed to the design of the in vivo EEG experiments. D.S.U., K.I. and S.C. performed in vivo EEG experiments. S.M.P. analyzed in vivo EEG data together with D.S.U., K.Y., S.C. A.G., L.Y. and J.Q.P. designed the overall project with input from Z.F. and G.F. J.Q.P. supervised all experiments of the project. A.G., S.M.P. and J.Q.P. drafted the manuscript with editing contribution from all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghoshal, A., Uygun, D.S., Yang, L. et al. Effects of a patient-derived de novo coding alteration of CACNA1I in mice connect a schizophrenia risk gene with sleep spindle deficits. Transl Psychiatry 10, 29 (2020). https://doi.org/10.1038/s41398-020-0685-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-0685-1
This article is cited by
-
IntelliSleepScorer, a software package with a graphic user interface for automated sleep stage scoring in mice based on a light gradient boosting machine algorithm
Scientific Reports (2023)
-
Sleep spindles in people with schizophrenia, schizoaffective disorders or bipolar disorders: a pilot study in a general population-based cohort
BMC Psychiatry (2022)
-
Alterations in TRN-anterodorsal thalamocortical circuits affect sleep architecture and homeostatic processes in oxidative stress vulnerable Gclm−/− mice
Molecular Psychiatry (2022)
-
Developmental oxidative stress leads to T-type Ca2+ channel hypofunction in thalamic reticular nucleus of mouse models pertinent to schizophrenia
Molecular Psychiatry (2022)
-
Association of polygenic risk for schizophrenia with fast sleep spindle density depends on pro-cognitive variants
European Archives of Psychiatry and Clinical Neuroscience (2022)
